

Abstract
Genetic fumarylacetoacetate hydrolase (Fah) deficiency is unique in that healthy gene-corrected hepatocytes have a strong growth advantage and can repopulate the diseased liver. Unfortunately, similar positive selection of gene-corrected cells is absent in most inborn errors of liver metabolism and it is difficult to reach the cell replacement index required for therapeutic benefit. Therefore, methods to transiently create a growth advantage for genetically modified hepatocytes in any genetic background would be advantageous. To mimic the selective pressure of Fah deficiency in normal animals, an efficient in vivo small molecule inhibitor of FAH, 4-[(2-carboxyethyl)-hydroxyphosphinyl]-3-oxobutyrate (CEHPOBA) was developed. Microarray analysis demonstrated that pharmacological inhibition of FAH produced highly similar gene expression changes to genetic deficiency. As proof of principle, hepatocytes lacking homogentisic acid dioxygenase (Hgd) and hence resistant to FAH inhibition were transplanted into sex-mismatched wild-type recipients. Time course analyses of 4–6 weeks of CEHPOBA administration after transplantation showed a linear relationship between treatment length and replacement index. Compared to controls, recipients treated with the FAH-inhibitor had 20–100-fold increases in liver repopulation. We conclude that pharmacological inhibition of FAH is a promising approach to in vivo selection of hepatocytes.
Introduction
Many inborn errors of metabolism are caused by deficiencies in hepatic enzymes, thereby making orthotopic liver transplant an effective therapy for these conditions.1 However, few patients benefit from this procedure due to the limited availability of donor organs, the high cost and significant morbidity and mortality associated with the procedure, as well as the long-term requirement for immune suppression.2 Most pediatric metabolic diseases are caused by a specific loss-of-function in only hepatocytes with otherwise normal liver anatomy. Thus, whole organ transplant is often unnecessary and restoration of enzymatic function through gene therapy or hepatocyte replacement would be more appropriate.
Hepatocyte transplantation is easily studied in animal models, including the murine models of the metabolic disease hereditary tyrosinemia type I (HTI). HTI is a tyrosine catabolism disorder caused by deficiency of fumarylacetoacetate hydrolase (Fah), the terminal enzyme in the pathway, which causes progressive liver disease and renal tubular dysfunction.3 In both HTI patients and Fah–/– mice, the hepatotoxin fumarylacetoacetate (FAA) and subsequent metabolites accumulate, causing cell cycle arrest and death in a cell-autonomous manner.3 Treatment with 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), an inhibitor that blocks the 4-hydroxyphenylpyruvate dioxygenase enzyme upstream of FAH, prevents FAA accumulation and rescues the phenotype.4 FAH+ cells have a strong selective advantage in an HTI liver that can be exploited in patients and animal models in vivo.5 Indeed, many patients with HTI have a somatic mosaic liver consisting of both FAH+ and FAH– hepatic tissue. These FAH+ nodules are generated by somatic reversion of the disease causing inherited mutation followed by clonal selection of reverted hepatocytes.6 The growth advantage of FAH-expressing cells in this system has been extensively utilized to select for hepatocytes modified by gene therapy including retroviral vectors,5 adenoviral vectors,7 adeno-associated virus,8 sleeping beauty transposase,9 and phiC31-integrase.10
To achieve therapeutic liver repopulation outside the context of the Fah–/– mouse for use in metabolic selection studies during cell transplantation, we previously generated a small molecule inhibitor of FAH, 4-[(2-carboxyethyl)-hydroxyphosphinyl]-3-oxobutyrate (CEHPOBA).11 CEHPOBA is a transition state analogue of the enzyme's substrate FAA and works at nanomolar concentrations with favorable kinetics (i.e., a long half-life that permits once daily administration). The Km of FAH for FAA is 3.5 µmol/l and the Ki of CEHPOBA is only 41 nmol/l. In addition, the crystal structure of FAH with CEHPOBA bound to the enzyme active site has been determined.12
Here we tested whether chronic CEHPOBA administration could select genetically resistant hepatocytes in wild-type mice in vivo. As proof of principle, Hgd–/– hepatocytes were used for transplantation because they are unable to convert tyrosine to FAA.13
Results
CEHPOBA administration efficiently blocks FAH enzyme activity in wild-type mice
In order to determine whether CEHPOBA could be used for long-term in vivo inhibition of FAH, several functional assays were performed. First, its effects were confirmed in an in vitro enzyme assay. Livers from wild-type mice were homogenized and FAH activity was measured with and without CEHPOBA added (Figure 1a). Untreated wild-type liver extracts had normal FAH enzyme activity, but the addition of the inhibitor completely blocked the degradation of FAA as expected. Next, we wished to determine how long the effect of a single injection of CEHPOBA would last in vivo. Wild-type mice were administered a single 1-µmol/g dose of CEHPOBA by intraperitoneal (i.p.) injection and the livers were harvested at various time points following treatment (Figure 1b). Hepatic FAH enzyme activity dropped precipitously immediately after administration of CEHPOBA, but the maximum inhibitory effect was seen after 4-hour. Reduced FAH activity levels were maintained for at least 72-hour. While the short-term effects of CEHPOBA were promising, the effects of its chronic administration were unknown. In order to be useful for selection, the effects of long-term dosing should closely mimic those of genetic Fah deficiency. To address this point, we performed microarray analysis on the livers of wild-type mice treated with CEHPOBA for 2 weeks and compared the patterns to Fah–/– mice. Four treatment cohorts of two mice each were assayed for differences in hepatic gene expression: Fah–/– mice off NTBC for 2 weeks to induce the gene expression changes typical of genetic HTI,14 Fah–/– mice kept on NTBC and thus protected from HTI-related damage, control untreated wild-type mice, and wild-type mice given daily CEHPOBA injections for 2 weeks. Livers were harvested from all eight mice and the labeled cRNA was analyzed by microarray. Gratifyingly, we found that just 2 weeks of CEHPOBA administration resulted in a gene expression pattern very similar to genetic Fah deficiency. Both CEHPOBA treatment and Fah deficiency resulted in massive changes to hepatic gene expression with considerable overlap (>80%) in the genes that were up- and downregulated. Cluster analysis clearly indicated that wild-type mice treated with CEHPOBA were most similar in hepatic gene expression to tyrosinemic Fah–/– mice off NTBC (Figure 2a). The heat map illustrates the levels of similarity and difference between the four treatment cohorts (Figure 2b). Several key genes affected by tyrosinemia were confirmed by quantitative PCR (Supplementary Table S1).
Figure 1.

Fumarylacetoacetate hydrolase (FAH) enzyme assay confirms 4-[(2-carboxyethyl)-hydroxyphosphinyl]-3-oxobutyrate (CEHPOBA) function both in vitro and in vivo. (a) Livers from adult wild-type mice analyzed for levels of FAH enzyme activity with and without CEHPOBA in vitro. Cytosolic liver homogenate was incubated with fumarylacetoacetate substrate and the rate of disappearance was measured spectrophotometrically. FAA degradation was blocked by CEHPOBA. (b) Wild-type mice were treated with a single injection of CEHPOBA at 1-µmole/g and the liver was assayed for the remaining FAH enzymatic activity.
Figure 2.

Microarray gene expression data illustrate the similarities between tyrosinemic mice and 4-[(2-carboxyethyl)-hydroxyphosphinyl]-3-oxobutyrate (CEHPOBA)-treated wild-type mice. (a) Microarray cluster analysis and (b) heat map comparing cohorts of Fah–/– mice on 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC) for 2 weeks, Fah–/– mice off NTBC for 2 weeks, untreated wild-type mice and wild-type mice treated with CEHPOBA daily for 2 weeks. Green = upregulated; red = downregulated relative to the control cohort of Fah–/– mice on NTBC. FAH, fumarylacetoacetate hydrolase.
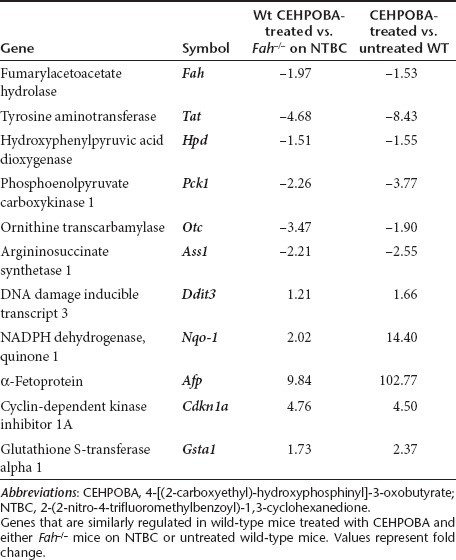
HTI patients and Fah–/– mice have a characteristic hepatic gene expression profile off NTBC.14 Notably, Fah expression is completely absent, cAMP-inducible genes (Tat, Pck1) are down-regulated along with urea cycle enzymes (Ass1, Otc), whereas prominent up-regulation is seen in genes for DNA damage (Ddit3), oxidative damage (Nqo-1), and liver proliferation (Afp). Importantly, wild-type mice given a 2-week course of CEHPOBA mimicked these prototypical gene expression changes (Table 1). In addition, p21 was upregulated, and it is known from other work that p21 is crucial for blocking hepatocyte cell division in Fah–/– mice15 and thus essential for the growth advantage of transplanted cells. Untransplanted control mice treated with only daily CEHPOBA for 2 weeks were unable to compensate for a 2/3 partial hepatectomy for >48-hour, indicating impaired cell proliferation (data not shown). Overall, chronic daily administration of a single dose of CEHPOBA resulted in a highly similar gene expression pattern to that seen in severe genetic Fah deficiency. This regimen therefore appeared suitable for the purpose of achieving in vivo growth selection of hepatocytes.
Table 1. Effects of CEHPOBA treatment on liver gene expression.
CEHPOBA selects for transplanted Hgd–/– hepatocytes in wild-type mice in vivo
To test the principle of transient pharmacological FAH deficiency to provide positive selection in an otherwise nonselective setting, the following in vivo experiment was devised. Adult female wild-type mice (Fah+/+) were transplanted with hepatocytes from male Hgd–/– donors and given a 4-6 week course of daily CEHPOBA injections. HGD is the third enzyme in the tyrosine catabolic pathway and its deficiency causes alkaptonuria, a condition that lacks any liver disease.3 Previous work has shown that Hgd–/– hepatocytes are protected from FAH-deficiency and are positively selected in the Fah–/– mouse model in vivo.13 Hgd–/– cells cannot convert tyrosine to the hepatotoxic substrate FAA. We hypothesized that CEHPOBA treatment would render both host and donor hepatocytes FAH deficient, resulting in the accumulation of FAA in the host wild-type hepatocytes and producing cell-cycle arrest. In contrast, Hgd–/– cells would be protected and be able to proliferate normally, providing them with a selective advantage. The donor contribution over time was quantitated using quantitative PCR for the Y-chromosome. As expected, donor hepatocytes were strongly selected when transplanted into genetically defective Fah–/– mice (data not shown). Hgd–/– hepatocytes were also selected in CEHPOBA-treated wild-type mice. The time course analysis demonstrated a linear relationship between the length of treatment and degree of repopulation. Compared to controls receiving transplant alone, CEHPOBA-treated transplant recipients had 20–100-fold increases in the degree of repopulation (Figure 3) as measured by percent Y-chromosome positivity by quantitative PCR.
Figure 3.

Kinetics of liver repopulation. The percentage of Y-chromosome donor marker was quantified by quantitative PCR (qPCR) from wild-type mice transplanted (Tx) with Hgd–/– hepatocytes, with or without a 4-6 week course of 4-[(2-carboxyethyl)-hydroxyphosphinyl]-3-oxobutyrate (CEHPOBA). Mean ± SD are shown, with the number of independent animals analyzed above each bar. Black, Tx alone; white, Tx+ CEHPOBA.
To ensure that results were not due to an immune-mediated factor, given that the Hgd–/– donor hepatocytes were derived from a mixed strain background, we repeated the experiment using immune-deficient NSG recipient mice. Female NSG recipients were transplanted with Hgd–/– donor hepatocytes and given a 3-week course of daily CEHPOBA. Compared to controls receiving transplant alone, CEHPOBA-treated transplant recipients again had a near tenfold increase in the degree of repopulation as measured by percent Y-chromosome positivity by quantitative PCR (Supplementary Table S2). This indicated that no immune-mediated factor was involved in the donor cell selection. To validate the PCR results by a different method, Y-chromosome FISH was also performed. Figure 4 shows representative Y chromosome FISH in control mice (male XY, female XX) (Figure 4a,c), CEHPOBA-treated transplanted NSG mice (female XX) (Figure 4b), and untreated transplanted NSG mice (female XX) (Figure 4d). Significant numbers of Y-chromosome positive hepatocytes could only be found in those transplant recipients who also received CEHPOBA treatment (Supplementary Table S2).
Figure 4.

Chromosomal and immunohistochemical staining of the liver. (a) Control Y-chromosome FISH in an untreated male wild-type mouse (blue, DAPI; pink, Y chromosome with white arrows to highlight Y-chromosome positive nuclei); (b) Y-chromosome stain from a female NSG mouse transplanted with male Hgd–/– hepatocytes and a 5 week course of 4-[(2-carboxyethyl)-hydroxyphosphinyl]-3-oxobutyrate (CEHPOBA); (c) control Y-chromosome FISH in an untreated female wild-type mouse; (d) Y-chromosome stain from a female NSG mouse transplanted with male Hgd–/– hepatocytes; (e) Hematoxylin and eosin (H&E) stain of liver from a female NSG mouse transplanted with male Hgd–/– hepatocytes and treated with a 5-week course of CEHPOBA; the liver architecture is normal; (f) H&E stain of kidney from a treated with a 5 week course of CEHPOBA; the kidney architecture is normal.
CEHPOBA treatment leaves no permanent hepatic or renal injury
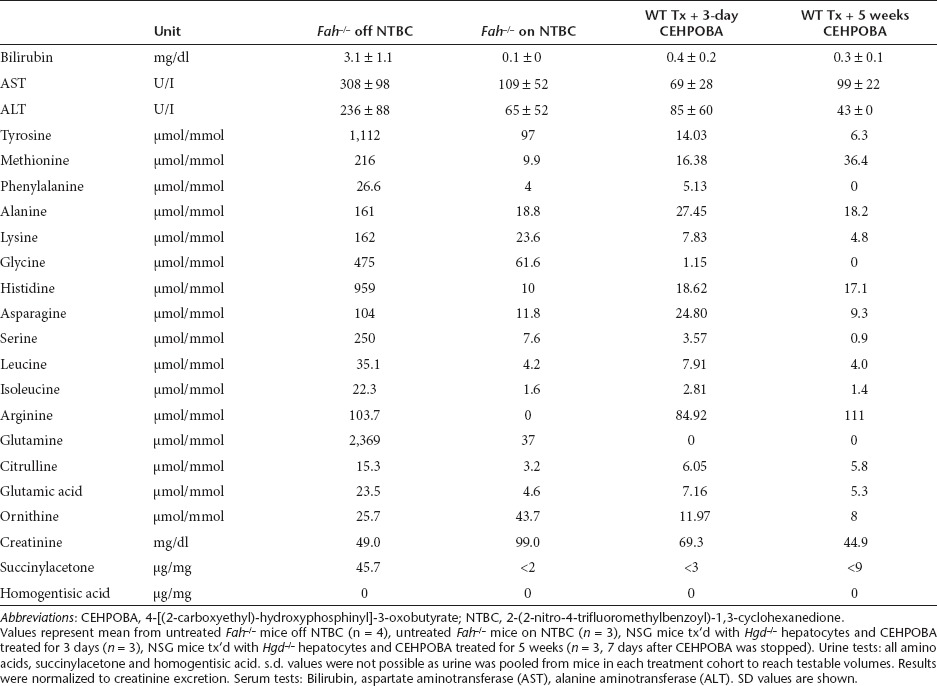
For a hepatic selection protocol to have any clinical potential, it should induce no lasting tissue damage/injury. In order to determine whether CEHPOBA was safe in this regard, chronically (6 weeks) drug-treated mice were analyzed 1 week after cessation of the injections. Histological analysis of inhibitor-treated mice (Figure 4e,f) showed a lack of permanent damage from CEHPOBA in both the liver and kidney. Serology and urinalysis to examine potential hepatorenal toxicity demonstrated that the engrafted hepatocytes could function and that CEHPOBA treatment lacked detectable long-term toxicity (Table 2). Additionally, two control nontransplanted mice treated with 2-weeks of CEHPOBA were monitored for 8 months and no evidence of hepatocellular carcinoma was found by pathological analysis (data not shown).
Table 2. Measures of functional restoration after cessation of CEHPOBA treatment.
Discussion
Clinical hepatocyte transplantation has many advantages over that of the whole organ:16 (i) the procedure is less invasive; (ii) cells from a single donor liver could be used for multiple recipients; (iii) human hepatocytes can be farmed in mice providing a constant reliable source of transplantable hepatocytes;17 (iv) hepatocytes can be cryopreserved with little loss in viability; and (v) cell suspensions may be less likely to induce an immune response than whole organs. In addition, cell transplantation can be achieved using the patient's own cells, whereby the cells are removed from the body, genetically manipulated, and then transplanted back into the patient's liver.18 Similarly, gene therapy directed at the liver has great potential for inborn errors of metabolism and could replace orthotopic transplantation for these disorders. For these approaches to be clinically successful, however, a critical percentage of host hepatocytes must be replaced or gene-corrected. This therapeutic threshold varies between different disorders and can be as low as 2% in the hemophilias19 and as high as 50% in the urea cycle disorders.20 In unmodified hepatocyte transplantation, replacement indices are well below 1%,21 and in gene therapy, very high vector doses are required to achieve transduction of high percentages of hepatocytes.22 rAAV vectors are currently the most promising for hepatic gene therapy,23 but this system has the additional problem that most of the rAAV remains episomal and is predicted to be lost over time.
The current disadvantages of cell and gene therapy for genetic liver diseases could be overcome if the healthy hepatocytes could be positively selected in vivo. A natural selective advantage exists in a subset of these disorders including HTI, Wilson's disease,24 progressive familial intrahepatic cholestasis25 and α1-antitrypsin deficiency.26 These examples clearly demonstrate that high levels of liver reconstitution can be achieved starting with very low numbers of gene-corrected cells. For bone marrow transplantation, conditioning regimens have been developed which permit a low number of transplanted donor cells to reconstitute the entire hematopoietic system. Several attempts have been made to develop similar regimens for the liver. Traditional hepatocyte-specific conditioning regimens such as acetaminophen, galactosamine, carbon tetrachloride, thioacetamide and Amanita mushroom poison do not work because they all cause severe acute hepatic failure.27 Successful liver repopulation takes weeks to months to achieve and patients simply cannot survive without hepatocellular function for this duration. Other more hepatocyte-specific conditioning regimens such as retrorsine, monocrotaline, and other pyrrolizidine alkaloids are useful in that they maintain sufficient liver function during conditioning, yet their potent DNA-damage and hepatocarcinogenicity render them inappropriate for the clinic.28
Recently, local hepatic irradiation has shown some potential for conditioning before hepatocyte transplantation.29 However, this approach requires pre-treatment of the host and is not suitable to selection in the context of in vivo gene therapy. An ideal in vivo hepatocyte selection system would involve a liver-specific toxin in combination with a protective genetic element. Such combinations have been described for both the hepatic and hematopoietic systems. The work of several groups (reviewed in ref. 30) has shown that genetic modifications to the DNA repair gene methylguanine methyltransferase (Mgmt) allows for genetic resistance to the chemotherapeutic O6-benzylguanine (O6BG). The combined approach of transducing bone marrow with a O6BG-resistant MGMT variant by retrovirus and then treating the O6BG-sensitive tumor to inhibit its wild-type MGMT activity, showed increases in the therapeutic index in mouse tumor xenograft models.31 Similarly, a study by Mitchell et al in 2000 elegantly showed the ability to selectively repopulate an apolipoprotein-E-deficient mouse liver using antiapoptotic protection with transgenic Fas/CD95-resistant hepatocytes expressing apolipoprotein-E.32 Daily injections of nonlethal doses of Jo2 antibody (a Fas agonist) provided a selective advantage for donor hepatocytes and resulted in normalization of the hypercholesterolemia and atherosclerosis in the mice. However, the use of an anti-apoptotic and cancer causing Bcl2 transgene in this system limits its clinical potential.
The tyrosine catabolic pathway is highly conserved in mammals and represents an intriguing target for the development of a drug-resistance gene system analogous to the O6BG/Mgmt combination. Genetic and pharmacologic studies have shown that deficiency of either Hpd33 or Hgd13 completely protects hepatocytes from FAH deficiency. In fact, pharmacological inhibition of Hpd is the basis for the US Food and Drug Administration-approved treatment of human HTI with NTBC.14 Here we show the successful development of a FAH-inhibitor that can achieve drug-induced transient HTI. In our current proof-of-principle study, we used Hgd–/– hepatocytes that are genetically resistant to FAH deficiency. The next step will be to test gene cassettes that can inhibit tyrosine catabolic enzymes upstream of Fah, i.e., tyrosine amino transferase (Tat), Hpd or Hgd. This could be achieved by short hairpin RNA -mediated knockdown of one or more of these genes. Short hairpin RNA vectors have successfully been used in the liver on numerous occasions.34,35
How safe would this approach be in humans? Most human HTI patients experience extended periods (weeks to years) without therapy prior to diagnosis. The effects of FAH deficiency are fully reversible if NTBC is started before 6 months of age and no tumor development has been observed in this group.36 Thus, short-term use of pharmacological FAH inhibitors like CEHPOBA has clinical potential and more detailed toxicology studies of the compound should be conducted in the future.
Materials and Methods
Mouse strains and animal husbandry. Mouse strains used were C57BL/6J and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ37 (herein referred to as WT and NSG mice, respectively) available from Jackson Labs (Bar Harbor, ME), FahΔexon5/FahΔexon5 (herein referred to as Fah–/– mice38) backcrossed ten generations onto a C57BL/6J background, and Hgdaku/Hgdaku (herein referred to as Hgd–/– mice39) on a mixed 129/Sv.C57BL/6J.NB background. All mice described were maintained on irradiated high fat low protein mouse chow (Lab Diet, Brentwood, MO, Cat# Picolab 5LJ5) ad libitum to decrease flux through the tyrosine pathway. NSG mice are immune-compromised and so were maintained on a rotating biweekly water schedule to prevent growth of various bacterial species. Beginning on the day of transplantation, NSG mice were maintained on 1 week of acidified water to prevent the growth of Pseudomonas aeruginosa. The following week, NSG mice were switched to 1 week of 8-mg/l SMX-TMP water (supplemented with 0.7 mol/l dextrose for palatability) for broad-spectrum bacterial protection. Fah–/– mice are maintained on 4-mg/l NTBC water as described.40 The Institutional Animal Care & Use Committee of Oregon Health & Science University approved all mouse procedures.
CEHPOBA. CEHPOBA was synthesized and purified at the Arthur F. Scott Laboratory of Chemistry at Reed College as previously described.11 All mice were given CEHPOBA at 1-µmol/g of a 0.22 mol/l stock solution by i.p. injection. CEHPOBA stocks were solubilized in sterile water, pH 7.8, and stored at –80 °C until use. Detailed pharmacokinetic data11 and crystal structures12 were described previously.
Hepatocyte transplantation. Donor hepatocytes were acquired from male Hgd–/– mice by collagenase liver perfusion as described,41 and 4.5 × 105 hepatocytes were transplanted into female NSG, WT, or Fah–/– recipients by intrasplenic injection as described.42 Pre- and postsurgical pharmacologics were administered as follows: ceftiofur (broad-spectrum cephalosporin antibiotic) was given at 4-mg/kg by i.p. injection immediately prior to surgery and for two days following surgery; anakinra (anti-inflammatory IL-1 receptor antagonist) was given at 75-mg/kg by intrasplenic injection with the cells at transplant and by i.p. injection 2 days postsurgery. CEHPOBA injections, if administered, began 3 days following transplant to allow mice to recover from surgery prior to selection. When mice receiving CEHPOBA lost >20% of their pretransplant weight, treatment was suspended for 3–7 days to allow mice to recover before further selection began. All Hgd–/– hepatocyte preparations were verified by transplantation into Fah–/– mice to ensure viability and engraftability. Procedures for 2/3 partial hepatectomy were performed as described.43
FAH enzyme assay. Livers of treated mice were harvested and snap frozen in liquid nitrogen until the assay was performed. Cytosolic liver homogenate was incubated with 250-µmol/l FAA substrate and the rate of disappearance was measured spectrophotometrically at 330-nm as described.17 Livers from untreated wild-type and Fah–/– mice were used for positive and negative assay controls.
Liver immunohistochemistry. In all experimental mice, a minimum of two liver sections from varying depths and randomly selected liver lobes were analyzed for Y-chromosome positivity and structural integrity by hematoxylin and eosin. Mouse Y chromosome FISH was performed on 4-µm paraffin sections using a Cy-3 labeled mouse Y-IDetect paint (ID Labs, Ontario, Canada) and counter-stained with DAPI (Abbott Molecular, Des Plaines, IL). Deparaffinization of paraffin slides was performed using the Vysis Paraffin Pretreatment Reagent Kit II (Abbott Molecular) using the VP2000 Automated Processor (Abbott Molecular) according to manufacturer's instructions. After these treatments, hybridization, washing, and counterstaining were performed according to the FISH probe manufacturer's instructions. Fluorescence was visualized on a CytoVysion image capture system (Applied Imaging, San Jose, CA) with a Nikon E800 microscope (Nikon, Melville, NY). Hematoxylin and eosin staining was completed as described,44 and hematoxylin and eosin images were taken on a DM IL LED microscope (Leica, Buffalo Grove, IL) using Leica LAS Image Analysis Software.
Biochemical parameters. Sterile urine was collected by cystocentesis at time of harvest. Quantitative urine amino acids were analyzed on a Waters ACQUITY UPLC using the Waters MassTrak Amino Acid Analysis System as described.45 Quantitative measurements of the urine organic acids succinylacetone and homogentisic acid were measured by trimethylsilyl derivatization followed by gas chromatography-mass spectrometry as described.46 The lowest limit of detection for HGA was 40-µg/mg creatinine. All urine parameters were normalized to creatinine excretion using the DetectX Urinary Creatinine Detection Kit Catalog #K002-H1 (Arbor Assays, Ann Arbor, MI). Blood for serology was collected by terminal cardiac puncture at time of harvest. Serum measurements of total bilirubin and transaminases were performed as described.14
Statistical analysis. Statistical analyses were conducted with GraphPad Prism software v.4.0 (GraphPad, San Diego, CA). Experimental differences were evaluated by Student two-tailed t-test assuming equal variance. P values <0.05 were considered statistically significant.
Y chromosome q-PCR. Total DNA was isolated from randomly dissected liver tissue with a MasterPure DNA Purification kit (Epicentre, Madison, WI). Genomic DNA (100-ng) was subjected to a two-step PCR amplification under the following conditions: 1 cycle 95 °C 3 minutes; 45 cycles 95 °C 15 seconds and 68 °C 40 seconds. Primers: Y-chr F: 5′-TCCTTGGGCTCTTCATT ATTCTTAAC-3′ Y-chr R: 5′-GAGAACCACGTTGGTTTGAGATG-3′ Actb F: 5′-AGAGGGAAATC GTGCGTGAC-3′ Actb R: 5′-CAATAGTGATGA CCTGGCCGT-3′. Dilutions of male genomic DNA into female genomic DNA were used to generate the standard curve. Results were normalized to β-actin expression. PCR was performed on an iQ5 Multicolor Real-Time PCR (Bio-Rad, Hercules, CA), using iQ5 Standard Edition Software, v.2.0.
Microarray analysis. The four treatment cohorts analyzed were Fah–/– on NTBC for 2 weeks, Fah–/– off NTBC for 2 weeks, untreated wild-type mice and wild-type mice treated with CEHPOBA daily for 2 weeks. Amplified cDNA was prepared from RNA from two mice in each cohort using the WT-Ovation Pico Amplification system (NuGEN Technologies, San Carlos, CA). Labeled target cRNA was prepared from total RNA from two mice in each cohort using the AMC One Cycle cDNA IVT Amplification/Labeling Kit (Affymetrix, Santa Clara, CA). Labeled samples were hybridized overnight to a Mouse Genome 430 2.0 GeneChip Array (Affymetrix). Arrays were washed, scanned, and median intensities of each element on the array were captured with Affymetrix GCOS software v.1.2. Quality control diagnostic plots were prepared for each array, and those failing to exhibit high-quality hybridizations were excluded. Microarray assays were performed in the Affymetrix Microarray Core of the OHSU Gene Microarray Shared Resource (Portland, OR). For statistical analysis, CEL files were processed with the RMA method47 in the Affymetrix Bioconductor package. Statistics were applied using false discovery rate48 adjusted P values from an empirical Bayes49 moderated t-test. The adjusted P value (q-Val) threshold used was 0.01 and the fold change threshold used was 2.0.50
SUPPLEMENTARY MATERIAL Table S1. Confirmatory qPCR for several key genes from microarray results. Table S2. Y-chromosome qPCR and FISH results in immune-deficient recipients.
Acknowledgments
We thank Angela Major (Texas Children's Hospital) for histology support, the Molecular Morphology Core (Texas Gulf Coast Digestive Diseases Center) for serological analysis, William Fleming (Oregon Health & Science University) for the NSG mice, and Xavier Montagutelli (Pasteur Institute of Paris) for the Hgd–/– mice. This work was supported by grants from the National Institute of Diabetes, Digestive & Kidney Diseases to M.G. (R01-DK48252), M.J.F. (R01-DK56338), and C.O.H (HD-057033), as well as the National Cancer Institute to N.K.P. (F31-CA130116). The funding organizations played no role in experimental design, data analysis, or manuscript preparation. This work was performed in Portland, OR and Houston, TX. The authors declared no conflict of interest.
Supplementary Material
Confirmatory qPCR for several key genes from microarray results.
Y-chromosome qPCR and FISH results in immune-deficient recipients.
REFERENCES
- Shneider BL. Pediatric liver transplantation in metabolic disease: clinical decision making. Pediatr Transplant. 2002;6:25–29. doi: 10.1034/j.1399-3046.2002.1p057.x. [DOI] [PubMed] [Google Scholar]
- Busuttil RW, Klintmalm GB, Lake JR, Miller CM., and, Porayko M. General guidelines for the use of tacrolimus in adult liver transplant patients. Transplantation. 1996;61:845–847. doi: 10.1097/00007890-199603150-00032. [DOI] [PubMed] [Google Scholar]
- Grompe M. The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin Liver Dis. 2001;21:563–571. doi: 10.1055/s-2001-19035. [DOI] [PubMed] [Google Scholar]
- Al-Dhalimy M, Overturf K, Finegold M., and, Grompe M. Long-term therapy with NTBC and tyrosine-restricted diet in a murine model of hereditary tyrosinemia type I. Mol Genet Metab. 2002;75:38–45. doi: 10.1006/mgme.2001.3266. [DOI] [PubMed] [Google Scholar]
- Overturf K, Al-Dhalimy M, Tanguay R, Brantly M, Ou CN, Finegold M.et al. (1996Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinaemia type I Nat Genet 12266–273. [DOI] [PubMed] [Google Scholar]
- Kvittingen EA, Rootwelt H, Brandtzaeg P, Bergan A., and, Berger R. Hereditary tyrosinemia type I. Self-induced correction of the fumarylacetoacetase defect. J Clin Invest. 1993;91:1816–1821. doi: 10.1172/JCI116393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overturf K, al-Dhalimy M, Ou CN, Finegold M, Tanguay R, Lieber A.et al. (1997Adenovirus-mediated gene therapy in a mouse model of hereditary tyrosinemia type I Hum Gene Ther 8513–521. [DOI] [PubMed] [Google Scholar]
- Paulk NK, Wursthorn K, Wang Z, Finegold MJ, Kay MA., and, Grompe M. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 2010;51:1200–1208. doi: 10.1002/hep.23481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E, Held PK, Noll M, Morcinek N, Al-Dhalimy M, Finegold M.et al. (2002In vivo correction of murine tyrosinemia type I by DNA-mediated transposition Mol Ther 6759–769. [DOI] [PubMed] [Google Scholar]
- Held PK, Olivares EC, Aguilar CP, Finegold M, Calos MP., and, Grompe M. In vivo correction of murine hereditary tyrosinemia type I by phiC31 integrase-mediated gene delivery. Mol Ther. 2005;11:399–408. doi: 10.1016/j.ymthe.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Bateman RL, Ashworth J, Witte JF, Baker LJ, Bhanumoorthy P, Timm DE.et al. (2007Slow-onset inhibition of fumarylacetoacetate hydrolase by phosphinate mimics of the tetrahedral intermediate: kinetics, crystal structure and pharmacokinetics Biochem J 402251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RL, Bhanumoorthy P, Witte JF, McClard RW, Grompe M., and, Timm DE. Mechanistic inferences from the crystal structure of fumarylacetoacetate hydrolase with a bound phosphorus-based inhibitor. J Biol Chem. 2001;276:15284–15291. doi: 10.1074/jbc.M007621200. [DOI] [PubMed] [Google Scholar]
- Manning K, Al-Dhalimy M, Finegold M., and, Grompe M. In vivo suppressor mutations correct a murine model of hereditary tyrosinemia type I. Proc Natl Acad Sci USA. 1999;96:11928–11933. doi: 10.1073/pnas.96.21.11928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grompe M, Lindstedt S, al-Dhalimy M, Kennaway NG, Papaconstantinou J, Torres-Ramos CA.et al. (1995Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I Nat Genet 10453–460. [DOI] [PubMed] [Google Scholar]
- Willenbring H, Sharma AD, Vogel A, Lee AY, Rothfuss A, Wang Z.et al. (2008Loss of p21 permits carcinogenesis from chronically damaged liver and kidney epithelial cells despite unchecked apoptosis Cancer Cell 1459–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grompe M. Principles of therapeutic liver repopulation. J Inherit Metab Dis. 2006;29:421–425. doi: 10.1007/s10545-006-0311-2. [DOI] [PubMed] [Google Scholar]
- Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E.et al. (2007Robust expansion of human hepatocytes in Fah–/–/Rag2–/–/Il2rg–/– mice Nat Biotechnol 25903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman M, Raper SE, Kozarsky K, Stein EA, Engelhardt JF, Muller D.et al. (1994Successful ex vivo gene therapy directed to liver in a patient with familial hypercholesterolaemia Nat Genet 6335–341. [DOI] [PubMed] [Google Scholar]
- Kay MA., and, High K. Gene therapy for the hemophilias. Proc Natl Acad Sci USA. 1999;96:9973–9975. doi: 10.1073/pnas.96.18.9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang H, Morizono H, Bell P, Jones D, Lin J.et al. (2012Sustained correction of OTC deficiency in spf(?ash) mice using optimized self-complementary AAV2/8 vectors Gene Ther 19404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox IJ, Chowdhury JR, Kaufman SS, Goertzen TC, Chowdhury NR, Warkentin PI.et al. (1998Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation N Engl J Med 3381422–1426. [DOI] [PubMed] [Google Scholar]
- Mingozzi F., and, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- Daya S., and, Berns KI. Gene therapy using adeno-associated virus vectors. Clin Microbiol Rev. 2008;21:583–593. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen KJ, Cheah DM, Wright PF, Gazeas S, Pettigrew-Buck NE, Deal YH.et al. (2004Liver cell transplantation leads to repopulation and functional correction in a mouse model of Wilson's disease J Gastroenterol Hepatol 191283–1290. [DOI] [PubMed] [Google Scholar]
- De Vree JM, Ottenhoff R, Bosma PJ, Smith AJ, Aten J., and, Oude Elferink RP. Correction of liver disease by hepatocyte transplantation in a mouse model of progressive familial intrahepatic cholestasis. Gastroenterology. 2000;119:1720–1730. doi: 10.1053/gast.2000.20222. [DOI] [PubMed] [Google Scholar]
- Lomas DA. The selective advantage of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2006;173:1072–1077. doi: 10.1164/rccm.200511-1797PP. [DOI] [PubMed] [Google Scholar]
- Tuñón MJ, Alvarez M, Culebras JM., and, González-Gallego J. An overview of animal models for investigating the pathogenesis and therapeutic strategies in acute hepatic failure. World J Gastroenterol. 2009;15:3086–3098. doi: 10.3748/wjg.15.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller L, Kasper P., and, Kaufmann G. The clastogenic potential in vitro of pyrrolizidine alkaloids employing hepatocyte metabolism. Mutat Res. 1992;282:169–176. doi: 10.1016/0165-7992(92)90091-u. [DOI] [PubMed] [Google Scholar]
- Yamanouchi K, Zhou H, Roy-Chowdhury N, Macaluso F, Liu L, Yamamoto T.et al. (2009Hepatic irradiation augments engraftment of donor cells following hepatocyte transplantation Hepatology 49258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino BP. Gene therapy to protect haematopoietic cells from cytotoxic cancer drugs. Nat Rev Cancer. 2002;2:431–441. doi: 10.1038/nrc823. [DOI] [PubMed] [Google Scholar]
- Koç ON, Reese JS, Davis BM, Liu L, Majczenko KJ., and, Gerson SL. DeltaMGMT-transduced bone marrow infusion increases tolerance to O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea and allows intensive therapy of 1,3-bis(2-chloroethyl)-1-nitrosourea-resistant human colon cancer xenografts. Hum Gene Ther. 1999;10:1021–1030. doi: 10.1089/10430349950018418. [DOI] [PubMed] [Google Scholar]
- Mitchell C, Mignon A, Guidotti JE, Besnard S, Fabre M, Duverger N.et al. (2000Therapeutic liver repopulation in a mouse model of hypercholesterolemia Hum Mol Genet 91597–1602. [DOI] [PubMed] [Google Scholar]
- Endo F, Kubo S, Awata H, Kiwaki K, Katoh H, Kanegae Y.et al. (1997Complete rescue of lethal albino c14CoS mice by null mutation of 4-hydroxyphenylpyruvate dioxygenase and induction of apoptosis of hepatocytes in these mice by in vivo retrieval of the tyrosine catabolic pathway J Biol Chem 27224426–24432. [DOI] [PubMed] [Google Scholar]
- Grimm D., and, Kay MA. Therapeutic short hairpin RNA expression in the liver: viral targets and vectors. Gene Ther. 2006;13:563–575. doi: 10.1038/sj.gt.3302727. [DOI] [PubMed] [Google Scholar]
- Czech MP, Aouadi M., and, Tesz GJ. RNAi-based therapeutic strategies for metabolic disease. Nat Rev Endocrinol. 2011;7:473–484. doi: 10.1038/nrendo.2011.57. [DOI] [PubMed] [Google Scholar]
- Holme E., and, Lindstedt S. Nontransplant treatment of tyrosinemia. Clin Liver Dis. 2000;4:805–814. doi: 10.1016/s1089-3261(05)70142-2. [DOI] [PubMed] [Google Scholar]
- Agliano A, Martin-Padura I, Mancuso P, Marighetti P, Rabascio C, Pruneri G.et al. (2008Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rgamma null mice generate a faster and more efficient disease compared to other NOD/scid-related strains Int J Cancer 1232222–2227. [DOI] [PubMed] [Google Scholar]
- Grompe M, al-Dhalimy M, Finegold M, Ou CN, Burlingame T, Kennaway NG.et al. (1993Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice Genes Dev 712A2298–2307. [DOI] [PubMed] [Google Scholar]
- Montagutelli X, Lalouette A, Coudé M, Kamoun P, Forest M., and, Guénet JL. aku, a mutation of the mouse homologous to human alkaptonuria, maps to chromosome 16. Genomics. 1994;19:9–11. doi: 10.1006/geno.1994.1004. [DOI] [PubMed] [Google Scholar]
- Overturf K, al-Dhalimy M, Ou CN, Finegold M., and, Grompe M. Serial transplantation reveals the stem-cell-like regenerative potential of adult mouse hepatocytes. Am J Pathol. 1997;151:1273–1280. [PMC free article] [PubMed] [Google Scholar]
- Grompe M, Jones SN, Loulseged H., and, Caskey CT. Retroviral-mediated gene transfer of human ornithine transcarbamylase into primary hepatocytes of spf and spf-ash mice. Hum Gene Ther. 1992;3:35–44. doi: 10.1089/hum.1992.3.1-35. [DOI] [PubMed] [Google Scholar]
- Ponder KP, Gupta S, Leland F, Darlington G, Finegold M, DeMayo J.et al. (1991Mouse hepatocytes migrate to liver parenchyma and function indefinitely after intrasplenic transplantation Proc Natl Acad Sci USA 881217–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell C., and, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc. 2008;3:1167–1170. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- Eckert JW, Buerkle CJ, Major AM, Finegold MJ., and, Brandt ML. In situ hybridization utilizing a Y chromosome DNA probe. Use as a cell marker for hepatocellular transplantation. Transplantation. 1995;59:109–111. doi: 10.1097/00007890-199501150-00019. [DOI] [PubMed] [Google Scholar]
- Slocum RH., and, Cummings JG. Wiley-Liss: New York; 1991. Techniques in Diagnostic Human Biochemical Genetics: A Laboratory Manual. [Google Scholar]
- Hoffmann G, Aramaki S, Blum-Hoffmann E, Nyhan WL., and, Sweetman L. Quantitative analysis for organic acids in biological samples: batch isolation followed by gas chromatographic-mass spectrometric analysis. Clin Chem. 1989;35:587–595. [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U.et al. (2003Exploration, normalization, and summaries of high density oligonucleotide array probe level data Biostatistics 4249–264. [DOI] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. J Roy Statis Soc B. 2002;64:479–498. [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- McCarthy DJ., and, Smyth GK. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics. 2009;25:765–771. doi: 10.1093/bioinformatics/btp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Confirmatory qPCR for several key genes from microarray results.
Y-chromosome qPCR and FISH results in immune-deficient recipients.