

Abstract
Antisense-mediated exon skipping for Duchenne muscular dystrophy (DMD) is currently tested in phase 3 clinical trials. The aim of this approach is to modulate splicing by skipping a specific exon to reframe disrupted dystrophin transcripts, allowing the synthesis of a partly functional dystrophin protein. Studies in animal models allow detailed analysis of the pharmacokinetic and pharmacodynamic profile of antisense oligonucleotides (AONs). Here, we tested the safety and efficacy of subcutaneously administered 2′-O-methyl phosphorothioate AON at 200 mg/kg/week for up to 6 months in mouse models with varying levels of disease severity: mdx mice (mild phenotype) and mdx mice with one utrophin allele (mdx/utrn+/−; more severe phenotype). Long-term treatment was well tolerated and exon skipping and dystrophin restoration confirmed for all animals. Notably, in the more severely affected mdx/utrn+/− mice the therapeutic effect was larger: creatine kinase (CK) levels were more decreased and rotarod running time was more increased. This suggests that the mdx/utrn+/− model may be a more suitable model to test potential therapies than the regular mdx mouse. Our results also indicate that long-term subcutaneous treatment in dystrophic mouse models with these AONs is safe and beneficial.
Keywords: antisense oligonucleotides, Duchenne muscular dystrophy, dystrophin, exon skipping, mouse models, therapy
Introduction
Antisense-mediated exon skipping has a lot of therapeutic potential for Duchenne muscular dystrophy (DMD), a severe, progressive neuromuscular disorder that affects one in 3,500 newborn boys.1,2 This disease is caused by mutations, mostly deletions of one or more exons, in the dystrophin-encoding DMD gene that disrupt the reading frame and give rise to prematurely truncated proteins that are non functional.3 Dystrophin is crucial for maintaining muscle fiber stability and lack of dystrophin leads to exercise-induced damage and continuous loss of muscle fibers and function and eventually premature death usually in the 3rd decade.4 The exon skipping approach aims to reframe the disrupted transcripts, allowing the production of internally deleted, but partially functional dystrophin proteins, as are found in Becker muscular dystrophy patients.2 This can be achieved with antisense oligonucleotides (AONs), small pieces of modified RNA that specifically hybridize to a target exon, hide it from the splicing machinery causing it not to be included in the mRNA. This approach is mutation-specific, but since deletions cluster in a hotspot region, skipping some exons applies to larger groups of patients. Exon 51 skipping would be beneficial for the largest group of patients (13%)5 and is currently developed in clinical trials. Initial trials using intramuscular injections of AONs of two chemistries [PRO051/GSK 2402968, a 2′-O-methyl phosphorothioate (2OMePS) and AVI-4658, a phosphorodiamidate morpholino oligomer (PMO)] resulted in local dystrophin restoration in the majority of muscle fibers at ~15–35% of wild-type levels.1,6,7 Both compounds have also been tested in systemic trials.8,9 For PRO051/GSK2402968 (2OMePS chemistry) 5 weekly subcutaneous injections at different doses was well tolerated and induced novel dystrophin expression was observed in 60–100% of muscle fibers in post-treatment biopsies of 10/12 DMD patients.9 Dystrophin levels increased in a dose-dependent manner to up to 15.6% of that in healthy muscle. All 12 patients were subsequently enrolled in an open label extension study receiving weekly doses of 6 mg/kg. After 12 weeks, a mean improvement of 35.2 m (±28.7) was observed in the 6-minute walk test (>65 meters in three patients).9 Although these results are very encouraging, they will need to be confirmed by blinded and placebo-controlled follow up studies, which are currently ongoing. Weekly intravenous injections of AVI-4658 (PMO chemistry) for 12 weeks were tested at doses up to 20 mg/kg.8 This revealed dystrophin restoration in seven patients, especially for the higher dose groups. In three very good responders up to 55% of fibers became dystrophin positive and up to 27% of wild-type levels were observed.
Due to AON, dystrophin mRNA transcript and protein turnover, lifelong repeated AON treatment will be required. Animal models have been instrumental in the optimization of systemic delivery of AONs and assessing their pharmacodynamic and pharmacokinetic properties.6,7,10,11,12,13,14,15,16 The dystrophin-negative mdx mouse model is most commonly used. However, the phenotype in mdx mice is less severe than in humans: mice have a more efficient regeneration, muscle function is only mildly impaired and lifespan nearly matches that of wild type mice.17 In mdx mice, antisense-mediated exon 23 skipping resulted in dystrophin restoration and improved muscle function.10,14,18 Due to the minimal phenotype, it is more difficult to establish functional efficacy in this model. In other animal models, such as dogs, loss of dystrophin leads to a severe phenotype that better resembles the human situation.13,19,20 However, dogs are 20–150 times heavier than mice, making systemic studies very expensive.
The mild phenotype in mdx mice might in part be due to overexpression of the dystrophin homologue utrophin. Indeed, mice that lack utrophin in addition to dystrophin (double knockout mice, mdx/utrn–/–) have a very severe phenotype, with a limited lifespan (generally 2–3 months).21 It has been possible to prolong the lifespan of these animals to >1 year using peptide-conjugated PMOs.22 However, due to the severity of this model, animals could only be rescued when treatment started before weaning at very high doses; while treatment that was initiated later, or treatment with lower doses had no effect.22 It has been reported that dystrophin-negative mice with a single utrophin allele (mdx/utrn+/–) have an intermediate phenotype, with higher levels of fibrosis than mdx and more impaired respiratory function.23,24 Our own studies show that muscle function is also more impaired than in dystrophin-negative mice with two utrophin alleles.25 Thus, this model might be more suitable than traditional mdx mice for therapeutic studies, as it would allow a better assessment of the effect of exon skipping on muscle function and quality.26
Toward long-term clinical trials we here tested weekly treatment with 2OMePS AONs at a high dose for a period of 6 months in mdx and mdx/utrn+/− mice. In both models treatment did induce exon skipping, restored dystrophin and improved muscle function and integrity, in the absence of toxic effects. The therapeutic effect was more pronounced in the mdx/utrn+/− mice.
Results
AON uptake and exon in mdx and mdx/utrn+/− mice
Exon 23 skipping would bypass the nonsense mutation present in the Dmd gene of the mdx and mdx/utrn+/− mouse models. We here used 2OMePS AONs targeting the 5′ splice, previously identified by the group of Steve Wilton.11 For 2OMePS AONs, exon skipping after systemic AON treatment of mdx mice can be achieved with a dose of 50 mg/kg/week, but the optimal dose appears to be 200 mg/kg/week,14 which would correspond to a dose of 16 mg/kg in humans after correcting for differences in body surface area between small and large animals (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm078932.pdf).
Mdx and the more severe mdx/utrn+/− mice were treated with weekly subcutaneous injections of 200 mg/kg AONs or saline for 24 weeks starting at 4 weeks of age (n = 7 per group). Treatment was well tolerated by all mice and body weights were similar for saline and AON-treated animals over time (data not shown). Mice were sacrificed 10 days after the last treatment. AON uptake by skeletal muscles, heart, diaphragm, liver, spleen, and kidney was assessed by an AON-specific ligation-hybridization assay (Figure 1a). This revealed higher uptake by kidney for the mdx/utrn+/− mice (P value <0.05), whereas AON tissue levels were comparable for diaphragm, gastrocnemius, tibialis anterior, heart, and liver.
Figure 1.

Biodistribution, RNA, and protein analysis of mdx/utrn+/– and mdx mice treated for 6 months. (a) Biodistribution analysis. (b) Reverse transcription-PCR (RT-PCR) analysis of muscle samples from mdx and mdx/utrn+/– mice. Exon 23 skipping could be confirmed for all samples. G is gastrocnemius, Q is quadriceps, TA is tibialis anterior, T is triceps, H is heart, D is diaphragm, C is negative control. The skip product is smaller than the wild-type fragment and thus binds less ethidium bromide, hampering visual estimation of skipping percentages. (c) Lab-chip analysis of exon skipping percentages. (d) Western blot analysis of gastrocnemius protein lysates of mdx mice treated for 3 or 6 months (3 m and 6 m, respectively) and mdx/utrn+/– mice treated for 6 months. NT, not treated. α-Actinin staining was used to confirm equal loading. (e) Quantification of dystrophin levels for gastrocnemius. Error bars show the SD. *P value <0.05.
Reverse transcription-PCR analysis of skeletal muscles, heart and diaphragm showed exon 23 skipping for all treated mice (Figure 1b), whereas no skipping was observed in saline-treated mice (data not shown). No significant differences were observed between skipping levels observed for mdx and mdx/utrn+/− mice for any of the muscles analyzed (Figure 1c). Exon skipping levels in heart were much lower (~5%) than in the other muscles (~15%), which has been reported before for 2OMePS AONs14,27,28,29 and PMOs.10,28
Exon skipping also resulted in dystrophin restoration as assessed by Western blot analysis (Figure 1d,e). Dystrophin levels in gastrosnemius muscles were comparable to those obtained after 3-month treatment with 2 weekly injections of 100 mg/kg in both mdx and mdx/utrn+/− mice (2–3% of wild-type levels).
Long-term AON treatment leads to improved muscle quality and function
Plasma creatine kinase (CK) levels are a marker for muscle fiber integrity and were assessed on a weekly basis. CK levels in untreated mdx/utrn+/− mice were higher over time than those of mdx mice (Figure 2a). Although the dystrophin levels observed in treated mice were modest, CK levels dropped very significantly in treated mice over time (P value <10–14 and 10–15, for mdx and mdx/utrn+/–, respectively; Figure 2b,c). No difference was observed between treated mdx and mdx/utrn+/– mice.
Figure 2.

Plasma creatine kinases (CKs). (a) Saline-treated mdx versus mdx/utrn+/– mice. CK levels are generally higher for mdx/utrn+/− mice. (b) Saline versus antisense oligonucleotide (AON)-treated mdx mice. (c) Saline versus AON-treated mdx/utrn+/– mice. For both models CK levels vary a lot, but starting at 6 weeks of treatment levels were lower in treated than untreated mice. In wild type mice, CK levels are below 500 U/l. Error bars show the SD.
Muscle function was assessed weekly by grip strength and rotarod analysis. No significant difference was observed at any time point or over time in grip strength between the different models or between the treated and untreated groups (data not shown). Without treatment, the mdx mice clearly outperformed mdx/utrn+/– mice in the rotarod analysis over time (P value <0.01, Figure 3a), corresponding to our previous results in younger mice.25 No difference could be observed between treated and untreated mdx mice (Figure 3b), whereas mdx/utrn+/–-treated mice performed significantly better than untreated mice (P value <0.01; Figure 3c).
Figure 3.

Rotarod running times. (a) Mdx/utrn+/– perform less than mdx mice. (b) No difference could be observed for treated and untreated mdx mice. (c) Treated mdx/utrn+/– clearly outperformed untreated mdx/utrn+/– mice (P value <0.01). Error bars show the SD.
Long-term treatment with 2OMePS AONs is well tolerated
As described above, long-term subcutaneous AON treatment did not result in any obvious changes in body weight during treatment. Plasma was collected at the end of treatment and organs harvested for toxicological analysis, which did not reveal any toxic effects. Liver weights did not differ between treated and untreated mice (Figure 4a) and bilirubin levels (a marker for liver damage/inflammation) were below the detection limit for all mice (data not shown). Alkaline phosphatase (ALP) is a marker for hepatobiliary function. ALP levels were increased in treated mice compared to untreated mice, although levels stayed within the normal range for all groups, but this difference was not significant (P value cut-off 0.01 after multiple testing correction) (Figure 4b). Glutamic oxaloacetic pyruvate transanimase and glutamate pyruvic transanimase are enzymes that are present in liver and muscle and leak into the bloodstream upon liver and muscle damage. Consequently, glutamic oxaloacetic pyruvate transanimase and glutamate pyruvic transanimase levels are elevated in dystrophic mouse models. Notably, levels in mdx mice are much higher than in the more severely affected mdx/utrn+/– mouse. This might be due to strain differences (C57/BL10 versus C57/BL6 backgrounds for mdx and mdx/utrn+/–, respectively). Levels did not further increase for treated mdx mice, while a nonsignificant increase was observed for treated mdx/utrn+/– mice (P value >0.01) (Figure 4c,d). The kidney weight was similar for all groups (Figure 4e), as were urea levels (a marker for kidney function) (Figure 4f). An increase in spleen weight could be observed for treated mdx mice, but this was not significant (Figure 4g). No difference in spleen weight was observed for mdx/utrn+/– mice or in hemoglobin levels for both models (Figure 4h). These results confirm that 6-month treatment with a high dose of 2OMePS AON is well tolerated.
Figure 4.
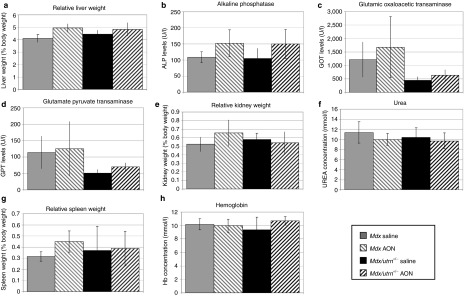
Toxicology analysis of treated and untreated mice. (a–d) Liver weight and liver damage and function markers. (e–f) Kidney weight and function marker. (g) Spleen weight. (h) Hemoglobin levels. Error bars show the SD.
Discussion
Antisense-mediated exon skipping is currently considered one of the most promising therapeutic approaches to treat DMD.1 Due to AON, mRNA transcript and dystrophin protein turnover, repeated treatment will be required. AONs are very small and are therefore generally cleared by the kidney. Due to the PS backbone, 2OMePS AONs will bind to serum proteins, which prevents renal clearance30 and increases the half-life in serum and tissue. We have previously shown that 2OMePS AONs are taken up efficiently by dystrophic muscle and that body wide exon skipping and dystrophin restoration can be achieved after subcutaneous treatment for up to 3 months in mdx mice in the absence of toxic effects.14,27 We here tested 6-month treatment with high doses (200 mg/kg/week) in two different dystrophic mouse models, i.e., the mdx and the slightly more severely affected mdx/utrn+/– model. Compared to the 6 mg/kg/week dose used in clinical trials for GSK2404968, a 2OMePS AON targeting exon 51, the 200 mg/kg/week dose required in mice seems high. However, a correction factor applies when translating doses between small and larger animals based on normalization to body surface area (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm078932.pdf). When applying this correction factor, a dose of 16 mg/kg would be predicted for humans, which is in the same order of magnitude as the 6 mg/kg used in the trials.9 The slightly higher corrected dose for mice might be explained by a higher clearance rate for 2OMePS AONs in mice, potential differences between pharmacokinetic and pharmacodynamic properties between exon 23 and exon 51 targeting AONs and the fact that a more efficient utrophin upregulation in mice leads to more stable muscle fibers and thus lower AON uptake.
We would like to propose the mdx/utrn+/– model as an easier model than the mdx/utrn–/– mouse, where breeding and husbandry is challenging. Still, the disease is more severe in the mdx/utrn+/– model than in the traditionally used mdx mouse model. Higher levels of fibrosis and a more impaired respiratory function have been reported by others24,31 and we recently reported that the mdx/utrn+/– model also performs worse than mdx mice in muscle function tests.25 Thus, assessment of treatment effects on muscle function can be easier picked up in this model. Indeed, while in untreated animals CK levels and rotarod running times were significantly more affected in mdx/utrn+/– mice, they were comparable for mdx and mdx/utrn+/– after 6 months AON treatment. Thus, the same amount of dystrophin was able to achieve a larger therapeutic effect in the more severe mdx/utrn+/– mice. While there is a clear benefit of these low dystrophin levels on muscle function and quality, it should be stressed that they do not fully normalize the phenotype. We have recently shown that levels of ~15–20% are probably needed for a more complete normalization of the phenotype in a mouse model with variable, low dystrophin levels due to skewed X-inactivation.32 Nevertheless, it is clear that even low levels of dystrophin already are beneficial.
Interestingly, we were unable to find a difference between treated and untreated mdx mice in rotarod running after 24-week treatment, while we have previously shown improvement in rotarod running in mdx mice after 8 weeks of AON treatment.14 As the analysis is done over time, a possible explanation for this discrepancy is the different duration of the experiments.
AON levels in muscles were comparable between the mouse models and highest in heart, while skipping levels were lowest for this tissue. However, the hybridization-ligation assay detects AON levels in the complete tissue and cannot discriminate between AON in the tissue and AON trapped in the interstitium. Based on the lower exon skipping levels observed in heart, we hypothesize more AONs are trapped in the interstitium for heart than skeletal muscles. Lower exon skipping levels in heart have been reported previously after systemic 2OMePS and PMO treatment,10,14,15,16,28,29,33,34 probably because the heart cells are not leaky like the skeletal muscle fibers and thus do not benefit from an enhanced uptake of AONs.14,28
When exon skipping and dystrophin levels in mdx mice were compared to those obtained after 4, 8, and 12 weeks of treatment, no further accumulation was observed, while an increase had been observed previously up to 12 weeks.14,27 This might be explained by a different dosing regime that was adopted to reduce stress and handing for the mice. Rather than 2 weekly subcutaneous injections of 100 mg/kg, these mice received 1 weekly injection of 200 mg/kg. It has recently been shown that for PMOs multiple low doses are more efficacious than a single high dose.15 This may also be the case for 2OMePS AONs.
Even though dystrophin levels were quite low in treated mice, significant improvements could be observed in CK levels for both strains and in rotarod running time for the mdx/utrn+/– mice. It has been reported previously that very low amounts of dystrophin have an effect on muscle quality.35,36 This might be related to additional functions of the dystrophin protein in signal transduction and regulating miRNA levels.
Long-term treatment was well tolerated by all mice. There were some slight increases in some markers for liver function and liver damage, but none were significant. In addition, the differences were generally only found for one strain (either mdx or mdx/utrn+/–). This suggests that 6-month treatment with high doses of 2OMePS AON by subcutaneous injections is well tolerated. However, patients will be treated even longer. AONs have thus far primarily been used to treat acute diseases and consequently, long-term safety data in humans is not yet available. Therefore, patients will have to be carefully monitored during long-term clinical trials that are currently ongoing for exon 51 skipping.
In conclusion, we show here that long-term subcutaneous treatment with 2OMePS AONs leads to exon skipping, dystrophin restoration, improved muscle quality and function in two DMD mouse models, in the absence of toxicity.
Materials and Methods
All animal experiments were approved by the Animal Ethics Committee of the Leiden University Medical Center.
AON treatment of mdx/utrn+/− and mdx mice. Mdx/utrn+/– and mdx mice (n = 7 per group) were treated with weekly subcutaneous injections of 200 mg/kg M23D (+2–18) a 2′-O-methyl phosphorothioate AON described previously11 or saline. Treatment started when mice were 4-week old and lasted for 24 weeks. Ten days after the last injection mice were sacrificed by cervical dislocation and muscles (gastrocnemius, quadriceps, tibialis anterior, triceps, heart, and diaphragm) and organs (liver, kidney, and spleen) were isolated and snap frozen in liquid nitrogen cooled 2-methylbutane and stored at –80 °C.
Hybridization-ligation assay. The assay for measuring the AON concentration in tissue samples is based on a previously published hybridization-ligation assay.37 All tissues and calibration curves of the analyzed AON were diluted 60 times in pooled control mdx mouse tissue lysates of the same type. The muscle samples were diluted 500 and 1,000 times, liver and kidney tissue 1,000 and 5,000 times. All analyses were performed in duplicate.
RNA isolation and reverse transcription-PCR. Muscles were minced in RNA-Bee (Campro Scientific, Veenendaal, the Netherlands) using MagNa Lyser green beads (Roche Diagnostics, Almere, the Netherlands) and the MagNa Lyser (Roche Diagnostics) according to the manufacturer's instructions. Total RNA was extracted and reverse transcription-PCR analysis was performed as described with a single round of PCR.38 PCR products were visualized on 2% agarose gels and quantified with the Agilent 2100 Bioanalyzer (Agilent, Amstelveen, the Netherlands).
Protein extraction and western blot. Muscles were homogenized in treatment buffer (100 mmol/l Tris–HCl pH 6.8 and 20% sodium dodecyl sulfate) using MagNa Lyser green beads in the MagNa Lyser (Roche Diagnostics). Protein concentration was determined using the BCA protein assay kit (Thermo Scientific, Etten Leur, the Netherlands), according to the manufacturer's instructions. Subsequently, the homogenate was completed to contain 75 mol/l Tris–HCl pH 6.8, 15% sodium dodecyl sulfate, 5% β-mercaptoethanol, 20% glycerol, and 0.001% bromophenol blue and boiled for 5 minutes. One hundred microgram was loaded on a 4–7% gradient polyacrylamide gel and run overnight at 4 °C. Gels were blotted to nitrocellulose BA83 (Whatman, Schleicher & Schuell, Germany) for 6 hours at 4 °C. Blots were blocked with 5% non-fat dried milk (Campina Melkunie, Maasdam, the Netherlands) in TBS followed by an overnight incubation with NCL-DYS1 (dilution 1:125; Novacastra, Leica Microsystems, Valkerswaard, the Netherlands) in TBS plus 0.05% Tween-20 to detect dystrophin. The fluorescent IRDye 800CW goat-anti-mouse IgG (dilution 1:5,000; Li-Cor, Lincoln, NE) was used as a secondary antibody. Blots were visualized and quantified with the Odyssey system and software (Li-Cor). α-Actinin (AB72592; Abcam, Cambridge, MA, dilution 1:7,500), was used as a loading control.
Plasma parameter assessment. Blood was collected weekly on Monday mornings in a Minicollect tube (0.8-ml Lithium Heparin Sep; Greiner Bio-One, Alphen aan den Rijn, the Netherlands) via a small cut at the end of the tail. CK levels were determined in plasma with Reflotron CK test strips in the Reflotron plus machine (Roche Diagnostics). Immediately before scarifying, blood was collected to assess plasma hemoglobin, ALP, urea, glutamic oxaloacetic transaminase, and glutamate pyruvate transaminase levels using the respective Reflotron test strips.
Functional analyses. Forelimb grip strength was assessed by means of a grip strength meter (Columbus Instruments, Columbus, OH). Mice were tested five times, with three consecutive measurements per trial (15 in total), and a 2-minute interval between trials. The three highest measured values were averaged to calculate the absolute strength, which was divided by the body weight in grams.
A rotarod test was performed weekly with the Rotarod (Ugo Basile, Comerio, Italy). Mice were placed on the rod that accelerated from 5 to 45 rotations per minute within 15 seconds. When a mouse ran for 500 seconds without falling from the rod, the test session was ended. Mice that fell off within 500 seconds were given a maximum of two more tries. The longest running time was used for analysis.
Statistical analysis. A two-tailed homoscedastic Student's t-test was performed in Microsoft Office Excel 2003 to compare AON tissue concentrations, exon skip levels, organ weights and plasma parameters between groups of mice. A P value <0.05 was considered significant for all tests, however for the analyses shown in Figure 4 a Bonferroni correction was applied for multiple testing and a P value <0.01 was considered significant. To assess whether CK levels, forelimb grip strength or rotarod running times differed significantly over time between groups linear regression analysis was performed with statistical software in R (version R.2.11.1), using either mouse strain or treatment as variables. Data from week 0 were excluded from the analysis assessing a potential effect of treatment as this time point was before treatment. A P value <0.05 was considered significant.
Acknowledgments
This work was funded by a grant from the Dutch Duchenne Parent Project and ZonMw (the Netherlands) and used the infrastructures of the Medical Center participates in the Center for Medical System Biology (CMSB) and the Center for Biomedical Genetics (CBG). LUMC is a partner in the FP6 TREAT-NMD network of excellence (LSHM-CT-2006–036825). H.H., A.A.-R. and J.v.D. report being coinventors on patents on exon skipping, licensed by LUMC to Prosensa Therapeutics, and being entitled to a share of royalties. J.v.D., T.G.K. and S.J.dK. report being employed by Prosensa Therapeutics. Grant support: This work was performed using funding of the Duchenne Parent Project (the Netherlands), ZonMw (the Netherlands) and TREAT-NMD (EU F6).
References
- Aartsma-Rus A., and, van Ommen GJ. Progress in therapeutic antisense applications for neuromuscular disorders. Eur J Hum Genet. 2010;18:146–153. doi: 10.1038/ejhg.2009.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ommen GJ, van Deutekom J., and, Aartsma-Rus A. The therapeutic potential of antisense-mediated exon skipping. Curr Opin Mol Ther. 2008;10:140–149. [PubMed] [Google Scholar]
- Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H., and, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2:90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen GJ.et al. (2009Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations Hum Mutat 30293–299. [DOI] [PubMed] [Google Scholar]
- Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C.et al. (2009Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study Lancet Neurol 8918–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M.et al. (2007Local dystrophin restoration with antisense oligonucleotide PRO051 N Engl J Med 3572677–2686. [DOI] [PubMed] [Google Scholar]
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K.et al. (2011Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study Lancet 378595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N.et al. (2011Systemic administration of PRO051 in Duchenne's muscular dystrophy N Engl J Med 3641513–1522. [DOI] [PubMed] [Google Scholar]
- Alter J, Lou F, Rabinowitz A, Yin H, Rosenfeld J, Wilton SD.et al. (2006Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology Nat Med 12175–177. [DOI] [PubMed] [Google Scholar]
- Mann CJ, Honeyman K, McClorey G, Fletcher S., and, Wilton SD. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J Gene Med. 2002;4:644–654. doi: 10.1002/jgm.295. [DOI] [PubMed] [Google Scholar]
- Yin H, Lu Q., and, Wood M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol Ther. 2008;16:38–45. doi: 10.1038/sj.mt.6300329. [DOI] [PubMed] [Google Scholar]
- Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S.et al. (2009Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs Ann Neurol 65667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk H, de Winter C, van Kuik P, Heuvelmans N, Sabatelli P, Rimessi P.et al. (2010Preclinical PK and PD studies on 2'-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model Mol Ther 181210–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malerba A, Thorogood FC, Dickson G., and, Graham IR. Dosing regimen has a significant impact on the efficiency of morpholino oligomer-induced exon skipping in mdx mice. Hum Gene Ther. 2009;20:955–965. doi: 10.1089/hum.2008.157. [DOI] [PubMed] [Google Scholar]
- Malerba A, Sharp PS, Graham IR, Arechavala-Gomeza V, Foster K, Muntoni F.et al. (2011Chronic systemic therapy with low-dose morpholino oligomers ameliorates the pathology and normalizes locomotor behavior in mdx mice Mol Ther 19345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain JS, Metzger J, Reyes M, Townsend D., and, Faulkner JA. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007;21:2195–2204. doi: 10.1096/fj.06-7353com. [DOI] [PubMed] [Google Scholar]
- Yin H, Moulton HM, Seow Y, Boyd C, Boutilier J, Iverson P.et al. (2008Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function Hum Mol Genet 173909–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClorey G, Moulton HM, Iversen PL, Fletcher S., and, Wilton SD. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006;13:1373–1381. doi: 10.1038/sj.gt.3302800. [DOI] [PubMed] [Google Scholar]
- Walmsley GL, Arechavala-Gomeza V, Fernandez-Fuente M, Burke MM, Nagel N, Holder A.et al. (2010A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skipping PLoS ONE 5e8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L.et al. (1997Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy Cell 90717–727. [DOI] [PubMed] [Google Scholar]
- Goyenvalle A, Babbs A, Powell D, Kole R, Fletcher S, Wilton SD.et al. (2010Prevention of dystrophic pathology in severely affected dystrophin/utrophin-deficient mice by morpholino-oligomer-mediated exon-skipping Mol Ther 18198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Rafael-Fortney JA, Huang P, Zhao XS, Cheng G, Zhou X.et al. (2008Haploinsufficiency of utrophin gene worsens skeletal muscle inflammation and fibrosis in mdx mice J Neurol Sci 264106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Cheng G, Lu H, Aronica M, Ransohoff RM., and, Zhou L. Impaired respiratory function in mdx and mdx/utrn(+/-) mice. Muscle Nerve. 2011;43:263–267. doi: 10.1002/mus.21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Putten M, Kumar D, Hulsker M, Hoogaars WM, Plomp JJ, van Opstal A.et al. (2012Comparison of skeletal muscle pathology and motor function of dystrophin and utrophin deficient mouse strains Neuromuscul Disord 22406–417. [DOI] [PubMed] [Google Scholar]
- Willmann R, Luca AD, Benatar M, Grounds M, Dubach J, Raymackers JM, TREAT-NMD Neuromuscular Network et al. Enhancing translation: guidelines for standard pre-clinical experiments in mdx mice. Neuromuscul Disord. 2012;22:43–49. doi: 10.1016/j.nmd.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk H, de Winter CL, van Ommen GJ, van Deutekom JC., and, Aartsma-Rus A. Development of antisense-mediated exon skipping as a treatment for duchenne muscular dystrophy. Ann N Y Acad Sci. 2009;1175:71–79. doi: 10.1111/j.1749-6632.2009.04973.x. [DOI] [PubMed] [Google Scholar]
- Heemskerk HA, de Winter CL, de Kimpe SJ, van Kuik-Romeijn P, Heuvelmans N, Platenburg GJ.et al. (2009In vivo comparison of 2'-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping J Gene Med 11257–266. [DOI] [PubMed] [Google Scholar]
- Lu QL, Rabinowitz A, Chen YC, Yokota T, Yin H, Alter J.et al. (2005Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles Proc Natl Acad Sci USA 102198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberbauer R, Schreiner GF., and, Meyer TW. Renal uptake of an 18-mer phosphorothioate oligonucleotide. Kidney Int. 1995;48:1226–1232. doi: 10.1038/ki.1995.406. [DOI] [PubMed] [Google Scholar]
- Zhou L, Rafael-Fortney JA, Huang P, Zhao XS, Cheng G, Zhou X.et al. (2008Haploinsufficiency of utrophin gene worsens skeletal muscle inflammation and fibrosis in mdx mice J Neurol Sci 264106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Putten M, Hulsker M, Nadarajah VD, van Heiningen SH, van Huizen E, van Iterson M.et al. (2012The effects of low levels of dystrophin on mouse muscle function and pathology PLoS ONE 7e31937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malerba A, Boldrin L., and, Dickson G. Long-term systemic administration of unconjugated morpholino oligomers for therapeutic expression of dystrophin by exon skipping in skeletal muscle: implications for cardiac muscle integrity. Nucleic Acid Ther. 2011;21:293–298. doi: 10.1089/nat.2011.0306. [DOI] [PubMed] [Google Scholar]
- Wu B, Lu P, Benrashid E, Malik S, Ashar J, Doran TJ.et al. (2010Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino Gene Ther 17132–140. [DOI] [PubMed] [Google Scholar]
- Cacchiarelli D, Martone J, Girardi E, Cesana M, Incitti T, Morlando M.et al. (2010MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway Cell Metab 12341–351. [DOI] [PubMed] [Google Scholar]
- Verhaart IE, Heemskerk H, Karnaoukh TG, Kolfschoten IG, Vroon A, van Ommen GJ.et al. (2012Prednisolone treatment does not interfere with 2'-O-methyl phosphorothioate antisense-mediated exon skipping in Duchenne muscular dystrophy Hum Gene Ther 23262–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RZ, Baker B, Chappell A, Geary RS, Cheung E., and, Levin AA. Development of an ultrasensitive noncompetitive hybridization-ligation enzyme-linked immunosorbent assay for the determination of phosphorothioate oligodeoxynucleotide in plasma. Anal Biochem. 2002;304:19–25. doi: 10.1006/abio.2002.5576. [DOI] [PubMed] [Google Scholar]
- Spitali P, Heemskerk H, Vossen RH, Ferlini A, den Dunnen JT, ‘t Hoen PA.et al. (2010Accurate quantification of dystrophin mRNA and exon skipping levels in duchenne muscular dystrophy Lab Invest 901396–1402. [DOI] [PubMed] [Google Scholar]