

Abstract
An emerging and important site of action for nitric oxide (NO) within cells is the mitochondrial inner membrane, where NO binds to and inhibits members of the electron transport chain, complex III and cytochrome c oxidase. Although it is known that inhibition of cytochrome c oxidase by NO is competitive with O2, the mechanisms that underlie this phenomenon remain unclear, and the impact of both NO and O2 partitioning into biological membranes has not been considered. These properties are particularly interesting because physiological O2 tensions can vary widely, with NO having a greater inhibitory effect at low O2 tensions (<20 μM). In this study, we present evidence for a consumption of NO in mitochondrial membranes in the absence of substrate, in a nonsaturable process that is O2 dependent. This consumption modulates inhibition of cytochrome c oxidase by NO and is enhanced by the addition of exogenous membranes. From these data, it is evident that the partition of NO into mitochondrial membranes has a major impact on the ability of NO to control mitochondrial respiration. The implications of this conclusion are discussed in the context of mitochondrial lipid:protein ratios and the importance of NO as a regulator of respiration in pathophysiology.
It has long been recognized that the interactions of nitric oxide (NO) with the mitochondrial respiratory chain may be important in mediating the biological effects of this signaling molecule. This control may be at the level of respiration (1, 2) or, as more recently recognized, inhibition of cytochrome c release in the early stages of apoptosis (1, 3–5). In a pathological context, NO-dependent regulation of mitochondrial respiration seems to contribute to a number of dysfunctions (6) which may include septic shock (7), cardiac hypertrophy, and heart failure (8–10). The best-characterized site of NO action in mitochondria is mitochondrial cytochrome c oxidase (complex IV), where NO competes with oxygen (O2) to bind at the binuclear CuB/cytochrome a3 site. This competition with O2 results in an inhibition of the enzyme, supporting the proposal that NO is an important physiological regulator of mitochondrial oxidative phosphorylation (1, 5, 11–14).
The preferential partitioning of both NO and O2 into biological membranes has been recognized as potentially important in regulating the effects of NO (15–17). NO is ≈8-fold more soluble in membranes than in water, whereas O2 is ≈3-fold more soluble. One site of particular significance for the interactions of NO and O2 is the mitochondrial inner membrane, where this phenomenon may affect the control of respiration. Indeed, the highly membranous nature of the mitochondrion suggests that the organelle may be a significant reservoir for NO within cells.
NO binding to metalloproteins, such as the ferrous heme group of soluble guanylate cyclase (sGC), is of extremely high affinity (18, 19). However, the effects of NO in initiating signaling by binding to sGC are transient, suggesting that reactions competing for NO are important in maintaining a steady-state concentration of the free radical. These reactions allow close modulation of NO bioactivity and responsiveness to the concentration of NO. Interestingly, the nature of these reactions competing for NO remains uncertain but could include oxyhemoglobin (20), lipoxygenases (21), or free radicals such as superoxide (22).
In the case of cytochrome c oxidase, the effectiveness of NO as an inhibitor is controlled both by binding to a reduced intermediate in the catalytic cycle (most likely CuB), and by competition with O2 for NO in free solution (5, 13, 14, 23). In the context of the mitochondrial inner membrane, the reaction between NO and O2 is potentially enhanced ≈300-fold as a consequence of the partitioning of both gases into the hydrophobic core of the phospholipid bilayer (15–17). In this study, we have asked the question, Does the reaction between NO and O2 in the mitochondrial inner membrane regulate the NO-dependent modulation of respiration?
Three possibilities have been examined. (i) At high O2 tensions, the consumption of NO in mitochondrial membranes is enhanced through an O2-dependent mechanism independent of electron transfer, and is competitive with cytochrome c oxidase inhibition. (ii) At low O2 tensions, the partitioning of NO into the inner membrane greatly enhances the potential of NO to modulate mitochondrial respiration. (iii) NO-dependent inhibition of respiration at low O2 tension may have a component that is caused by other NO-derived metabolites. To investigate these possibilities, we examined both the respiration and NO consumption of rat liver mitochondria and submitochondrial particles exposed to various concentrations of NO, NO scavengers, and liposomes, at several O2 tensions. It is evident that the reaction between NO and O2 in the mitochondrial membrane has a significant impact on the inhibition of cytochrome c oxidase and the consequent control of respiration. The implications of these results for the effect of NO as a regulator of mitochondrial respiration are discussed.
Materials and Methods
Male Sprague–Dawley rats, 250 g in weight, were obtained from Harlan Breeders (Indianapolis) and handled in accordance with National Institutes of Health guidelines. Food and water were available ad libitum. All biochemicals were obtained from Fluka, except 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO), which was obtained from Alexis (San Diego). Human hemoglobin was prepared as described (24).
Liver mitochondria were prepared as described (25). Submitochondrial particles (SMPs) were prepared by sonication of mitochondria (60 mg of protein) in 4 ml of SMP buffer (300 mM sucrose/5 mM Mops/1 mM EGTA/5 mM KH2PO4, pH 7.4 at 4°C) followed by centrifugation at 20,000 × g and one pellet washing. Final resuspension was in 1 ml of buffer. Protein concentration was determined by using the Folin phenol reagent against a standard curve constructed with BSA (26). Purity of SMPs was >99%, as determined by using citrate synthase activity (measured spectrophotometrically; ref. 27) as a marker of mitochondrial matrix contamination. Cytochrome c oxidase activity was determined as described (28).
Oxygen and NO were measured simultaneously with Clark-type electrodes (Instech, Plymouth Meeting, PA and World Precision Instruments, Sarasota, FL, respectively) in a 4-ml sealed chamber magnetically stirred at 37°C. Mitochondria or SMPs (0–2 mg/ml) were incubated in respiration buffer composed of 120 mM KCl/3 mM Hepes/1 mM EGTA/25 mM sucrose/5 mM MgCl2/5 mM KH2PO4, pH 7.4. Data were collected by a digital recording device (Dataq Instruments, Akron, OH) connected to a PC. NO was added from saturated solutions prepared as described (29). The concentration of NO was determined by using spectrophotometric monitoring of oxyhemoglobin to MetHb conversion as described (30). Low O2 tensions were achieved by gassing the chamber with nitrogen.
The NO electrode was calibrated by bolus additions of NO (0.1–3 μM) to the chamber at low O2 tension (20%) to establish the [NO]–electrode current relationship. Under these conditions, no significant NO consumption caused by the reaction with O2 occurred over the time course for establishing a stable current for the amount of NO added.
Liposomes were made by rehydrating thin films of soybean asolectin at 37°C in mitochondrial respiration buffer (100 mg of lipid per ml) under an N2 atmosphere, followed by sonication of the resultant multilamellar liposomes to yield unilamellar liposomes.
Kinetic analysis of concentration against time curves for NO consumption were performed by calculating the time over which 90% of the NO added was consumed; these times are referred to as t0.9. This analysis was selected to allow comparison between NO consumption pathways between buffer and mitochondria through potentially different mechanisms. Many studies have reported a second-order dependence in NO and third-order overall for the reaction between NO and O2 (15, 31). However, data traces for NO consumption obtained in this study did not conform well to either first-order or second-order kinetics. A key variable in this process was found to be the setting of the baseline. A small (<5%) change in the baseline did yield good fits to second-order kinetics and rate constants (3.2 × 108 M−1⋅s−1), similar to reported values for the reaction of NO with O2 in buffer at 37°C. To overcome the technical difficulty of establishing an accurate (1–2%) baseline with the polarographic electrodes, and to gain a more accurate determination of the properties that govern NO consumption in mitochondria, we calculated the t0.9 value as described above. All experiments were performed a minimum of three times, and data are presented as means ± SEM. Statistical significance was determined with the Student's t test, with P < 0.05 representing significance. Curve fitting was performed with origin software (Microcal Software, Northampton, MA).
Results
Consumption of NO by Mitochondria.
To determine whether NO is consumed by mitochondria in the absence of respiration, a series of 1 μM additions of NO were made to mitochondria, and the decay was monitored with the NO electrode. An initial rapid consumption of NO occurs during the mixing and response time of the electrode (≈5–10 s) resulting in a level of detectable NO of ≈400–500 nM in the buffer alone (Fig. 1A). It is clear from Fig. 1B that in the presence of mitochondria, the initial peak height is reduced, and the rate of NO consumption is substantially more rapid than that in buffer alone. These data are quantified as the time taken to consume 90% of the initial NO added (t0.9)—in this case 1 μM. It should be noted that the initial measured concentrations of NO are approximately 1/2 that added and recorded at low O2 tension. This result is readily simulated from the known rate constants for the reaction of NO with O2 under these conditions.
Figure 1.

Comparison of NO decay in buffer, mitochondria, and SMPs. NO (1 μM) was added to respiration buffer (A), mitochondria (B), and SMPs (C), as indicated by the arrows, at 90% O2. Data are representative of three similar experiments. Numbers alongside trace identifiers are t0.9—the time for 90% of NO decay to occur (i.e., for NO to decay to 10% of its peak value).
Mitochondria contain millimolar concentrations of molecules reactive with NO in the matrix and outer membrane (32). Some of these reactions may be direct (e.g., with metalloproteins), and some may require other reactants such as oxygen or metals (e.g., with glutathione). To test a role for these components, SMPs that lack the outer membrane and matrix were prepared. These SMPs were incubated in the chamber such that the final concentration of cytochrome c oxidase was the same as in the intact mitochondrial incubations. Addition of NO to SMPs resulted in rates of NO consumption that were indistinguishable from those of intact mitochondria (Fig. 1 B vs. C), demonstrating that the inner membrane is the major site of NO consumption within the organelle.
Three features of the NO consumption in intact mitochondria are also evident in the SMPs. (i) NO is consumed at a faster rate than in buffer alone. (ii) Each sequential addition of NO results in a similar decay profile; i.e., it is not saturable over this concentration range (up to 5 μM). (iii) Respiratory substrate is not required for NO consumption. To confirm that the NO consumption is indeed substrate independent, mitochondria were also incubated in the presence of respiratory inhibitors. Table 1 shows that antimycin A did not affect NO consumption rates. In contrast, rotenone and cyanide accelerated the rate of NO consumption to a modest extent. It is important to note that this result is opposite to that predicted if NO was interacting with a redox center in the substrate-free mitochondria, because the inhibitors increase rather than inhibit NO consumption. A possible mechanism for this fact would be consumption of NO caused by increased superoxide in the presence of these inhibitors as reported in the literature (33, 34). In this case, the reducing equivalents for superoxide generation could derive from small levels of endogenous substrate or partially reduced redox centers. However, it is clear that such contributions to the consumption of NO by nominally substrate-free mitochondrial preparations are minor. Furthermore, experiments in SMPs (not shown) indicated no significant effect of any of the mitochondrial inhibitors tested in Table 1 on NO consumption. Free radical generation either at the respiratory chain or through lipid peroxidation could also lead to NO consumption (21, 35). However, this mechanism is unlikely because, as shown in Table 1, neither Cu/Zn superoxide dismutase nor the lipid-soluble antioxidant butylated hydroxytoluene changed NO consumption by mitochondria.
Table 1.
NO consumption by mitochondria incubated with inhibitors and antioxidants
| Experimental conditions | t0.9, (% of control) |
|---|---|
| Rotenone (50 μM) | 87.0 ± 3.2* |
| KCN (50 μM) | 80.4 ± 7.3* |
| Antimycin A (30 μM) | 101.7 ± 6.0 |
| Cu/Zn SOD (1,000 units/ml) | 98.1 ± 1.1 |
| BHT (50 μM) | 101.6 ± 4.0 |
Mitochondria were incubated with compounds as listed. SOD, superoxide dismutase; BHT, butylated hydroxytoluene. NO consumption was measured using the NO electrode as detailed in Materials and Methods. Values are presented as the time for 90% of NO decay to occur (t0.90), as a percentage of that in the absence of added inhibitor/antioxidant. Data are means ± SEM of at least three independent experiments.
, P < 0.05 vs. mitochondria alone.
Oxygen Sensitivity of NO Consumption in Mitochondria.
As discussed above, the reaction of NO with O2 is a possible mediator of NO consumption within mitochondria. To investigate this process in more detail, NO was added to buffer alone at both high and low O2 tensions (Fig. 2A). As expected, the consumption of NO at 20% O2 was substantially inhibited compared with that at 100% O2. A similar effect was also observed in the presence of mitochondria, consistent with an O2-dependent component to substrate-free mitochondrial consumption of NO (Fig. 2). Fig. 2B shows the quantitation of these data from a number of similar experiments. The effect of O2 on mitochondrial NO consumption seems less marked because the starting NO is lower. Proportionally in both cases, lowering the O2 tension from 100% to 20% resulted in a 30–40% inhibition of NO consumption. In the next series of experiments, the effect of changing mitochondrial concentration on the rate of NO decomposition was determined. These data (Fig. 3) show that with increasing concentrations of mitochondria, the amplitude of the NO signal decreases, and the rate of NO consumption increases (as indicated by a decrease in t0.9).
Figure 2.

Oxygen-dependent consumption of NO by mitochondrial membranes. NO (1 μM) was added to mitochondria at high (80%) and low (20%) O2 tensions. (A) Averaged decay profiles for buffer alone (thin lines) and mitochondria at 1 mg/ml (thick lines), both at low O2 (solid lines) and high O2 (dashed lines). (B) t0.9 values for the curves in A. Data are means ± SEM from at least three independent experiments.
Figure 3.

Dependence of NO Decay on mitochondrial concentration. NO (1 μM) was added to buffer alone, or mitochondria suspended at 0.2, 1, and 2 mg/ml, at 80% O2. (A) Averaged decay profiles for buffer alone (solid line) and mitochondria (dotted and dashed lines). (B) t0.9 values for the curves in A. Data are means ± SEM from at least three independent experiments.
Inhibition of Mitochondrial Respiration by NO.
Several other studies have reported that the NO inhibition of mitochondrial respiration is oxygen sensitive (1, 5, 11–13, 36). However, the mechanisms contributing to this effect remain unclear. To address this issue, bolus additions of NO were made to respiring mitochondria at either high (80%) or low (30%) O2, as shown in Fig. 4A. From these data, corresponding values of NO concentration and respiration rate were determined and used to construct an inhibition curve (Fig. 4B). The rate of O2 consumption is decreased by NO at both O2 tensions but is substantially greater at the lower O2 tension. This observation is consistent with a greater fractional occupation of cytochrome c oxidase binding sites by NO at the lower O2 tension. The gradient for the inhibition of respiration by NO is, however, essentially the same for both O2 concentrations. This fact indicates that other reactions competing with NO determine the available NO for inhibition of cytochrome c oxidase. It is interesting to note that the relationship between NO and the inhibition of respiration is essentially constant over the range of NO concentrations examined. This finding is unusual because it would be anticipated that as the NO concentration decreases, O2 consumption would return to control values. This behavior arises from the fact that NO inhibition of respiration is partial, and the O2 concentration is decreasing concomitantly with the decrease in NO (Fig. 4A). These data further support the modulatory effects of NO on O2 consumption and the effect of O2 on the bioavailability of NO in biological membranes.
Figure 4.

Oxygen sensitivity of NO inhibition of respiration. NO (1 μM) was added to mitochondria respiring in state 3, at either high (80%) or low (30%) O2 tension. (A) Oxygen and NO traces for high O2 (solid lines) and low O2 (dashed lines) incubations. NO was added as indicated by the arrows. (B) The effect of NO concentration on inhibition of respiration. Data were calculated from several experiments of the type shown in A, at high O2 (○) and low O2 (■).
The previous data imply that NO inhibits mitochondrial respiration at all O2 tensions, and that persistent inhibition is not caused by the formation of other NO metabolites. To test for this possibility, NO was added to mitochondria respiring in state 3 (ADP turnover). Once inhibition of respiration was established, oxyhemoglobin (oxyHb) or PTIO was added to scavenge NO. The results shown in Fig. 5 indicate that the NO-inhibited rate of respiration is restored completely to the pre-NO rate immediately upon the addition of oxyHb or PTIO. These results indicate that the species responsible for inhibition is NO and not a metabolite. The reversibility of this reaction throughout the time course of inhibition again suggests that reactions outside the enzyme active site that consume NO have a modulatory effect on the extent of inhibition at cytochrome c oxidase. These data also show that NO can be removed from the enzyme during turnover, suggesting that cross talk between other NO-consuming reactions in the cell and control of respiration will also occur. In addition, the PTIO data indicate that NO is a more potent inhibitor than NO2, because this NO scavenger converts NO to NO2.
Figure 5.

Effects of NO scavengers on the inhibition of mitochondrial respiration by NO. Mitochondria were suspended at 0.5 mg/ml, and state 3 respiration was initiated by the addition of ADP (0.6 mM), as indicated by the arrow. (A) NO (1 μM) was added as indicated by the arrow. Trace annotations: (i) NO alone, (ii) NO followed by PTIO (100 μM) as indicated by the arrow, (iii and iv) NO followed by oxyhemoglobin (7.5 μM) as indicated by the arrow, and (v) mitochondria alone, no additions other than ADP. Traces are representative of at least three similar experiments. (B) Respiration rates in the presence of NO alone, or NO plus scavengers. Values are means ± SEM from at least three independent experiments.
Effects of Liposomes on NO Inhibition of Respiration.
As a further test of the potential of phospholipid membranes to provide an environment for NO consumption, the effects of adding exogenous membranes to respiring mitochondria in the presence of NO were determined. In the first series of experiments, NO was added to mitochondria at ≈70% O2 and, after inhibition was established, liposomes were introduced into the chamber. The introduction of liposomes resulted in an initial rapid decrease in NO concentration, consistent with partitioning into the lipid phase (Fig. 6). The first phase was followed by accelerated NO consumption, which was accompanied by a more rapid recovery of the inhibited respiration rate than in the absence of liposomes. Identical results were obtained whether liposomes were added at the start of the incubation, before the addition of mitochondria or NO (not shown). These data show that the addition of exogenous membrane increases the rate of NO consumption, and that directly impacts the inhibition of respiration by NO. Indeed, the effects of membranes are strikingly similar to those of NO scavengers, supporting the hypothesis that the removal of NO from the binding site in cytochrome c oxidase is based on a shift in the equilibrium between free and bound NO brought about by consumption outside the enzyme's active site. To confirm this hypothesis, the data shown in Fig. 6A were analyzed to yield NO/inhibition curves (Fig. 6B). It is clear that when normalized for NO concentration, inhibition does not change the potency of NO as an inhibitor of cytochrome c oxidase. This result is predicted if the sole effect of adding the excess membrane is to change the equilibrium between free NO and enzyme-bound NO by decreasing the concentration in the mitochondrial inner membrane.
Figure 6.

Effects of exogenous membrane on the NO inhibition of mitochondrial respiration. (A) Mitochondria were incubated at 1 mg/ml in state 3 as described in Materials and Methods. NO (1 μM) was added as indicated by the arrow. After inhibition of respiration, liposomes (1 mg of phospholipid per ml) were added as indicated by the arrow, and the resultant NO and O2 fluxes were monitored. Solid line, control (no liposomes); dashed line, with liposomes. Traces are representative of at least three similar experiments. (B) Inhibition of respiration by NO with and without liposomes was calculated from several experiments of the type shown in A. Data shown are shown in the absence (●) or presence (○) of liposomes.
Discussion
The reversible binding of NO to ferrous heme groups in proteins seems to be responsible for many of its biological actions (20). Known examples are the activation of soluble guanylate cyclase (sGC; refs. 18 and 19), interactions with hemoglobin (20, 30), and as recently proposed, the inhibition of mitochondrial respiration (1, 2). Although the affinity of NO for ferrous heme is extremely high, the binding of NO to these molecules is transient (20). In the case of both sGC and cytochrome c oxidase, this finding is thought to be caused by a competition for NO between the ferrous heme and various NO-consuming pathways in the cell. Although the reversible binding of NO to these heme proteins is an important feature, we know relatively little about what these pathways might be.
Because the inner mitochondrial membrane constitutes one of the most compact and concentrated hydrophobic structures within the cell, we decided to determine whether NO could be consumed by these membranes and what impact it would have on the modulation of mitochondrial respiration through inhibition of cytochrome c oxidase. The primary observation in this article is that the reaction of NO with O2 in mitochondrial membranes, independent of respiratory substrates, can modulate the inhibition of respiration by NO. Thus, the NO available to bind to the reduced intermediate of the enzyme is modulated by the O2 concentration surrounding the binuclear O2 binding site buried in the hydrophobic phase of the membrane. Interestingly, the O2 is thought not to compete directly with NO for the same intermediates in the catalytic cycle.
It is important to recognize that whereas we have chosen to concentrate on the reaction of NO with O2 as the main mechanism of NO consumption within mitochondria, this reaction may not be the only mechanism by which NO is consumed. A number of routes for consumption of NO by mitochondria have been tentatively proposed. However, the mechanisms remain poorly defined and the impact on the control of respiration remains unexplained. One route for NO consumption that has been identified is inhibited by cyanide and azide, and has been ascribed to cytochrome c oxidase (13, 37, 38). Indeed, it has been shown that NO directly reduces Cu to Cu
to Cu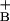 to form NO+ which then becomes hydrated to HNO2 (13, 36). This result raises the possibility that the reaction between NO and O2 in the mitochondrial inner membrane, independent of the enzyme, could account for NO consumption (15, 37). It has also been proposed that NO can react with at least one other redox-active species present in mitochondria associated with complex III (33). However, the fact that the consumption observed here is nonsaturable (Fig. 1) with up to five 1 μM additions precludes any reaction with compounds present at a concentration lower than 5 μM in mitochondria. Under these conditions, the concentration of ubiquinol is ≈5 nmol/mg protein (33), making it unlikely that this reaction is significant in our system.
to form NO+ which then becomes hydrated to HNO2 (13, 36). This result raises the possibility that the reaction between NO and O2 in the mitochondrial inner membrane, independent of the enzyme, could account for NO consumption (15, 37). It has also been proposed that NO can react with at least one other redox-active species present in mitochondria associated with complex III (33). However, the fact that the consumption observed here is nonsaturable (Fig. 1) with up to five 1 μM additions precludes any reaction with compounds present at a concentration lower than 5 μM in mitochondria. Under these conditions, the concentration of ubiquinol is ≈5 nmol/mg protein (33), making it unlikely that this reaction is significant in our system.
The data shown here suggest that NO inhibition of mitochondrial respiration can be rapidly reversed by two structurally distinct NO scavengers (Fig. 5). This result confirms that under these conditions, the inhibition of respiration depends on NO itself and not an NO metabolite such as an S-nitrosothiol. It is important to recognize that these data do not preclude the possibility that in a cellular setting under conditions of low glutathione, inhibition of respiratory complexes such as complex I by S-nitrosation may occur (6). Indeed, it is possible that the partitioning of NO into mitochondrial membranes enhances such reactions. Furthermore, these data do not preclude the intermediate formation of S-nitrosothiols which then lead to NO release. However, these reactions require reducing equivalents and are unlikely to occur at rates sufficient to account for the rapid and complete reversal of NO-dependent inhibition observed after addition of NO scavengers.
One implication of these data is that the lipid:protein ratio and composition will have an impact on the inhibition of mitochondrial respiration by NO. Indeed, it has been known for some time that mitochondrial lipid composition and the lipid:protein ratio vary greatly between tissues and species (39, 40). More recently, the concept that mitochondria may contain an NO synthase could imply a necessary association with the mitochondrial membrane for control of respiration (41, 42). Taken together, these possibilities suggest that mitochondria in different tissues may be differentially sensitive to NO at identical O2 concentrations. It is possible that modulation of mitochondrial lipid composition (by dietary, hormonal, or other means) may also provide a mechanism for regulating the potency of NO as an inhibitor of respiration. The results also suggest that the presence of other biological membranes may affect the ability of NO to inhibit respiration; examples include multiple layers of myelination on neurons and the extensive Golgi networks within secretory cells.
Another interesting phenomenon that may play a role in regulating the availability of NO is the structural organization of mitochondrial membranes. Organization of the inner membrane has been shown to vary drastically between respiring and nonrespiring mitochondria (respiring mitochondria are more compact; ref. 43). Such changes in three dimensional structure may facilitate or inhibit NO and O2 diffusion/partitioning, thereby affecting the regulation of respiration by NO.
These data also have potential implications for other NO-signaling pathways (e.g., soluble guanylate cyclase), because the reaction between NO and O2 in the membrane will impact on NO availability at distal nonmembrane sites within the cell. Membrane partitioning of NO will, in turn, affect the equilibria of NO binding to diverse targets and result in the reversibility of NO signaling, which will increase as a function of the proximity and availability of biological membranes. Some of these aspects have also been appreciated in recent studies (15–17).
In summary, the regulation of O2 and NO gradients by the controlled production of NO by enzymes at specific sites in the cell and the inhibition of respiration at cytochrome c oxidase represent a regulatory mechanism linking cell signaling to metabolism. As with most complex regulatory processes, control is evident at several levels. The first level is the acute interplay between NO and O2 that has been alluded to by other investigators (15). The second level is the potential for the mitochondrial NO-signaling system to participate in cell differentiation, growth, and apoptosis. The third level is exemplified by these studies, and suggests that the modulation of membrane composition at key sites of NO action is a further critical factor.
Acknowledgments
P.S.B. is funded by an American Heart Association postdoctoral fellowship. R.P.P. is a Parker B. Francis Fellow in Pulmonary Research and acknowledges support from American Heart Association Southeastern Affiliate Grant 0060328B. P.G.A. is funded by National Institutes of Health Grant RO1HL58895. V.M.D.-U. is funded by the American Diabetes Association, and National Institutes of Health Grants RO1ES/HL10167 and RO1HL58031.
Abbreviations
- PTIO
2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide
- SMPs
submitochondrial particles
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Cleeter M W, Cooper J M, Darley-Usmar V M, Moncada S, Schapira A H. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 2.Brown G C. FEBS Lett. 1995;369:136–139. doi: 10.1016/0014-5793(95)00763-y. [DOI] [PubMed] [Google Scholar]
- 3.Brookes P S, Salinas E P, Darley-Usmar K, Eiserich J P, Freeman B A, Darley-Usmar V M, Anderson P G. J Biol Chem. 2000;275:20474–20479. doi: 10.1074/jbc.M001077200. [DOI] [PubMed] [Google Scholar]
- 4.De Nadai C, Sestili P, Cantoni O, Lievremont J P, Sciorati C, Barsacchi R, Moncada S, Meldolesi J, Clementi E. Proc Natl Acad Sci USA. 2000;97:5480–5485. doi: 10.1073/pnas.070062397. . (First Published May 2, 2000; 10.1073/pnas.070062397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torres J, Darley-Usmar V, Wilson M T. Biochem J. 1995;312:169–173. doi: 10.1042/bj3120169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clementi E, Brown G C, Feelisch M, Moncada S. Proc Natl Acad Sci USA. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singer M, Brealey D. Biochem Soc Symp. 1999;66:149–166. doi: 10.1042/bss0660149. [DOI] [PubMed] [Google Scholar]
- 8.Brookes P S, Zhang J, Dai L, Zhou F, Parks D A, Darley-Usmar V M, Anderson P G. J Mol Cell Cardiol. 2001;33:69–82. doi: 10.1006/jmcc.2000.1276. [DOI] [PubMed] [Google Scholar]
- 9.Brown G C. Mol Cell Biochem. 1997;174:189–192. [PubMed] [Google Scholar]
- 10.Loke K E, Laycock S K, Mital S, Wolin M S, Bernstein R, Oz M, Addonizio L, Kaley G, Hintze T H. Circulation. 1999;100:1291–1297. doi: 10.1161/01.cir.100.12.1291. [DOI] [PubMed] [Google Scholar]
- 11.Brown G C. Acta Physiol Scand. 2000;168:667–674. doi: 10.1046/j.1365-201x.2000.00718.x. [DOI] [PubMed] [Google Scholar]
- 12.Giuffre A, Sarti P, D'Itri E, Buse G, Soulimane T, Brunori M. J Biol Chem. 1996;271:33404–33408. doi: 10.1074/jbc.271.52.33404. [DOI] [PubMed] [Google Scholar]
- 13.Sarti P, Giuffre A, Forte E, Mastronicola D, Barone M C, Brunori M. Biochem Biophys Res Commun. 2000;274:183–187. doi: 10.1006/bbrc.2000.3117. [DOI] [PubMed] [Google Scholar]
- 14.Okada S, Takehara Y, Yabuki M, Yoshioka T, Yasuda T, Inoue M, Utsumi K. Physiol Chem Phys Med NMR. 1996;28:69–82. [PubMed] [Google Scholar]
- 15.Liu X, Miller M J, Joshi M S, Thomas D D, Lancaster J R. Proc Natl Acad Sci USA. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lancaster J R. Nitric Oxide. 1997;1:18–30. doi: 10.1006/niox.1996.0112. [DOI] [PubMed] [Google Scholar]
- 17.Thomas D D, Liu X, Kantrow S P, Lancaster J R., Jr Proc Natl Acad Sci USA. 2001;98:355–360. doi: 10.1073/pnas.011379598. . (First Published December 26, 2000; 10.1073/pnas.011379598) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ignarro L J, Adams J B, Horwitz P M, Wood K S. J Biol Chem. 1986;261:4997–5002. [PubMed] [Google Scholar]
- 19.Ignarro L J, Degnan J N, Baricos W H, Kadowitz P J, Wolin M S. Biochim Biophys Acta. 1982;718:49–59. doi: 10.1016/0304-4165(82)90008-3. [DOI] [PubMed] [Google Scholar]
- 20.Cooper C E. Biochim Biophys Acta. 1999;1411:290–309. doi: 10.1016/s0005-2728(99)00021-3. [DOI] [PubMed] [Google Scholar]
- 21.O'Donnell V B, Taylor K B, Parthasarathy S, Kuhn H, Koesling D, Friebe A, Bloodsworth A, Darley-Usmar V M, Freeman B A. J Biol Chem. 1999;274:20083–20091. doi: 10.1074/jbc.274.29.20083. [DOI] [PubMed] [Google Scholar]
- 22.Kojda G, Harrison D. Cardiovasc Res. 1999;43:562–571. doi: 10.1016/s0008-6363(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 23.Torres J, Cooper C E, Sharpe M, Wilson M T. J Bioenerg Biomembr. 1998;30:63–69. doi: 10.1023/a:1020559528124. [DOI] [PubMed] [Google Scholar]
- 24.Antonini E, Brunori M. Haemoglobin and Myoglobin and Their Reaction with Ligands. Amsterdam: Elsevier; 1971. p. 24. [Google Scholar]
- 25.Rickwood D, Wilson M T, Darley-Usmar V M. In: Mitochondria, A Practical Approach. Darley-Usmar V M, Rickwood D, Wilson M T, editors. Oxford: IRL; 1987. pp. 3–5. [Google Scholar]
- 26.Lowry O H, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 27.Shepherd J A, Garland P B. Methods Enzymol. 1969;13:11–19. [Google Scholar]
- 28.Wharton D C, Tzagoloff A. Methods Enzymol. 1967;10:245–250. [Google Scholar]
- 29.Beckman J S, Wink D A, Crow J P. In: Methods in Nitric Oxide Research. Feelisch M, Stamler J S, editors. New York: Wiley; 1996. pp. 64–65. [Google Scholar]
- 30.Kharitonov V G, Bonaventura J, Sharma V S. In: Methods in Nitric Oxide Research. Feelisch M, Stamler J S, editors. New York: Wiley; 1996. pp. 39–44. [Google Scholar]
- 31.Kharitonov V G, Sundquist A R, Sharma V S. J Biol Chem. 1994;269:5881–5883. [PubMed] [Google Scholar]
- 32.Meredith M, Reed D. J Biol Chem. 1982;257:3747–3753. [PubMed] [Google Scholar]
- 33.Poderoso J J, Lisdero C, Schopfer F, Riobo N, Carreras M C, Cadenas E, Boveris A. J Biol Chem. 1999;274:37709–37716. doi: 10.1074/jbc.274.53.37709. [DOI] [PubMed] [Google Scholar]
- 34.Packer M A, Porteous C M, Murphy M P. Biochem Mol Biol Int. 1996;40:527–534. doi: 10.1080/15216549600201103. [DOI] [PubMed] [Google Scholar]
- 35.O'Donnell V B, Chumley P H, Hogg N, Bloodsworth A, Darley-Usmar V M, Freeman B A. Biochemistry. 1997;36:15216–15223. doi: 10.1021/bi971891z. [DOI] [PubMed] [Google Scholar]
- 36.Boveris A, Costa L E, Poderoso J J, Carreras M C, Cadenas E. Ann N Y Acad Sci. 2000;899:121–135. doi: 10.1111/j.1749-6632.2000.tb06181.x. [DOI] [PubMed] [Google Scholar]
- 37.Torres J, Sharpe M A, Rosquist A, Cooper C E, Wilson M T. FEBS Lett. 2000;475:263–266. doi: 10.1016/s0014-5793(00)01682-3. [DOI] [PubMed] [Google Scholar]
- 38.Borutaite V, Brown G C. Biochem J. 1996;315:295–299. doi: 10.1042/bj3150295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daum G. Biochim Biophys Acta. 1985;822:1–42. doi: 10.1016/0304-4157(85)90002-4. [DOI] [PubMed] [Google Scholar]
- 40.Porter R K, Hulbert A J, Brand M D. Am J Physiol. 1996;271:R1550–R1560. doi: 10.1152/ajpregu.1996.271.6.R1550. [DOI] [PubMed] [Google Scholar]
- 41.Giulivi C, Poderoso J J, Boveris A. J Biol Chem. 1998;273:11038–11043. doi: 10.1074/jbc.273.18.11038. [DOI] [PubMed] [Google Scholar]
- 42.Ghafourifar P, Richter C. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- 43.Andrews P M, Hackenbrock C R. Exp Cell Res. 1975;90:127–136. doi: 10.1016/0014-4827(75)90365-1. [DOI] [PubMed] [Google Scholar]