

Abstract
Animal and human studies of enzyme replacement therapy (ERT) for Pompe disease (PD) have indicated that antibodies (Abs) generated against infused recombinant human α-glucosidase (rhGAA) can have a negative impact on the therapeutic outcome and cause hypersensitivity reactions. We showed that parenteral administration of anti-CD3 Abs into mice can reduce the titer of anti-human GAA Abs in wild-type mice administered the enzyme. Mice that had been treated with anti-CD3 Abs and then subjected to a secondary challenge with rhGAA showed a lower increase in Ab titers than control mice. Moreover, the administration of anti-CD3 Abs also reduced the levels of pre-existing Abs. Treatment with anti-CD3 Abs also prevented a lethal hypersensitivity reaction and reduced the Ab titers in a mouse model of PD. Mice treated with anti-CD3 Abs showed reduced numbers of CD4+ and CD8+ cells, and an increased ratio of CD4+CD25+/CD4+ and CD4+CD25+FoxP3+/CD4+ cells. When the CD4+CD25+ cells were depleted using anti-CD25 Abs, the observed reduction in Abs against the enzyme by anti-CD3 Abs was abrogated. This suggests that CD4+CD25+ cells are important for the immune suppressive activity of anti-CD3 Abs. In summary, anti- CD3 Abs are useful for inducing immune tolerance to ERT for PD.
Introduction
Pompe disease (PD) (also known as glycogen storage disease II [MIN 232300]) is a lysosomal storage disease (LSD) characterized by a deficiency of acid α-glucosidase (GAA) activity. Because of this deficiency, glycogen accumulates progressively in the heart and skeletal muscles with the resultant presentation of cardiomyopathy and muscle weakness. PD can be divided into two clinical entities: infantile- and late-onset PD. Patients with infantile-onset PD present with hypertrophic cardiomyopathy, hypotonia, muscle weakness, respiratory failure, feeding problems, and failure to thrive within the first few months of life. The disease progresses rapidly, resulting in premature death typically in the first year of life if left untreated. Late-onset PD (child and adult type) has a variable clinical presentation. The onset of clinical signs can occur as early as the first year of life and as late as the seventh decade of life. Patients with late-onset PD present with muscle weakness and respiratory failure, but not cardiac symptoms. Until 2006, there were no therapies to target the underlying basis of PD. The only available treatment was supportive therapy for heart and respiratory failure. In 2006, enzyme replacement therapy (ERT) with recombinant human GAA (rhGAA) (aglucosidase alfa) (Myozyme; Genzyme) was approved for treating this disease in many countries. Lumizyme (aglucosidase alfa; Genzyme) was also approved for late-onset PD in the United States in 2010. Both enzymes harbor the same protein sequence, but have a slightly different carbohydrate composition. In a clinical trial involving infants, patients who were not undergoing ventilation were treated with biweekly infusions of rhGAA at either 20 or 40 mg/kg.1 A nontreated historical cohort was used as the control group.2 The treated patients lived longer and the proportion of ventilation-free patients was larger compared with the historical cohort. These observations clearly indicated that rhGAA was effective in treating infantile-onset PD. On the basis of these results, rhGAA was approved; however, until recently, there has been no research that clearly shows the effectiveness of rhGAA for late-onset PD. Recently, a randomized control trial was carried out in late-onset PD patients, and ERT was associated with an improved walking distance and stabilization of pulmonary function over an 18-month period.3 From these findings, ERT for PD would appear to be effective for both infantile- and late-onset types. Although ERT has been shown to be effective in treating PD patients, some challenges remain. One of these challenges is the immune response to the infused enzyme. Animal and human studies of ERT for PD have indicated that the formation of antibodies (Abs) against rhGAA can reduce the efficacy of treatment.1,4,5,6,7,8,9 Kishnani et al. retrospectively studied infantile-onset PD in patients with an onset at <6 months of age who received ERT.7 They divided the patients into two groups: cross-reactive immune material (CRIM) negative and CRIM positive. The baseline demographics and disease-related characteristic were comparable between the two groups. Immunoglobulin G (IgG) Abs to rhGAA developed earlier and titers were higher and more sustained in the CRIM-negative group. The CRIM-negative patients also exhibited poorer clinical outcomes. They concluded that the effect of CRIM status on outcome appears to be mediated by Ab responses to the infused rhGAA. More recently, some CRIM-positive infantile-onset PD patients also developed high titers of Abs against rhGAA and had poorer clinical outcomes, similar to the CRIM-negative infantile-onset PD patients.9 From these observations, we argue that the induction of tolerance against rhGAA would be highly desirable to improve therapeutic outcome and prevent hypersensitivity reactions.
There are reports describing approaches to inducing immune tolerance to rhGAA. These include the administration of methotrexate,10 anti-CD20 Abs,11,12 anti-IgE Abs,13 adeno-associated vector-expressing GAA into the thymus,14 genetically modified GAA-expressing self bone marrow cells,15,16 oral rhGAA,17 and adeno-associated vector containing a liver-specific promoter at the beginning of therapy.18,19,20 In addition, Lipinski et al. reported that immune tolerance could also be induced by a gradual escalation of the administered dose of rhGAA.21 The ideal agents for the induction of tolerance should be antigen-specific, present with minimal side-effects, and have a long-lasting effect; however, methotrexate, anti-CD20 Abs, and anti-IgE Abs do not meet these requirements. Recently, Waters et al. reported that parenteral administration of anti-CD3 Abs could induce immune tolerance to coagulation factor supplementation treatment.22 This induction of tolerance was long-lasting and, once it was established, tolerance was maintained for a sustained period; moreover, they suggested that this tolerance was probably antigen-specific. From this observation, we decided to test whether parenteral administration of anti-CD3 Abs could also induce tolerance to rhGAA for PD. Here, we investigated whether parenteral administration of anti-CD3 Abs can reduce the host immune response to infused rhGAA in mice.
Results
Immune response to rhGAA in wild-type (Balb/c and C57BL/6) and PD model (B6;129-Gaatm1Rabn/J) mice
In order to determine whether there was a difference in the immune reaction between wild-type (7–8-weeks-old Balb/c and C57BL/6) and PD model (23-27-weeks-old B6;129-Gaatm1Rabn/J) mice, rhGAA (10 mg/kg) was administered to three groups of mice once a week for a total of 4 doses. One week after the final administration of rhGAA, the anti-rhGAA Ab titers were analyzed. As shown in Supplementary Figure S1, Ab titers against rhGAA in PD mice were significantly higher than in the control groups (Balb/c and C57BL/6) (P ≤ 0.05, Kruskal–Wallis test followed by post-hoc Dunn's test), whereas the Ab titers in the Balb/c mice were not significantly different to those in the C57BL/6 mice. We started this experiment using seven Balb/c, six C57BL/6, and eight PD model mice. Following the repeated administration (four times) of rhGAA, three of the eight PD model mice died from anaphylactic shock, and the Ab titers of these mice could not be assayed; however, the Ab titers in these expired mice were probably very high. Thus, if we had been able to include the data of the Ab titers from these expired mice, the Ab titers of the PD model mice would probably be much higher than the presented data. In addition, significant levels of Abs against rhGAA also developed in both groups of wild-type mice following the repeated administration of rhGAA, but they rarely died from anaphylaxis. Therefore, to prevent loss of mice from anaphylaxis, we used wild-type mice in most of the following experiments, unless otherwise stated.
Prevention of Ab formation by anti-CD3 Abs in wild-type mice
After the intravenous administration of anti-CD3 Abs (10 µg per dose) for five consecutive days, rhGAA (10 mg/kg) was administered to the mice once a week for a total of four doses. One week after the final administration of rhGAA, the anti-rhGAA Ab titers were analyzed. Anti-CD3 Abs significantly reduced the formation of Abs in BALB/c (P = 0.0034; Figure 1a) and C57BL/6 mice (P = 0.0002; Figure 1b). Therefore, we performed the following experiments using BALB/c mice, unless otherwise stated. To assess the duration of the effects of the anti-CD3 Abs, rhGAA (10 mg/dose once a week for a total of four doses) was readministered at 11 weeks after the first rhGAA challenge. The anti-rhGAA Ab titers were analyzed at 1 week after the final infusion of rhGAA. As shown in Figure 1c, the Ab titers just before the second rhGAA challenge were reduced in both hamster IgG-treated groups. After the second challenge, the Ab titers in the anti-CD3 Ab-treated group were significantly lower than in the hamster IgG-treated control group (P = 0.0061; Figure 1c). This observation indicated that the immune-suppressive effect of the anti-CD3 Abs was long-lasting.
Figure 1.

Prevention of antibody (Ab) formation by anti-CD3 Abs in wild-type mice. Anti-rhGAA Ab formation as a result of four doses of recombinant human α-glucosidase (rhGAA) was significantly reduced by anti-CD3 Abs in (a) wild-type BALB/c (P = 0.0034) and (b) C57BL/6 mice (P = 0.0002). In BALB/c mice, Ab formation due to a second challenge of rhGAA was also significantly lower in the anti-CD3 Ab-administered group (P =0.0061). (c) “Pre” refers to the initial day of rhGAA administration. “Post” refers to 1 week after the final administration of rhGAA. The individual Ab values are shown as mean values and SEM.
Immune-suppressive effect of anti-CD3 Abs against pre-existing anti-rhGAA Abs in wild-type mice
We also tested the effect of anti-CD3 Abs against pre-existing anti-rhGAA Abs. BALB/c mice were immunized once weekly for a total of four doses with rhGAA followed by the administration of anti-CD3 Abs or hamster IgG as a control for five consecutive days. Three days after the final administration of the anti-CD3 Abs, the Ab titers against rhGAA were measured. The study design is shown in Figure 2a. The Abs against rhGAA were elevated to the same level in both groups following immunization with rhGAA (indicated as post-immune by rhGAA, P = 0.7132), but the Ab titers in the anti-CD3 Ab group were significantly lower than in the hamster IgG-treated group (indicated as post-hamster IgG or post-anti-CD3 Abs, respectively, P = 0.0373; Figure 2b).
Figure 2.

Suppressive effect of anti-CD3 antibodies (Abs) against pre-existing anti-recombinant human α-glucosidase (rhGAA) Abs in wild-type mice (BALB/c). The study design and immunization schedule are shown in a. (b)”Preimmune by rhGAA” refers to the initial day of rhGAA administration (# in a). “Postimmune by rhGAA” refers to 1 week after the final administration of rhGAA and the initial day of hamster immunoglobulin G (IgG) or anti-CD3 Ab administration (## in a). “Post-hamster IgG” or “Post-anti-CD3 Abs” refers to 3 days after the final administration of hamster IgG or anti-CD3 Abs, respectively (### in a). After rhGAA immunization, the Ab titers increased to the same extent in both the hamster- and anti-CD3 Ab-treated groups (P = 0.7132); however, after the administration of anti-CD3 Abs, the Ab titers were significantly reduced compared with the hamster IgG-treated group (P = 0.0373). The individual Ab values are shown as mean values and SEM.
Prevention of lethal hypersensitivity reactions and Ab titers in PD model mice
PD mice repeatedly administered rhGAA frequently died because of hypersensitivity reactions. We tested whether the anti-CD3 Abs can prevent this lethal hypersensitivity reaction. As in the studies using the wild-type mice, anti-CD3 Abs or control hamster IgG were administered to PD mice. Then, rhGAA was administered intravenously at 20 mg/kg once every other week for up to 20 injections. The experimental schedule is shown in Figure 3a and the Kaplan-Meier survival curve is shown in Figure 3b. Only 1 of 11 mice survived in the cohort treated with hamster IgG. By contrast, 10 of 12 mice survived in the anti-CD3 Ab-treated group. The anti-CD3 Ab-treated mice survived for a significantly longer period than those injected with hamster IgG (P = 0.0002, log-rank test).
Figure 3.

Prevention of the lethal hypersensitivity reaction and antibody (Ab) titers in Pompe disease (PD) mice. PD mice that were administered repeated doses of recombinant human α-glucosidase (rhGAA) usually died as a result of a lethal hypersensitivity reaction. The immunization schedule is shown in (a). The PD mice that received anti-CD3 Abs survived for a longer period than the hamster immunoglobulin G (IgG)-treated group (log-rank test, P = 0.0002) (b). “0” refers to the initial day of rhGAA administration.
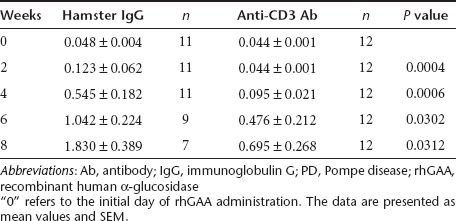
Anti-rhGAA IgG Ab levels were analyzed at various time points. As shown in Figure 4a, the Ab titers against rhGAA in most of the hamster IgG-treated group (n = 11) increased rapidly, and the animals typically died due to a hypersensitivity reaction. The Ab titers against rhGAA in 2 of the mice were only elevated slightly and these animals survived for a longer period. In one mouse (indicated by *), the Ab titer increased moderately during the early period, but this mouse died at 22 weeks after treatment (4 hours after the final rhGAA injection). Typically, the lethal hypersensitivity reactions occurred within 1 hour of the rhGAA injection, so it is likely that this mouse died from another cause. One mouse (indicated by **) survived for a longer period and its Ab titer declined. In contrast, in the anti-CD3 Ab-treated group (n = 12), the titers in most of the mice (10 of 12 mice) were elevated less than in the hamster IgG-treated group and these levels eventually declined (Figure 4b). It is interesting that four mice did not exhibit any increase in their Ab titer. After 8 weeks, only 2 mice were still alive in the hamster IgG-treated group, so we were unable to perform a statistical comparison of the Ab titers. Therefore, the Ab titers were compared at 2, 4, 6, and 8 weeks after ERT. The Ab titers were significantly lower in the anti-CD3 Ab group at all time points (Table 1). However, this immune-suppressive effect seemed to be less than in the wild-type mice.
Figure 4.

Titers of Abs against recombinant human α-glucosidase (rhGAA) following the repeated administration of rhGAA in individual Pompe disease (PD) mice. The antibody (Ab) titers of individual mice that received rhGAA repeatedly are shown. The Ab titers of the hamster group are shown in (a) and those of the anti-CD3 Ab group are shown in (b). “0” refers to the initial day of rhGAA administration.
Table 1. Statistical comparison of the anti-rhGAA Ab titers following the repeated administration of rhGAA in PD mice at 2, 4, 6, and 8 weeks after rhGAA treatment.
The effect of anti-CD3 Abs on the lymphocyte population
Anti-CD3 Abs (10 µg) or the same dose of hamster IgG were administered to BALB/c mice for five consecutive days. Three days after the last administration of anti-CD3 Abs or hamster IgG, spleen cells were harvested and analyzed using flow cytometry for CD4+, CD8+, CD4+CD25+, and CD4+CD25+FoxP3+ cells with fluorescently labeled Abs against cell surface and intracellular markers. The administration of anti-CD3 Abs significantly reduced the numbers of CD4+ (P = 0.0079; Figure 5a) and CD8+ (P = 0.0079; Figure 5b) cells and increased the ratio of CD4+CD25+/CD4+ (P = 0.0286; Figure 5c) and CD4+CD25+FoxP3+/CD4+ (P = 0.0286; Figure 5d) cells. We analyzed the levels of CD4+ and CD8+ cells in peripheral blood up to 48 days after administration of Anti-CD3 Abs or hamster IgG, and found that level of CD4+ cells did not recover to control levels for up to 36 days after the initiation of anti-CD3 Ab treatment (Figure 6a,b). Representative examples of flow cytometry plots for CD4+CD25+FoxP3+/CD4+ and CD4+CD25+/CD4+ cells are shown in Supplementary Figures S2 and S3.
Figure 5.

The effect of anti-CD3 antibodies (Abs) on effector T cells and regulatory T cells in the spleen. The lymphocyte population in the spleen from mice that received hamster immunoglobulin G (IgG) or anti-CD3 Abs is shown. The anti-CD3 Abs significantly reduced the number of (a) CD4+ and (b) CD8+ cells and increased the ratio of (c) CD4+CD25+/CD4+ and (d) CD4+CD25+FoxP3+/CD4+ cells.
Figure 6.

Effect of anti-CD3 Abs on the number of CD4+ and CD8+ cells in the peripheral blood. The levels of (a) CD4+ and (b) CD8+ cells in peripheral blood from mice that received hamster immunoglobulin G (IgG) or anti-CD3 Abs are shown. The administration of anti-CD3 Abs for five consecutive days reduced the levels of CD4+ and CD8+ cells in the peripheral blood at various time points for up to 36 days after the initiation of anti-CD3 antibody (Ab) or hamster IgG treatment. “0” refers to the initial day of anti-CD3 Abs or hamster IgG administration. *Significant difference between the two groups and “NS” indicates no significant difference between the two groups.
Depletion of CD4+CD25+ cells
Because anti-CD3 Ab treatment increased the ratio of CD4+CD25+/CD4+ and CD4+CD25+FoxP3+/CD4+T cells, we speculated that CD4+CD25+ cells may play an important role in the immune-suppressive activity of the anti-CD3 Abs. We depleted CD4+CD25+ cells by administering anti-CD25 Abs to BALB/c mice and tested whether the suppressive effect of anti-CD3 Abs on the formation of Abs against rhGAA was abrogated. Depletion of CD4+CD25+ cells was confirmed by flow cytometry using anti-CD4-FITC and anti-CD25-PE clone 7D4 (data not shown). When CD4+CD25+ cells were depleted by anti-CD25 Abs, the Ab titers were increased after rhGAA challenge (group 1, Figure 7). When the isotype control of the anti-CD25 Ab was administered, the increase in Abs against rhGAA was prevented (group 2, Figure 7). If neither anti-CD25 nor isotype control Abs were administered, anti-CD3 Abs prevented the increase in Abs, but hamster IgG did not (groups 3 and 4, respectively, Figure 7). This observation indicated that CD4+CD25+ cells play an important role in preventing the formation of Abs against rhGAA by anti-CD3 Abs during ERT for PD.
Figure 7.

Depletion of CD4+CD25+ cells. Group 1: CD4+CD25+ cells were depleted by the administration of anti-CD25 Abs and tolerance was induced by the administration of anti-CD3 Abs. Group 2: As a control for group 1, isotype control anti-CD25 Abs were administered prior to tolerance induction by anti-CD3 Abs. Group 3: Tolerance was induced by anti-CD3 Abs without any pretreatment. Group 4: As a control for group 3, hamster immunoglobulin G (IgG) was administered instead of anti-CD3 Abs without any pretreatment. “Pre” refers to the initial day of recombinant human α-glucosidase (rhGAA) administration. “Post” refers to 1 week after the final administration of rhGAA. The individual antibody (Ab) values are shown as mean values and SEM.
Discussion
Clinical studies of ERT for LSDs have highlighted some challenges associated with this approach. One of these challenges is the immune response to the infused enzyme. There are several preclinical reports, including from our laboratory,23,24 indicating that such Abs can have a negative impact on therapeutic efficacy in patients and in mouse models of LSDs. However, it is very difficult to make this conclusion in the clinic as LSDs are very rare and clinically heterogeneous disorders. Moreover, in some LSDs, the clinical manifestations are slow to develop, making correlations between Abs and clinical improvement due to ERT difficult. However, PD, especially the infantile-onset form, although rare and clinically heterogeneous, is a relatively rapidly progressing disease. Thus, among the various LSDs, it may be the most suitable to study the influence of Ab formation on clinical outcome following ERT. As mentioned above, several studies have demonstrated that Abs against rhGAA might have a negative impact on the efficacy of ERT for PD. To overcome this obstacle, several methods have been tested for the induction of immune tolerance against rhGAA in mouse models and human PD patients. Among them, we chose the oral administration of rhGAA17 and anti-CD3 Abs because these methods might induce an antigen-specific and long-lasting tolerance. In this study, we focused on the administration of anti-CD3 Abs.
We showed that the parenteral administration of anti-CD3 Abs before ERT effectively reduced Ab formation against the infused enzyme in mice. Moreover, anti-CD3 Abs reduced the titers of pre-existing Abs against rhGAA. This is a very important observation as most CRIM-negative PD patients usually develop high Ab titers against rhGAA;7,9 therefore, in the clinical setting, anti-CD3 Abs should be administered prior to the initiation of ERT. By contrast, it is very difficult to predict whether or not CRIM-positive patients will develop high Ab titers. It is not practical for all CRIM-positive PD patients to receive anti-CD3 Abs before ERT. Perhaps, once CRIM-positive patients develop high Ab titers, anti-CD3 Abs should be administered because they also reduce the levels of pre-existing Abs.
Anti-CD3 Abs appeared to be more effective in the wild-type mice than in the PD model mice. This may be due to the complete absence of GAA in this mouse model of PD, resulting in a robust immune reaction against rhGAA, compared with the wild-type mice. Therefore, the anti-CD3 Ab dose used for the wild-type mice may not be suitable for the PD mice, and dose adjustment of anti-CD3 Abs might be necessary for the PD mice. However, even in the PD mice, anti-CD3 Abs prevented the lethal hypersensitivity reaction and reduced the Ab titers against rhGAA. Only 1 mouse out of 11 survived in the control hamster IgG-administered group. The Ab titer against rhGAA in the mouse that survived increased, but it then declined over time, even when ERT was continued without any immune-suppressive treatment (including anti-CD3 Ab administration). Even if the decrease in the number of CD4+ and CD8+ cells and the increase in the ratio of CD4+CD25+ /CD4+ and CD4+CD25+ FoxP3+/CD4+ cells following the administration of anti-CD3 Abs are transient, once lethal hypersensitivity has been induced, which occurred rapidly following ERT and was prevented by the administration of anti-CD3 Abs, tolerance might be induced by repeated intravenous infusions of rhGAA.
An important issue to consider is whether the generation of Abs against rhGAA inhibits the effects of ERT. To clarify this issue, we infused PD mice with 10 mg/kg rhGAA once a week (for a total of five infusions) and with or without anti-CD3 Ab administration before ERT. The day after the final administration of rhGAA in both groups, the mice were killed and GAA activity in the liver was analyzed (Supplementary Figure S4). We examined GAA activity in the liver of two nontolerant hamster IgG-administered group mice exhibiting the highest Ab titers and three tolerant anti-CD3 Ab-administered group mice exhibiting no Abs. As a control, GAA activity in liver from untreated PD model mice was also examined (n = 3). GAA activity was not significantly different in the liver of the nontolerant hamster IgG-administered group mice exhibiting high Ab titer and the tolerant anti-CD3 Ab-administered group mice exhibiting no Ab. One reason for this observation is that the mice that exhibited very high Ab titers died after the third or fourth infusion of rhGAA. Consequently, we were unable to measure the GAA activity in these mice. We only assayed GAA activity in the mice that survived, and these animals had relatively low Ab titers. Therefore, in this study, we were not able to show the effect of anti-CD3 Abs on clinical outcome through the induction of immune tolerance. However, many reports have shown that the induction of tolerance improves the therapeutic effect of ERT in PD patients. So there is no doubt that the induction of tolerance is essential to maximize the effect of ERT in PD patients.
Here, we did not study the mechanism of tolerance induction by the anti-CD3 Abs in detail. The mechanism by which anti-CD3 Abs induce tolerance in protein-supplement therapy was previously studied extensively using mouse models of hemophilia.22 The authors concluded that the induction of immune tolerance occurred through the generation of distinct regulatory T cells (Tregs), including CD4+CD25+ cells, and by T cells polarized toward a Th1 immune response. Regarding the induction of Tregs by anti-CD3 Abs, their results were consistent with ours. Although the mechanism of Treg induction by the anti-CD3 Abs is not clear, it has recently become clear that anti-CD3 Abs increase the levels of CD25 by inducing its expression on peripheral effector CD4+CD25- cells, rather than by expanding thymically derived CD4+CD25+ Tregs.25,26 In addition to the expansion of CD4+CD25+ Tregs, the levels of CD4+ cells were low in anti-CD3 Ab-administered mice for a long period, even though their levels gradually increased. Taking this observation into account, both the expansion of Tregs and the reduction of CD4+ effector cells are probably necessary to induce tolerance in anti-CD3-administered mice. In other words, the ratio of regulatory to effector cells is probably an important component of tolerance in this setting. Recently, the immune responses following ERT for PD were reported to be caused by the activation of T cells.27 In this regard, it is logical to use anti-CD3 Abs to induce immune tolerance.
The clinical efficacy and safety of anti-CD3 Abs have been studied in various autoimmune diseases including autoimmune diabetes,28 ulcerative colitis,29 and Crohn's disease.30 The results in some diseases are very encouraging, but not all. In general, the induction of immune tolerance was difficult in situations in which there is an existing immune response, such as autoimmune diseases. In the case of ERT for PD, we can induce tolerance prior to the establishment of an immune reaction using anti-CD3 Abs. Thus, it is probably easier to induce tolerance in PD using anti-CD3 Ab treatment than in autoimmune diseases in which the immune reaction is already established. As such, we believe that the administration of anti-CD3 Abs prior to ERT is a very promising strategy to maximize the efficacy of ERT for PD.
The Abs used in this study were anti-murine CD3 antigens. We do not know whether anti-human CD3 Abs, which have been used in clinical trials, will be as effective as the anti-murine CD3 Abs. In this regard, Kuhn et al. developed a good animal model that expresses human CD3 antigens and was used to demonstrate the efficacy of antihuman CD3 Abs against autoimmune insulin-dependent diabetes.31 This animal model can be used as a preclinical study tool to assess the use of anti-CD3 Abs for immune tolerance induction therapy in ERT for PD.
In conclusion, the administration of anti-CD3 Abs is a very promising strategy to induce immune tolerance to ERT for PD to maximize its efficacy and prevent hypersensitivity reactions.
Materials and Methods
Animals. Wild-type BALB/C and C57BL/6 mice and PD mice (B6;129-Gaatm1Rabn/J) were used in this study. Both types of wild-type mice were purchased from Sankyo Labo Service (Tokyo, Japan). The PD mice were a generous gift from Dr N. Raben (NIH, Bethesda, MD). In this study, we generally used wild-type mice because most of the PD mice that received rhGAA repeatedly died due to a hypersensitivity reaction at an early stage if there was no immune-suppressive treatment. Therefore, it was difficult to compare the Ab titers in treated and untreated mice. All animal experiments were reviewed and approved by the Animal Care Committee of The Jikei University School of Medicine.
Anti-CD3 Abs. Anti-CD3 Abs (clone 145-2C11) were obtained from Bioexpress (Lebanon, NH). We used whole IgG anti-CD3ε or non-FcR-binding anti-CD3ε F(ab′)2 Abs. Both Abs reduced the Ab titers against rhGAA to the same extent in a preliminary experiment (data not shown). However, the latter Ab had fewer side-effects than the former. As a control, we used hamster whole IgG or F(ab′)2 (Bioexpress).
Assay for rhGAA-specific IgG. IgG Abs against rhGAA were assayed using an enzyme-linked immunosorbent assay (ELISA). Briefly, 96-well plates were coated with 1 µg of rhGAA in phosphate-buffered saline (PBS) overnight at 4 °C. The plates were blocked by adding 100 µl PBS/1% bovine serum albumin and incubating for 5 h at room temperature. After this step, the wells were washed with PBS/0.05% Tween 20. The serum samples from mice were diluted 20,000-fold with PBS/1% bovine serum albumin, and 100 µl diluted serum was added to each well and incubated for 1 h at room temperature. The plate was then washed with the same buffer and reacted with 100 µl 5,000-fold diluted peroxidase-conjugated anti-mouse IgG Ab (Kirkegaard & Perry Labs., Gaithersburg, MA). After incubation for 30 minutes at room temperature, the plates were washed again and color was generated by the addition of 3,3′,5′,5′-tetramethylbenzidine substrate reagent (Promega, Madison, WI) for 10 minutes. The reaction was stopped by adding 100 µl 0.6 N H2SO4, and the optical density was measured at 450 nm using an ARVOMX/Light plate reader (PerkinElmer, Waltham, MA). The Ab titer was reported as the value of the optical density at 450 nm. To compare the Ab titers between the two groups, all of the samples were assayed in one 96-well plate to avoid variability among the experiments.
Flow cytometry. Abs specific for CD4-FITC (clone H129.19), CD8-FITC (53-6.7), CD25-PE (clone PC61), CD25-PE (clone 7D4), and FoxP3-Alexa Fluor 647 (MF23) were used (BD Biosciences, San Jose, CA). Single-cell suspensions were pooled from the spleens or peripheral blood of the mice. The cells were incubated with an Fc blocker (CD16/32); BD Biosciences) for 15 minutes and then stained for the expression of surface markers for 30 minutes at 4 °C. To detect FoxP3 expression, the cells were permeabilized using a Cytofix/Cytoperm Kit (BD Biosciences), incubated with the Fc blocker and surface maker stain as described earlier, and stained with anti-FoxP3-Alexa Fluor 647 for 30 minutes at room temperature. The data were acquired on a MACSQuant Analyzer (Miltenyi Biotec, Bergisch Gladbach, Germany) and analyzed using MACSQuantify Software (Miltenyi Biotec).
Induction of tolerance by anti-CD3 Abs in wild-type mice. We administered 10 µg of anti-CD3 Abs intravenously for five consecutive days to each 10-week-old Balb/c or C57BL/6 mouse. As a control, the same dose of hamster IgG was administered. Three days after the final administration of the anti-CD3 Abs or hamster IgG, the mice received 10 mg/kg rhGAA intravenously once every week for a total of four doses. One week after the final administration of rhGAA, blood was collected from the retro-orbital plexus. Eleven weeks after the first administration of the anti-CD3 Abs, Balb/c mice were challenged with 10 mg/kg rhGAA intravenously once every week for a total of four doses. One week after the final administration of rhGAA, Abs against rhGAA were assayed using ELISA.
Effect of anti-CD3 Abs against pre-existing Abs in wild-type mice. To immunize 10-week-old BALB/c mice, 10 mg/kg rhGAA was administered intravenously once every week for a total of four doses. One week after the final administration of rhGAA, 10 µg anti-CD3 Abs were administered for five consecutive days. As a control, the same amount of hamster IgG was administered. Three days after the final administration of the anti-CD3 Abs or hamster IgG, anti-rhGAA Abs were assayed using ELISA.
Prevention of the hypersensitivity reaction to ERT in PD mice. PD mice develop a lethal hypersensitivity against rhGAA when it was infused repeatedly; therefore, we tested whether anti-CD3 Abs inhibit this reaction. We administered 10 µg anti-CD3 Abs for 5 consecutive days to PD mice (9–18 weeks old). As a control, the same dose of hamster IgG was administered. Three days after the final administration of the anti-CD3 Abs or hamster IgG, the mice received 20 mg/kg rhGAA intravenously once every other week for a total of 20 doses. We defined lethal hypersensitivity as death within 30 minutes after the intravenous administration of rhGAA. The occurrence of a lethal hypersensitivity reaction was monitored and Ab titers were also assayed at various time points.
Depletion of CD4+CD25+ cells. Wild-type BALB/c mice were injected intraperitoneally with 1 mg anti-CD25 Abs (clone PC61; Bioexpress) or isotype control Abs (rat IgG1; Bioexpress) on days 0 and 5; on days 3–7, they received 10 µg/day anti-CD3 Abs or control hamster IgG. Three days after the final injection of the anti-CD3 Abs, the mice were killed and splenocytes were analyzed for the expression of CD4 and CD25 using anti-CD4-FITC and anti-CD25-PE clone 7D4. An identical treatment regimen was performed, but at 3 days after the anti-CD3 or hamster IgG injections (day 10), the mice received the first of 4 weekly rhGAA immunizations (10 mg·kg–1·day–1). The subsequent immune response to rhGAA was determined at 1 week after the final rhGAA immunization using ELISA.
Statistical analysis. The results are expressed as the mean ± SEM. Statistical differences between two groups were determined using the Mann–Whitney test, and differences between three groups were determined by analysis of variance with the Kruskal–Wallis and Dunn's multiple comparison tests. In addition, we calculated the risk of lethal hypersensitivity to rhGAA infusion using the Kaplan–Meier method, while the log-rank test was used to assess the differences between the study groups. P values <0.05 were considered to be significant.
SUPPLEMENTARY MATERIAL Figure S1. Immune response to rhGAA in wild-type (Balb/c and C57BL/6) and PD model mice. Figure S2. Representative flow cytometry plot demonstrating that anti-CD3 Abs increased the ratio of CD4+CD25+/CD4+ cells. Figure S3. Representative flow cytometry plot demonstrating that anti-CD3 Abs increased the ratio of CD4+CD25+FoxP3+/CD4+ cells. Figure S4. GAA activity in the liver from nontolerant or tolerant PD mice.
Acknowledgments
The authors thank Nina Raben (NIH, Bethesda, MD) for providing the PD mouse. We also wish to thank Seng H. Cheng (Genzyme) for critically reviewing the manuscript. This research was partially supported by a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Science, Sports, and Culture of Japan. T.O., H.I., and Y.E. have active research support from the Genzyme. These activities have been fully disclosed and are managed under a Memorandum of Understanding with the Conflict of Interest Resolution Board of The Jikei University School of Medicine. This work was performed at the Department of Gene Therapy, Institute of DNA Medicine, Research Center for Medical Sciences/Department of Pediatrics, The Jikei University School of Medicine, 3-25-8 Nishishinbashi, Minato-ku, Tokyo 105-8461, Japan.
Supplementary Material
Immune response to rhGAA in wild-type (Balb/c and C57BL/6) and PD model mice.
Representative flow cytometry plot demonstrating that anti-CD3 Abs increased the ratio of CD4+CD25+/CD4+ cells.
Representative flow cytometry plot demonstrating that anti-CD3 Abs increased the ratio of CD4+CD25+FoxP3+/CD4+ cells.
GAA activity in the liver from nontolerant or tolerant PD mice.
REFERENCES
- Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL.et al. (2007Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease Neurology 6899–109. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F., and, Corzo D, Infantile-Onset Pompe Disease Natural History Study Group A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–676. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ.et al. (2010A randomized study of alglucosidase alfa in late-onset Pompe's disease N Engl J Med 3621396–1406. [DOI] [PubMed] [Google Scholar]
- Raben N, Danon M, Gilbert AL, Dwivedi S, Collins B, Thurberg BL.et al. (2003Enzyme replacement therapy in the mouse model of Pompe disease Mol Genet Metab 80159–169. [DOI] [PubMed] [Google Scholar]
- Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL.et al. (2001Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial Genet Med 3132–138. [PubMed] [Google Scholar]
- Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J.et al. (2006Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease J Pediatr 14989–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S.et al. (2010Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants Mol Genet Metab 9926–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries JM, van der Beek NA, Kroos MA, Ozkan L, van Doorn PA, Richards SM.et al. (2010High antibody titer in an adult with Pompe disease affects treatment with alglucosidase alfa Mol Genet Metab 101338–345. [DOI] [PubMed] [Google Scholar]
- Banugaria SG, Prater SN, Ng YK, Kobori JA, Finkel RS, Ladda RL.et al. (2011The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease Genet Med 13729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph A, Munroe K, Housman M, Garman R., and, Richards S. Immune tolerance induction to enzyme-replacement therapy by co-administration of short-term, low-dose methotrexate in a murine Pompe disease model. Clin Exp Immunol. 2008;152:138–146. doi: 10.1111/j.1365-2249.2008.03602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn NJ, Messinger YH, Rosenberg AS., and, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe's disease. N Engl J Med. 2009;360:194–195. doi: 10.1056/NEJMc0806809. [DOI] [PubMed] [Google Scholar]
- Messinger YH, Mendelsohn NJ, Rhead W, Dimmock D, Hershkovitz E, Champion M.et al. (2012Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease Genet Med 14135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach M, Klein A, Köhli-Wiesner A, Veraguth D, Scheer I, Balmer C.et al. (2010CRIM-negative infantile Pompe disease: 42-month treatment outcome J Inherit Metab Dis 33751–757. [DOI] [PubMed] [Google Scholar]
- Chu Q, Moreland RJ, Gao L, Taylor KM, Meyers E, Cheng SH.et al. (2010Induction of immune tolerance to a therapeutic protein by intrathymic gene delivery Mol Ther 182146–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douillard-Guilloux G, Richard E, Batista L., and, Caillaud C. Partial phenotypic correction and immune tolerance induction to enzyme replacement therapy after hematopoietic stem cell gene transfer of alpha-glucosidase in Pompe disease. J Gene Med. 2009;11:279–287. doi: 10.1002/jgm.1305. [DOI] [PubMed] [Google Scholar]
- van Til NP, Stok M, Aerts Kaya FS, de Waard MC, Farahbakhshian E, Visser TP.et al. (2010Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype Blood 1155329–5337. [DOI] [PubMed] [Google Scholar]
- Ohashi T, Iizuka S, Shimada Y, Eto Y, Ida H, Hachimura S.et al. (2011Oral administration of recombinant human acid a-glucosidase reduces specific antibody formation against enzyme in mouse Mol Genet Metab 10398–100. [DOI] [PubMed] [Google Scholar]
- Sun B, Bird A, Young SP, Kishnani PS, Chen YT., and, Koeberl DD. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am J Hum Genet. 2007;81:1042–1049. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Kulis MD, Young SP, Hobeika AC, Li S, Bird A.et al. (2010Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine pompe disease Mol Ther 18353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Li S, Bird A, Yi H, Kemper A, Thurberg BL.et al. (2010Antibody formation and mannose-6-phosphate receptor expression impact the efficacy of muscle-specific transgene expression in murine Pompe disease J Gene Med 12881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski SE, Lipinski MJ, Burnette A, Platts-Mills TA., and, Wilson WG. Desensitization of an adult patient with Pompe disease and a history of anaphylaxis to alglucosidase alfa. Mol Genet Metab. 2009;98:319–321. doi: 10.1016/j.ymgme.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Waters B, Qadura M, Burnett E, Chegeni R, Labelle A, Thompson P.et al. (2009Anti-CD3 prevents factor VIII inhibitor development in hemophilia A mice by a regulatory CD4+CD25+-dependent mechanism and by shifting cytokine production to favor a Th1 response Blood 113193–203. [DOI] [PubMed] [Google Scholar]
- Ohashi T, Iizuka S, Ida H., and, Eto Y. Reduced alpha-Gal A enzyme activity in Fabry fibroblast cells and Fabry mice tissues induced by serum from antibody positive patients with Fabry disease. Mol Genet Metab. 2008;94:313–318. doi: 10.1016/j.ymgme.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Ohashi T, Sakuma M, Kitagawa T, Suzuki K, Ishige N., and, Eto Y. Influence of antibody formation on reduction of globotriaosylceramide (GL-3) in urine from Fabry patients during agalsidase beta therapy. Mol Genet Metab. 2007;92:271–273. doi: 10.1016/j.ymgme.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Belghith M, Bluestone JA, Barriot S, Mégret J, Bach JF., and, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–1208. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- You S, Leforban B, Garcia C, Bach JF, Bluestone JA., and, Chatenoud L. Adaptive TGF-beta-dependent regulatory T cells control autoimmune diabetes and are a privileged target of anti-CD3 antibody treatment. Proc Natl Acad Sci USA. 2007;104:6335–6340. doi: 10.1073/pnas.0701171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banati M, Hosszu Z, Trauninger A, Szereday L., and, Illes Z. Enzyme replacement therapy induces T-cell responses in late-onset Pompe disease. Muscle Nerve. 2011;44:720–726. doi: 10.1002/mus.22136. [DOI] [PubMed] [Google Scholar]
- Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ, Jr, Protégé Trial Investigators et al. Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378:487–497. doi: 10.1016/S0140-6736(11)60931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart DC, Targan SR, Dignass AU, Mayer L, van Assche G, Hommes DW.et al. (2010Prospective randomized open-label multicenter phase I/II dose escalation trial of visilizumab (HuM291) in severe steroid-refractory ulcerative colitis Inflamm Bowel Dis 16620–629. [DOI] [PubMed] [Google Scholar]
- van der Woude CJ, Stokkers P, van Bodegraven AA, Van Assche G, Hebzda Z, Paradowski L, Initiative on Crohn's and Colitis, The Netherlands et al. Phase I, double-blind, randomized, placebo-controlled, dose-escalation study of NI-0401 (a fully human anti-CD3 monoclonal antibody) in patients with moderate to severe active Crohn's disease. Inflamm Bowel Dis. 2010;16:1708–1716. doi: 10.1002/ibd.21252. [DOI] [PubMed] [Google Scholar]
- Kuhn C, You S, Valette F, Hale G, van Endert P, Bach JF.et al. (2011Human CD3 transgenic mice: preclinical testing of antibodies promoting immune tolerance Sci Transl Med 368ra10. [DOI] [PubMed] [Google Scholar]
- Kyosen SO, Iizuka S, Kobayashi H, Kimura T, Fukuda T, Shen J.et al. (2010Neonatal gene transfer using lentiviral vector for murine Pompe disease: long-term expression and glycogen reduction Gene Ther 17521–530. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Immune response to rhGAA in wild-type (Balb/c and C57BL/6) and PD model mice.
Representative flow cytometry plot demonstrating that anti-CD3 Abs increased the ratio of CD4+CD25+/CD4+ cells.
Representative flow cytometry plot demonstrating that anti-CD3 Abs increased the ratio of CD4+CD25+FoxP3+/CD4+ cells.
GAA activity in the liver from nontolerant or tolerant PD mice.