

Abstract
Arginase deficiency is characterized by hyperargininemia and infrequent episodes of hyperammonemia. Human patients suffer from neurological impairment with spasticity, loss of ambulation, seizures, and severe mental and growth retardation. In a murine model, onset of the phenotypic abnormality is heralded by weight loss beginning around day 15 with death occurring typically by postnatal day 17 with hyperargininemia and markedly elevated ammonia. The goal of this study was to address the development of a gene therapy approach for arginase deficiency beginning in the neonatal period. Lifespan extension, body weight, circulating amino acids and ammonia levels were examined as outcome parameters after gene therapy with an adeno-associated viral vector expressing arginase was administered to mice on the second day of life (DOL). One-hundred percent of untreated arginase-deficient mice died by DOL 24, whereas 89% of the adeno-associated virus (AAV)-treated arginase deficient mice have survived for >8 months. While animals at 8 months demonstrate elevated glutamine levels, ammonia is less than three times that of controls and arginine levels are normal. These studies are the first to demonstrate that AAV-based therapy for arginase deficiency is effective and supports the development of gene therapy for this and the other urea cycle disorders.
Introduction
Arginase deficiency, an autosomal recessive disorder, is a rare metabolic disease resulting from a loss of arginase I (ARG1), the final enzyme in the urea cycle, which is the major pathway for the detoxification of ammonia in mammals. ARG1 is expressed most prevalently in hepatocytes and red blood cells. In the liver, in coordination with the other enzymes of the urea cycle, sequestration of nitrogen as urea occurs.1 ARG1 hydrolyzes arginine into ornithine which can then re-enter the urea cycle as urea is then excreted as waste.
Neonatal and early infantile presentation of ARG1 deficiency with severe hyperammonemia does occur but is rare;2,3 ARG1 deficiency usually presents later in life beginning in late infancy to the second year of life with microcephaly, spasticity, seizures, clonus, loss of ambulation (often manifesting as a spastic diplegia that may be indistinguishable from cerebral palsy), seizures and failure to thrive associated with hyperargininemia.4 The neurologic manifestations seen in arginase deficiency may arise from the accumulation of arginine and its metabolites or may result from hyperargininemia, in which several guanidino compounds (neurotoxins) increase; the exact cause is not known. The neurological impairment and developmental regression are associated with corticospinal5 and pyramidal tract deterioration, and severe mental and growth retardation but they usually avoid the catastrophic hyperammonemic crises characteristic of the other urea cycle disorders and thus tend to survive much longer.1 The lack of frequent episodes of hyperammonemia is probably due to an increase in the second form of arginase, arginase II, which compensates for the lack of arginase I;1 this enzyme is expressed in extrahepatic tissues, mainly in the kidney and brain.1,6 Currently long-term therapy rests on provision of a low-protein diet and administration of sodium benzoate and sodium phenylbutyrate. While these dietary and pharmaceutical interventions can partially alleviate ARG1 deficiency, there is no completely effective therapy available today.
ARG1-deficient mice were previously generated in our laboratory by replacing exon 4, the active site, of the ARG1 gene with the neomycin resistance gene.7 No ARG1 RNA was found on northern blot nor was any crossreacting material detected. These mice completely lack liver arginase I activity. In contrast to the human disease in which patients can survive into adulthood, NIH-Swiss mice with ARG1 deficiency typically die between postnatal days 14 and 21 (average day 17) with severe hyperammonemia. Plasma ammonia levels of ARG1-deficient mice in metabolic crisis are increased greater than tenfold and their livers are abnormal with histopathologic features similar to those seen in human arginase-deficient patients who died with hyperammonemia later in life.7
Early postnatal gene therapy has the potential to ameliorate genetic abnormalities before the development of phenotypic disease. However, such therapy may have greater challenges than therapy of post-mitotic tissues. Unlike neonates, where the rate of hepatocellular proliferation is much higher and may affect episomal vector genomes,8,9 rapid cellular proliferation in adults is uncommon and individual hepatocytes in the adult mouse liver are replaced once every 180–400 days.10,11 In this report, we describe the therapeutic efficacy of adeno-associated virus (AAV)-mediated gene transfer to correct the biochemical defect in a neonatal murine model of arginase I deficiency. The treated ARG1-deficient mice have survival comparable to littermate controls and while demonstrating less fat than littermates are otherwise phenotypically indistinguishable. Enzymatic activity is sustained to at least 9 months as demonstrated by an examination of plasma ammonia levels and amino acid profiles. Long-term survival of the animals was attained with intact fertility. Successful development of this model supports future consideration of this therapy for human patients and will also allow for examination of the effect of arginase I deficiency on the brain and for study of the role of excess arginine in neurodegeneration.
Results
Gene therapy rescues the lethal ARG1-deficient mouse
ARG1−/− day of life (DOL) two mice received an intravenous injection of 3 × 1013 vector genome copies per kilogram (gc/kg) of rAAVrh10-mAI and 1 × 1013 gc/kg when a weight of 10 g was achieved (typically around DOL 20). Littermate controls were uninjected (n = 23). Of the ARG1−/− mice followed long-term 16 (total followed n = 18) survived to beyond 8 months (88.9%) (Figure 1) (P < 0.0001 when compared to survival of untreated ARG−/− mice). Of the two that were found dead (one on DOL 109 and the other on DOL 159), neither demonstrated any abnormalities on necropsy (P = 0.09 when comparing littermate controls with AAV-treated ARG1−/− mice). A single ARG1+/– mouse littermate control was euthanized on DOL 97 for ethical reasons due to a tail abnormality. In contrast, 100% of the untreated ARG1−/− (n = 37) mice died by DOL 24 (average day of death was 17). In another control group, DOL 2 ARG1−/− (n = 7) were injected intravenously with 3 × 1013 vector gc/kg with AAV rh10 carrying a reporter gene (luciferase) and all perished by DOL 21 (average day of death was 16) (P = 0.43 when compared with untreated ARG−/− mice). Untreated ARG1−/− mice and rAAV-luciferase-treated ARG1−/− animals were either found dead or were in extremis and then euthanized. Other ARG1−/− mice treated with rAAV rh10-mARG1 gene therapy and sacrificed for blood collection and tissue harvesting had no gross pathological abnormalities. On gross examination by a blinded observer, treated ARG1−/− mice were indistinguishable in activity from littermate controls. Brains of AAV-rh10mAI-treated ARG1−/− mice at day 20 demonstrated no gross or histopathologic abnormalities (see Supplementary Figure S1). Eight-month-old mice (n = 4 treated ARG1−/− mice and n = 3 littermate controls) were euthanized at 8 months followed by histopathological examination of the liver by a blinded pathologist to eliminate bias. No evidence of hepatocellular carcinoma was present. None of the livers from treated mice showed any evidence of fibrosis. Other cell types (Kupffer, Ito, and endothelial cells) were unremarkable.
Figure 1.

Rescue of ARG1−/− mice. Survival in days between the untreated ARG1−/− mice (n = 37), intravenous-injected rAAVrh10-Luciferase 3 × 1013 gc/kg ARG1−/− mice (n = 7), intravenous-injected rAAVrh10-mAI 3 × 1010 (DOL #2) + rAAVrh10-mAI 1 × 1013 (DOL #20) gc/kg (n = 18) ARG1−/− mice, and untreated littermate controls (n = 23). The group of rAAVrh10-mAI-treated ARG1−/− mice exhibited a substantial improvement in survival relative to untreated and rAAVrh10-luciferase-treated ARG1−/−mice. (*P = 0.098 for rAAVrh10-mAI treated compared to untreated ARG1−/+ littermate control mice; P < 0.0001 for rAAVrh10-mAI untreated ARG1−/− mice compared to rAAVrh10-mAI-treated ARG1−/−mice; P = 0.43 for untreated ARG1−/− mice compared to rAAVrh10-Luciferase-treated ARG1−/−mice.). DOL, day of life; rAAV, recombinant adeno-associated virus; gc, genome copy.
Gene therapy results in improvement of growth retardation
ARG1−/− mice were not distinguishable from ARG1+/– or ARG1+/+ littermates at birth. As untreated ARG1−/− mice develop over time, a growth disparity becomes apparent as the rate of weight gain between ARG1−/− (blue line) and littermate controls grows (orange line) (Figure 2a). Eventually, often at day 13–16, the ARG1−/− mice stop gaining weight and the growth disparity is obvious. Once lack of weight gain begins, the animals typically begin to lose weight over the next 2–3 days followed by death (Figure 2b). ARG1−/− mice treated by intravenous injection of 3 × 1013 gc/kg of rAAVrh10-mARG1 on DOL 2 (green line) were grossly indistinguishable in size from littermate controls (Figure 2c); however as adults they did not achieve the adult body weight of their gender matched littermates (Figure 2d). Computed axial tomographic imaging of the mice demonstrated that treated ARG1−/− animals (n = 3) lack the extravisceral fat of littermate controls (n = 3); skeletal muscle, brain and viscera are of similar size between groups. The axial skeleton is slightly smaller in the treated ARG1−/− mice however, there is no evidence of microcephaly [Figure 2e (A–D)]. AAV-vector administered ARG1−/− neonates expressing the reporter gene luciferase provided no benefit in weight gain or survival (red line).
Figure 2.

Growth of mice after rAAVrh10-mARG1 gene therapy. (a) Improvement in growth comparing treated and untreated ARG1−/− mice with littermate controls. The graph shows the daily weights of AAVrh10-mAI-treated ARG1−/− mice compared to untreated ARG1−/− mice, littermate controls and adeno-associated virus (AAV)-reporter gene-injected mice. The rAAVrh10-mAI-treated ARG1−/− mice demonstrate substantial growth improvement compared to the untreated ARG1−/− mice. Error bars represent SD. (b) A littermate control ARG1+/– mouse (bottom) with an untreated ARG1−/− mouse at day of life 11; the untreated arginase affected animal is unable to right itself and cannot ambulate. (c) A treated ARG1−/− mouse (bottom) compared to a littermate control mouse (top) at day of life 17 demonstrate the size and development. (d) An ARG1−/− mouse (bottom) that received rAAVrh10-mAI intravenously is next to an ARG1+/– littermate control at 8 months of age. (e) Computed axial tomographic imaging of an ARG1+/– littermate control (left, A) compared to a treated ARG1−/− mouse (right, B) at 4 months of age. The treated animal is of similar size to the littermate control except that the amount of nonvisceral (closed arrows) and visceral fat (open arrows) is substantially less in the ARG1−/− mice (right, D) compared to the ARG1+/– littermate control (left, C). (In 2B, C, and D, red marks on tails are for identification.) (†designates death). rAAV, recombinant adeno-associated virus.
Expression of arginase I after treatment with rAAVrh10-mARG1
Arginase expression in liver from treated ARG1−/− mice was examined by immunohistochemical evidence in liver sections, western blot to examine protein content, and using quantitative PCR to measure mRNA levels. Hepatic enzyme in treated ARG1−/− mice demonstrated levels similar to that of littermates on DOL 7. Additional studies were performed at DOL 20, 4 months, and 8 months demonstrated persistent but diminished expression of the hepatic ARG1 transgene and protein expression; this, however, was readily detectable by quantitative PCR (Table 1), immunohistochemistry (Figure 3) and western blot (Figure 4).
Table 1. Hepatic viral copy number determination by PCR and arginase expression by qPCR after rAAV-mARG1 administration.
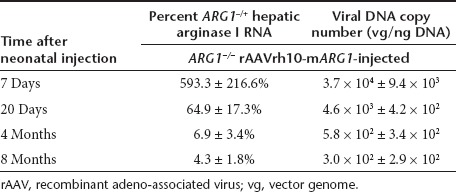
Figure 3.

Arginase expression in hepatocytes by immunohistochemistry after recombinant adeno-associated virus (rAAV)-mARG1 administration. Representative sections of immunohistochemical detection of AAV-mediated ARG1 expression in liver. Images demonstrate expression of arginase in liver of treated and untreated mice by DAB staining (a,b,c,g,h,i) and fluorescence (d,e,f,j,k,l). For fluorescent images, red is arginase expression, green is albumin (as a hepatocyte marker), and blue is DAPI nuclear staining. (a,d) Day of life 7 ARG1+/+ untreated; (b and e) day of life 7 ARG1−/− untreated; (c,f) Day of life 7 ARG1−/− AAV-mARG1-treated; (g,j) Day of life 20 AAV-mARG1−/− treated; (h,k) day of life 4 months AAV-mARG1−/− treated; (i,l) Day of life 8 months AAV-mARG1−/− treated (all DAB images are at 320× magnification; for fluorescent images, d,e,f, and j are at 200× magnification, k and l are at 400× magnification.
Figure 4.

Arginase expression by western blot after rAAV-mARG1 administration. Western blot of total liver extracts (25 µg/well) were prepared from ARG1+/+, ARG1–/+, and untreated ARG1−/− mice (all at day of life 7) to compare with AAV-mARG1-treated mice at day of life 7, 20, 4 months, and 8 months. The same blots were probed with an anti-β-actin antibody to control for loading. While 7 and 20 day samples show easily visible bands, at 4 and 8 months bands are visible but diminished.
Gene therapy results in improvement of urea cycle function
Circulating amino acids and ammonia were measured in mice (≥4 animals of each group at each time point). Ammonia levels became elevated early in life in affected animals (Figure 5a). While littermate controls demonstrate ammonia levels of 164.2 ± 71.3 at DOL 7 (n = 17), affected ARG1−/− demonstrate ammonia levels of 283.7 ± 82.8 (P = 0.03) (n = 8); AAV-treated ARG1−/− mice demonstrate ammonia levels of 183.3 ± 144.1 (n = 4) (P = 0.67 when compared to littermate controls). Ammonia levels are substantially elevated in untreated ARG1−/− mice by day 19–23 when they are in extremis (n = 7): 1,829.6 ± 1,073.4 whereas littermate controls demonstrate ammonia values of 124.5 ± 65.7 (n = 11) (P = 8.0 × 10–11); treated ARG1−/− mice demonstrate ammonia levels of 101.6 ± 18.4 (n = 4) (P = 2.4 × 10–14 when compared to untreated mice; P = 0.90 when compared with littermate controls). Beginning at month 2, ammonia levels of treated ARG−/− mice remain 2.5–4 times that of littermate controls through 9 months of life (P < 0.002 from 2 to 7 months).
Figure 5.

Metabolic improvements after rAAV-mARG1 treatment. Plasma metabolite levels were measured at times points of 7 days, 20 days (for untreated ARG1−/− mice, when they became ill), 2 months, 4 months, 5 months, 6 months, 7 months, 8 months, and 9 months after birth. The three presented groups are: untreated ARG−/− (black circle), littermate ARG–/+ controls (white square), and AAV-treated ARG−/− mice (gray triangle). Error bars represent the SD. The rAAV-mARG1-treated mice show a substantial improvement in amino acids and ammonia compared to the untreated ARG1−/− mice. (a) Ammonia, (b) citrulline, (c) arginine, (d) glutamine, (e) ornithine, (g) glutamate. All measurements are in micromoles per liter. (For ammonia, citrulline, arginine, glutamine, and glutamate. *designates P < 0.01; for ornithine. *designates P < 0.05.).
By DOL 7, untreated ARG1−/− mice have dysfunction of the urea cycle demonstrated by elevated citrulline, arginine, and glutamine with depressed ornithine (Figure 5b–f); arginosuccinic acid is detected (13.1 ± 1.4 µmol/l) which is not detected in treated ARG1−/− mice or littermate controls at any time point. The plasma arginine levels in the treated ARG1−/− mice were significantly lower than in the untreated ARG1−/− mice. By day 19–23 when these mice are in extremis, there is marked elevation in glutamine, glutamate, citrulline, and ornithine with abnormalities in multiple other amino acids (data not shown) and the further elevation of arginosuccinic acid (35.3 ± 19.2 µmol/l versus 0.0 in treated ARG1−/− mice and littermate controls). AAV-treated ARG1−/− mice demonstrate an amino acid profile similar to that of littermate controls where plasma arginine levels in the treated ARG1−/− mice are not significantly different than the plasma arginine levels in the littermate control mice (P > 0.1 for 2 months and older). However, from 2–8 months of age, glutamine levels in the treated ARG1−/− mice slowly rise and remain >1,000 by 4 months of age (littermate controls 479.3 ± 45.7) (P < 0.01 for 2 months and older) while glutamate levels remain normal (P > 0.2 for all time points). During this same time period, citrulline levels become mildly elevated (<2 times littermate controls) (P < 0.006 for 2 months and older) and ornithine levels become mildly depressed (~2/3rds that of littermate controls) (P < 0.05 for 4 months and older).
rAAVrh10-mARG1 treatment results in fertility
While not completely defined, arginase is believed to play a role in male fertility. While untreated ARG1−/− mice die before reaching sexual maturity, it is unclear if they have the capability of normal spermiogenesis. A treated ARG1−/− male (ID# K4) was mated with a wild-type female resulting in 11 pups, all confirmed to be heterozygotes as expected. Two treated ARG−/− males (ID#s R8 and S2) were mated with heterozygous females resulting in 5 litters (R8 sired 2 litters and S2 sired 3 litters) of 20 ARG1−/− and 26 ARG−/+ pups. Finally a treated ARG1−/− male (ID# I2) and a treated ARG1−/− female (ID# G3) were mated that resulted in 10 live-born pups; genotyping revealed that all were ARG1−/− as expected.
Discussion
Arginase I deficiency is thought to be the least common of the urea cycle disorders and results in hyperargininemia. Hyperargininemia in humans is characterized clinically by progressive mental impairment, spasticity, growth retardation, and periodic episodes of hyperammonemia; while uncommon, it can result in neonatal lethality. Our earlier studies with a neonatally administered helper-dependent adenoviral vector expressing murine arginase I demonstrated that short-term phenotypic correction of this disease was possible in mice; however due to loss of vector with hepatocyte division, mice die by day 27 of life.12 In the untreated murine model on the NIH-Swiss background, animals die on average by day 17 of life with hyperammonemia, hyperargininemia, neurologic injury, and in extremis. While the primary function of ARG1 in the liver is to catalyze the urea cycle, it is believed that the same reaction in extrahepatic sites directs metabolites into other, quite different, pathways. In this setting, arginine and its hydrolytic products become substrates for the synthesis of a number of important neurotransmitters, secondary signaling molecules and DNA support molecules, including glutamate, γ-aminobutyric acid, proline, creatine, nitric oxide, citrulline, and polyamines;1,13,14 while it is not known, elevation of these compounds may in part explain the characteristic neurological findings in human arginase 1-deficient patients.
The studies performed in this series of investigations were designed to test the efficacy of neonatally administered recombinant AAV (rAAV)-mediated gene therapy in the murine model of arginase deficiency as potential therapy for this incompletely treated disorder in humans. In considering development of gene replacement therapy approaches for inborn errors of metabolism, correction of genetic diseases during neonatal development offers several potential advantages over adult gene transfer. First, neonatal gene therapy may allow for the correction or prevention of genetic diseases before the onset of pathologic processes. Early gene transfer and long-term expression of therapeutic proteins during development may limit or abrogate the pathologic consequences of genetic mutations. Second, highly proliferative stem cells may be more efficiently transduced than are more quiescent adult stem cell populations. Organs inaccessible later in life may also be more readily transduced in the early postnatal period. Finally, neonatal immune immaturity (particularly of B cells) may result in blunted immune responses to the vector or the transgene, and may induce B cell immune tolerance.15,16,17
Earlier studies conducted in our laboratory demonstrated that AAV serotype rh10 led to greater maintenance of copy number in the liver18 and led to the selection of this serotype as a gene delivery vector for these studies. Knowing that the copy number would decline rapidly following the initial neonatal injection, animals received a second injection when they reached a weight of 10 g (about day 20 of life). We had previously determined that this was possible in mice as an injection on the second DOL did not result in an immune response to vector capsid proteins;19,20 thus no neutralizing antibody response would prevent vector readministration.
The observed results demonstrate that these injections were sufficient to rescue arginase 1-deficient mice from lethality before weaning and to at least 9 months of age (length of study). In addition, the effects of the AAV-based therapy extend beyond rescue from mortality and include weight gain, correction of hyperargininemia, improvement in hyperammonemia, and establishment of fertility. Skeletal imaging has demonstrated normal calvarial development and treated animals are able to tolerate a diet while appearing to have normal activity levels all while having mildly elevated ammonia and amino acids, compared to heterozygous littermate controls; these levels are decreased compared to the markedly abnormal untreated arginase 1-deficient mice. As complete restoration of all plasma amino acids and metabolites did not occur, interestingly plasma arginine levels were not different from littermate controls; glutamine, however, was substantially elevated demonstrating that transamination and transamidation are necessary to keep ammonia levels under control.
Consistent with our prior studies20 and that of others,9,21 the AAV vector produced persistent expression in the liver that was detectable to at least 8 months by immunohistochemistry and mRNA studies. The rapid growth of the liver resulted in the dilution and degradation of episomal vector DNA with copy number decline9,20 along with the level of arginase expression declining over time. It is likely that the general stability of expression detected between 4 and 8 months is because the liver in mice typically reaches its adult size by 5 weeks of age.18 While the AAV-based gene therapy was not completely efficacious in regards to the urea cycle and circulating amino acids, the administrations of vector resulted in improvement in function of the urea cycle with normal appearing and behaving mice with low morbidity and mortality. We have not excluded the possibility of arginase expression in other tissues; however it is not known if extrahepatic expression would result in effective ureagenesis. Future studies will address this issue. While the levels of arginase expression may be relatively low in the liver, the cohort of arginase 1-deficient mice have survived beyond 8 months demonstrating that even low levels of hepatic arginase 1 expression are sufficient to provide acceptable metabolic homeostasis and prevent mortality. In addition, treated mutant animals appear well with no obvious neurological or behavioral abnormality; the difference that is obvious however is that they are lean compared with littermate controls.
These studies demonstrate that a preclinical model of arginase deficiency, the second murine model of a urea cycle defect treated with AAV-based gene therapy (OTC deficiency22,23,24 being the first), benefits this disorder of ammonia detoxification as the murine model can be rescued by continued and low-level hepatic arginase 1 expression preventing neurological sequelae and death. While human patients with this disorder demonstrate multisystem dysfunction including growth and often mental retardation and spastic diplegia, these studies suggest that such abnormalities in general may be prevented with AAV-based gene therapy for arginase deficiency and warrant future studies.
Materials and Methods
Virus production. The vector contains the cytomegalovirus enhancer/chicken β-actin promoter, cloning sites for the insertion of a complementary DNA, the WPRE element (provided by Thomas Hope, University of Illinois, Chicago, IL), and the rabbit β-globin polyA signal with terminal repeats from AAV serotype 2 flanking the expression cassette. Either the murine ARG1 or luciferase cDNA was subcloned into the vector and packaged into rAAVrh10, purified by cesium chloride centrifugation, and titered by quantitative PCR as previously described.18
Mouse procedures. The targeted ARG1 allele contains a deletion in exon 4 of the ARG1 gene.7 All mice were housed under specific pathogen-free conditions; food and water were provided ad libitum. All mice were kept according to the National Institutes of Health guidelines and all experimental procedures were conducted in accordance with guidelines for the care and use of research animals at our institution. Newborn pups on the second DOL were injected with 3.0 × 1013 gc/kg of AAVrh10 CBA-mARG1-WPRE diluted in pharmaceutical grade saline by the superficial facial vein. The injections were performed in a total volume of 50 µl. At 10 g weight (typically 20 days of life) mice were administered 1.0 × 1013 gc/kg of AAVrh10 CBA-mARG1-WPRE by tail vein in 200 µl of pharmaceutical saline. Male and female mice were equally represented throughout the study. After the vector injection, scheduled blood sampling was taken from retro-orbital plexus. Serum was frozen immediately and stored at –80 °C until analysis.
PCR genotyping. Genomic DNA was prepared from tail tip by standard methods. Anion-exchange column-purified genomic DNA was subjected to PCR for genotyping. Primer sets for wild-type gene: KO reverse primer: Exon 5 reverse 5′-ACGATGTCTTTGGCAGATATGC -3′ and wild-type forward primer: mAI forward 5′-AACCAGCACCTCTAAGGTCTATGG-3′. Primer sets for KO: wild-type/KO reverse primer: Exon 5 reverse 5′-ACGATGTCTTTGGCAGATATGC -3′ and KO forward primer: Neo forward 5′-GCCCATTCGACCACCAAG-3′. Cycle parameters: Denaturation: 94 °C for 30 seconds, annealing: 60 °C for 30 seconds, Elongation: 72 °C for 3 minutes for 40 cycles using DNA polymerase (Takara, Mountain View, CA, Catalog #RR006A).
Biochemical and ammonia analysis. Serum amino acid analysis was performed on a Biochrom 30 HPLC amino acid analyzer. In brief, 30–50 µl of serum was mixed with equal volumes of Biochrom Seraprep and Lithium dilution buffer. Protein was precipitated by centrifugation and 10 µl supernatant was injected into the analyzer. Physiological amino acid standard (Sigma-Aldrich, St Louis, MO) was used to calibrate and determine analyte concentration. Results analysis was performed using EZchrom Elite software. Ammonia was determined in serum samples, by reductive amination of 2-oxoglutarate and oxidation of NADPH, employing a commercial kit (Sigma-Aldrich) using 20 µl of serum for each sample tested.
Immunohistochemistry for arginase expression. Tissues were removed from euthanized animals and placed in 4% paraformaldehyde for 18–24 hours. After rinsing with tap water for 15 minutes tissues were placed in 70% ethanol followed by routine processing and embedding in paraffin. Tissues on slides were deparaffinized and rehydrated with ethanol and xylene by routine procedures. Slides were removed from tap water and placed in a microwaveable vessel filled with sodium citrate buffer (10 mmol/l sodium citrate, 0.05% Tween 20, pH 6.0) for antigen retrieval. Tissues were then permeabilized with 1× TBS + 0.2% Triton X-100 for 5–10 minutes followed by 1× TBS + 0.025% Triton X-100 for 5 minutes. Tissues were blocked with protein blocker (Dako Cat# X0909) + 0.1% Tween 20 for 20 minutes. Primary antibody was applied to the sections: Rabbit arginase I (H-52) antibody (Santa Cruz Biotechnology, Santa Cruz, CA; catalog number sc-20150) diluted in protein blocker plus 0.1% Tween 20 at 1:50 ratio and incubated overnight at 4 °C. After rinsing twice for 5 minutes with 1× TBS 0.025% Triton the slides were incubated in 0.3% H2O2 in TBS for 15 minutes followed by application of the secondary antibody (Goat anti-rabbit IgG-HRP [Santa Cruz Biotechnology, catalog number sc-2301) (1:100 in protein blocker + 0.1% Tween 20)] and incubated for 1 hour at room temperature. After rinsing three times with 1× TBS, slides were developed with ImmPACT DAB kit (Vector, Laboratories, Burlingame, CA; Cat# SK-4105) for 3 minutes at room temperature. Slides were counterstained with hematoxylin and coverslipped. For fluorescent imaging, the anti-arginase antibody was followed by a sheep polyclonal anti-rabbit IgG, fragment-Cy3 (Sigma, Catalog No. 2306). As a generalized hepatocyte marker, cells were stained with a goat polyclonal anti-mouse serum albumin antibody (Abcam, Cambridge, MA; Catalog No. ab19194) diluted in protein blocker plus 0.1% Tween 20 at 1:50 ratio (incubated for 3 hours) followed by a donkey polyclonal antibody to goat IgG FITC conjugated (Abcam, Catalog No. Ab6881) at 1:800 dilution.
Real-time PCR for arginase expression. Total RNA was extracted from liver tissue using the RNeasy Kit (Qiagen, Valencia, CA) per the manufacturer's instructions. cDNA synthesis was by reverse transcription using the Thermoscript RT-PCR kit (Invitrogen/Life Technologies, Grand Island, NY) per the manufacturer's instructions. Determination of expression of exogenous arginase I by AAV compared to endogenous GAPDH expression was accomplished by real-time PCR. Samples were performed in duplicate using SsoAdvanced SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA). Primers were as follows: arginase forward primer: 5′-GGA AGC ATC TCT GGC CAC GCC-3′ Reverse Primer: 5′-TCC CAG AGC TGG TTG TCA GGG G-3′ GAPDH forward primer: 5′-ACT CCA CTC ACG GCA AAT TC-3′ reverse primer: 5′-TCT CCA TGG TGG TGA AGA CA-3′. Reaction conditions were 40 cycles of denaturation: 95 °C for 5 seconds and annealing/extension: 64 °C for 30 seconds.
Vector copy number analysis. At regular intervals, mice were euthanized and liver tissue was removed. Genomic DNA was extracted using the DNAEasy Kit (Qiagen) and quantitated by nanodrop (Implen, Westlake Village, CA). Using real-time quantitative PCR (MyIQ2; Bio-Rad Laboratories) of the HGH polyadenylation signal (Forward primer: 5′AATCTTGGCTCACTGCAATCTCCG3′, Reverse primer: 5′CATGCAT GCCTGGAATCCCAACAA3′) with SsoFast EvaGreen detection (Bio-Rad Laboratories), quantification of vector genomes in DNA was determined in duplicate as previously described.19
Western blotting. Tissue samples were homogenized with a 2-ml tissue grinder in RIPA buffer (Sigma-Aldrich) in the presence of Halt protease inhibitor cocktail (Pierce Biotechnology, Rockford, IL). Twenty-five micrograms of centrifuged clarified total protein were used in western analysis and probed with polyclonal rabbit antisera against the murine ARG1 enzyme (Santa Cruz Biotechnology, catalog number SC-20150). β-Actin was used as a loading control and was detected by immunoblotting (rabbit affinity-purified polyclonal anti-β-actin antibody; Biolegend, San Diego, CA, catalog number 11143). The anti-ARG1 antibody was used at a dilution of 1:500, and the anti-β-actin antibody was used at a dilution of 1:500. Horseradish peroxidase–conjugated goat anti-rabbit IgG (Santa Cruz, Biotechnology, catalog number sc-2301) was used as the secondary antibody and was visualized by enhanced chemiluminescent detection (Pierce Biotechnology).
Histology. Mice were euthanized by inhalation of isoflurane. For light microscopy, livers and brains were carefully dissected out and immersion-fixed in 4% formalin for 48 hours followed by immersion in 70% alcohol solution. Tissue was then coronally sectioned after routine processing and embedding in a single paraffin block using standard techniques. 4-µm sections of the block were routinely cut, stained with hematoxylin-eosin and analyzed using an Olympus BX40 microscope and images were captured with cellSens software (Olympus, Center Valley, PA).
Imaging protocol. All mice were imaged in a multimodality chamber designed to allow maintaining the mice on isoflurane anesthesia at 1–2% during imaging and provide heating to maintain the mice's body temperature.25 The chamber also enables reproducible positioning to <1 mm, thus minimizing any attenuation variability due to animal positioning. Mice were imaged in a MicroCAT II small animal CT system (Siemens Preclinical Solutions, Knoxville, TN). Exposure settings were 70 kVp, 500 mAs, 500 ms exposure time and 360° rotation in 1° steps with 2.0 mm aluminum filtration. Images were reconstructed using a modified Feldkamp process to a cubic voxel size of 0.20 mm.
Statistical analysis. Survival curves were computed in each group of mice using the Kaplan–Meier method and compared across groups using the log rank test. Each continuous lab measure was log10 transformed prior to the analysis, since the data on the original scale were skewed but the log transformed data followed a roughly normal distribution. Since younger animals are euthanized at each time, for age <4 months, groups are independent across time. Therefore, for age <4 months, between-group mean comparisons across age were carried out using the fixed effects ANOVA model assuming independent observations. Since older animals are measured repeatedly across time, for age ≥4 months, between group mean comparisons across age were carried out using the repeated measures ANOVA model taking into account that observations within the same animal are not independent. P values of < 0.05 were considered significant. Calculations were made using SAS version 9.2 (SAS Institute, Cary, NC).
SUPPLEMENTARY MATERIAL Figure S1. Brain sections of day 20 mice.
Acknowledgments
The authors thank Waldemar Ladno for assistance with CT imaging of mice, Daniela Markovic for assistance with the statistical evaluation, and both the Semel Insitute for Neuroscience and the Intellectual and Developmental Disabilities Research Center at UCLA for their support. This work was supported by grants from the National Institutes of Health (5K08HD057555-05 and 1R01NS071076-02A1). The authors declared no conflict of interest.
Supplementary Material
Brain sections of day 20 mice.
REFERENCES
- Iyer R, Jenkinson CP, Vockley JG, Kern RM, Grody WW., and, Cederbaum S. The human arginases and arginase deficiency. J Inherit Metab Dis. 1998;21 Suppl 1:86–100. doi: 10.1023/a:1005313809037. [DOI] [PubMed] [Google Scholar]
- Jain-Ghai S, Nagamani SC, Blaser S, Siriwardena K., and, Feigenbaum A. Arginase I deficiency: severe infantile presentation with hyperammonemia: more common than reported. Mol Genet Metab. 2011;104:107–111. doi: 10.1016/j.ymgme.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picker JD, Puga AC, Levy HL, Marsden D, Shih VE, Degirolami U.et al. (2003Arginase deficiency with lethal neonatal expression: evidence for the glutamine hypothesis of cerebral edema J Pediatr 142349–352. [DOI] [PubMed] [Google Scholar]
- Prasad AN, Breen JC, Ampola MG., and, Rosman NP. Argininemia: a treatable genetic cause of progressive spastic diplegia simulating cerebral palsy: case reports and literature review. J Child Neurol. 1997;12:301–309. doi: 10.1177/088307389701200502. [DOI] [PubMed] [Google Scholar]
- Oldham MS, VanMeter JW, Shattuck KF, Cederbaum SD., and, Gropman AL. Diffusion tensor imaging in arginase deficiency reveals damage to corticospinal tracts. Pediatr Neurol. 2010;42:49–52. doi: 10.1016/j.pediatrneurol.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vockley JG, Jenkinson CP, Shukla H, Kern RM, Grody WW., and, Cederbaum SD. Cloning and characterization of the human type II arginase gene. Genomics. 1996;38:118–123. doi: 10.1006/geno.1996.0606. [DOI] [PubMed] [Google Scholar]
- Iyer RK, Yoo PK, Kern RM, Rozengurt N, Tsoa R, O'Brien WE.et al. (2002Mouse model for human arginase deficiency Mol Cell Biol 224491–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J.et al. (2005Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart Nat Biotechnol 23321–328. [DOI] [PubMed] [Google Scholar]
- Cunningham SC, Dane AP, Spinoulas A, Logan GJ., and, Alexander IE. Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol Ther. 2008;16:1081–1088. doi: 10.1038/mt.2008.72. [DOI] [PubMed] [Google Scholar]
- Fausto N., and, Campbell JS. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech Dev. 2003;120:117–130. doi: 10.1016/s0925-4773(02)00338-6. [DOI] [PubMed] [Google Scholar]
- Magami Y, Azuma T, Inokuchi H, Kokuno S, Moriyasu F, Kawai K.et al. (2002Cell proliferation and renewal of normal hepatocytes and bile duct cells in adult mouse liver Liver 22419–425. [DOI] [PubMed] [Google Scholar]
- Gau CL, Rosenblatt RA, Cerullo V, Lay FD, Dow AC, Livesay J.et al. (2009Short-term correction of arginase deficiency in a neonatal murine model with a helper-dependent adenoviral vector Mol Ther 171155–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson CP, Grody WW., and, Cederbaum SD. Comparative properties of arginases. Comp Biochem Physiol B, Biochem Mol Biol. 1996;114:107–132. doi: 10.1016/0305-0491(95)02138-8. [DOI] [PubMed] [Google Scholar]
- Singh R, Pervin S, Karimi A, Cederbaum S., and, Chaudhuri G. Arginase activity in human breast cancer cell lines: N(omega)-hydroxy-L-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 2000;60:3305–3312. [PubMed] [Google Scholar]
- Fan X, Ang A, Pollock-Barziv SM, Dipchand AI, Ruiz P, Wilson G.et al. (2004Donor-specific B-cell tolerance after ABO-incompatible infant heart transplantation Nat Med 101227–1233. [DOI] [PubMed] [Google Scholar]
- West LJ, Morris PJ., and, Wood KJ. Neonatal induction of tolerance to cardiac allografts. Transplant Proc. 1994;26:207–208. [PubMed] [Google Scholar]
- West LJ., and, Tao K. Acceptance of third-party cardiac but not skin allografts induced by neonatal exposure to semi-allogeneic lymphohematopoietic cells. Am J Transplant. 2002;2:733–744. doi: 10.1034/j.1600-6143.2002.20807.x. [DOI] [PubMed] [Google Scholar]
- Hu C, Busuttil RW., and, Lipshutz GS. RH10 provides superior transgene expression in mice when compared with natural AAV serotypes for neonatal gene therapy. J Gene Med. 2010;12:766–778. doi: 10.1002/jgm.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Cela RG, Suzuki M, Lee B., and, Lipshutz GS. Neonatal helper-dependent adenoviral vector gene therapy mediates correction of hemophilia A and tolerance to human factor VIII. Proc Natl Acad Sci USA. 2011;108:2082–2087. doi: 10.1073/pnas.1015571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C., and, Lipshutz GS. AAV-based neonatal gene therapy for hemophilia A: long-term correction and avoidance of immune responses in mice. Gene Ther. 2012. [DOI] [PMC free article] [PubMed]
- Chandler RJ., and, Venditti CP. Long-term rescue of a lethal murine model of methylmalonic acidemia using adeno-associated viral gene therapy. Mol Ther. 2010;18:11–16. doi: 10.1038/mt.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham SC, Spinoulas A, Carpenter KH, Wilcken B, Kuchel PW., and, Alexander IE. AAV2/8-mediated correction of OTC deficiency is robust in adult but not neonatal Spf(ash) mice. Mol Ther. 2009;17:1340–1346. doi: 10.1038/mt.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham SC, Kok CY, Dane AP, Carpenter K, Kizana E, Kuchel PW.et al. (2011Induction and prevention of severe hyperammonemia in the spfash mouse model of ornithine transcarbamylase deficiency using shRNA and rAAV-mediated gene delivery Mol Ther 19854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L, Wang, H, Morizono, H, Bell, P, Jones, D, Lin, J.et al. (2011Sustained correction of OTC deficiency in spf(ash) mice using optimized self-complementary AAV2/8 vectors Gene Ther [DOI] [PMC free article] [PubMed]
- Suckow C, Kuntner C, Chow P, Silverman R, Chatziioannou A., and, Stout D. Multimodality rodent imaging chambers for use under barrier conditions with gas anesthesia. Mol Imaging Biol. 2009;11:100–106. doi: 10.1007/s11307-008-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Brain sections of day 20 mice.