

Abstract
Ammonia is metabolized by the liver and has established neurological effects. The current study examined the possibility that ammonia contributes to the neurotoxic effects of methamphetamine (METH). The results show that a binge dosing regimen of METH to the rat increased plasma and brain ammonia concentrations that were paralleled by evidence of hepatotoxicity. The role of peripheral ammonia in the neurotoxic effects of METH was further substantiated by the demonstration that the enhancement of peripheral ammonia excretion blocked the increases in brain and plasma ammonia and attenuated the long-term depletions of dopamine and serotonin typically produced by METH. Conversely, the localized perfusion of ammonia in combination with METH, but not METH alone or ammonia alone, into the striatum recapitulated the neuronal damage produced by the systemic administration of METH. Furthermore, this damage produced by the local administration of ammonia and METH was blocked by the GYKI 52466 [4-(8-methyl-9H-1,3-dioxolo[4,5-h][2,3]benzodiazepin-5-yl)-benzamine hydrochloride], an AMPA receptor antagonist. These findings highlight the importance of ammonia derived from the periphery as a small-molecule mediator of METH neurotoxicity and more broadly emphasize the importance of peripheral organ damage as a possible mechanism that mediates the neuropathology produced by drugs of abuse and other neuroactive molecules.
Introduction
Much research has been directed toward understanding the fundamental processes of addiction to methamphetamine (METH) and the mechanistic underpinnings of its neurotoxicity. The causes of the neurotoxicity are conventionally thought to originate in the brain; however, due to the metabolic dependence of the brain on peripheral organ function and the high susceptibility of the brain to alterations in systemic physiology, peripheral organ damage may contribute significantly to the neurological damage caused by METH.
METH produces relatively specific damage to dopamine and 5-HT terminals (Ricaurte et al., 1980; Wagner et al., 1980; Seiden et al., 1988). Established mechanisms include excitotoxicity, oxidative damage, inflammation, and metabolic compromise (Yamamoto and Raudensky, 2008), all of which are assumed to be initiated by factors in the CNS. However, little consideration is given to peripheral organ damage. An early report on the clinical presentation of intravenous METH abusers noted hepatitis as the second most common presenting complaint (Smith and Fischer, 1970). However, since that finding, little more has been studied along these lines until a clinical report appeared demonstrating that METH causes acute liver failure in the absence of viral hepatitis (Kamijo et al., 2002). Thus, there is a high potential for METH to produce hepatotoxicity (Smith and Fischer, 1970; Kamijo et al., 2002; Wijetunga et al., 2003; Ago et al., 2006). Surprisingly, it is unknown whether liver damage mediates the neurobiological actions of METH despite knowledge that it can compromise CNS function, as evidenced in hepatic encephalopathy. Because the liver is essential for ammonia metabolism and elimination, hyperammonemia is a key pathological feature of hepatic encephalopathy and is responsible for the neurological sequelae depicted by motor and cognitive deficits (Felipo and Butterworth, 2002).
Ammonia, much like METH, causes neuronal damage via excitotoxicity, oxidative stress, and inflammation. Ammonia decreases the expression of the astrocytic glutamate transporter, EAAT-1, thereby increasing extracellular glutamate, depolarizing glutamate receptors, and producing excitotoxicity (Fan and Szerb, 1993; Chan and Butterworth, 1999; Chan et al., 2000). Ammonia also increases glutamate release from astrocytes (Görg et al., 2010) and ammonia neurotoxicity is attenuated by glutamate receptor antagonists (Sáez et al., 1999; Kosenko et al., 2003). Ammonia also causes oxidative damage through alterations in oxidative metabolism (Hawkins et al., 1973; McCandless and Schenker, 1981), increases superoxide radicals, and elevates glutathione peroxidase, superoxide dismutase, and catalase activities (Kosenko et al., 1997). Moreover, ibuprofen attenuates the behavioral consequences of the neuronal damage caused by ammonia, suggesting a role for inflammation in ammonia neurotoxicity (Cauli et al., 2007).
The parallels between the mechanisms underlying ammonia and METH neurotoxicities prompted our hypothesis that METH causes liver damage and parallel elevations in plasma and brain ammonia concentrations, which converge with the neuropharmacological effects of METH to produce neuronal damage. To this end, we assessed hepatocellular damage, plasma and brain ammonia, and the direct effects of METH and/or ammonia on striatal dopamine and 5-HT content. To further substantiate a causative role for ammonia, peripheral ammonia excretion was enhanced during and after METH and the long-term effects on dopamine and 5-HT content were examined.
Materials and Methods
Drug treatments.
Male Sprague Dawley rats received METH (10 mg/kg, i.p., every 2 h × 4) or saline (1 ml/kg, i.p., every 2 h × 4) (Sigma-Aldrich). This dose of METH was chosen because it produces long-term neuronal damage similar to what is seen in studies of human METH users (McCann et al., 1998). In lactulose experiments, rats received lactulose (5.3 g/kg, Pharmaceutical Associates) or vehicle (8 mL/kg every 12 h, 100 mg/ml galactose and 80 mg/ml lactose, Fisher Scientific) via oral gavage every 12 h for 2 d before METH treatment to the day before rats were killed. Lactulose enhances ammonia excretion and attenuates increases in plasma ammonia and the neurological symptoms of hepatic encephalopathy (Jia and Zhang, 2005; Nicaise et al., 2008; Al Sibae and McGuire, 2009). All rats were killed by rapid decapitation 2 h after the end of treatment or 24 h or 7 d after the start of the METH or saline treatment. Temperature transponders (IPTT-300 transponder, BMDS) were subcutaneously implanted into the rats 2 d before the beginning of each experiment to allow for equilibration. This method of remote temperature monitoring minimizes stress to the rats produced by repeated rectal recordings. All treatments were performed in accordance with the National Institutes of Health Guide for Care and Use of Rats. All treatments have also been approved by the University of Toledo Institutional Animal Care and Use Committee.
Hematoxylin and eosin staining.
Liver tissue was fixed overnight in formalin and embedded in paraffin. Tissue sections 4 mm thick were stained using the Leica Autostainer XL for hematoxylin and eosin.
Aspartate aminotransferase, alanine aminotransferase, and ammonia determination.
Serum was prepared by allowing trunk blood to coagulate after collection. Samples were then centrifuged at 3000 × g for 10 min to separate serum. Plasma was prepared using BD Microtainer Plasma Separation Tubes centrifuged at 10,000 × g for 2 min. Serum was analyzed for the liver enzymes aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels. When hepatocytes are damaged, hepatocellular cytosolic enzymes are released into the blood and can be quantified as a measure of cellular damage. Plasma was analyzed for ammonia on a UniCel DxC800 Synchron Clinical System (Beckman Coulter).
In vivo microdialysis.
Intracranial probes for use in microdialysis were constructed using PE 20 tubing (Becton Dickinson), silica tubing (OD, 150 μm; Polymicro Technologies), 26 ga stainless steel hypodermic tubing (Small Parts), Hollow Fiber Microdialysis Membrane (active membrane, 4 mm; 13,000 molecular weight cutoff, 216 μm outer diameter; Spectrum Laboratories), 2 ton waterproof epoxy, and tygon microbore tubing (Yamamoto and Pehek, 1990). Surgeries were performed on the morning of the day before drug treatment. Rats were anesthetized using xylazine (5 mg/kg) and ketamine (75 mg/kg) and the probe was stereotaxically lowered into the striatum. For brain ammonia measurements 1 d later and after a 1 h equilibration period, baseline samples were collected every hour for 3 h at a flow rate of 1.5 μl/min. Samples were collected every hour during drug treatment until 2 h after the last drug injection. For reverse dialysis experiments, METH (100 μm), ammonium chloride (570 μm), and/or GYKI 52466 [4-(8-methyl-9H-1,3-dioxolo[4,5-h][2,3]benzodiazepin-5-yl)-benzamine hydrochloride, 100 μm, Sigma-Aldrich] were reverse dialyzed at a flow rate of 1.5 μl/min for 8 h. Probe placement was verified histologically. For reverse dialysis experiments, 450 μm thick sections containing the probe tract was cut using a cryostat and tissue adjacent to the tract (1 mm) was dissected while the tissue was kept frozen.
Brain ammonia determination.
Ammonia concentrations in microdialysate were determined using the Sigma-Aldrich Ammonia Assay Kit. This assay uses l-glutamate dehydrogenase to measure ammonia concentration as a stoichiometric product of NADPH levels via the reaction: α-ketoglutarate + NH4+ + NADPH → l-Glu + NADP+ + H2O. NADPH levels were monitored by measuring absorbance at 340 nm using a Molecular Devices Spectramax microplate spectrophotometer. In each determination, we used an internal standard made of 10 μg/ml ammonium sulfate, and we used only assays in which the concentration of the internal standard was within 5% of the true concentration of ammonia.
HPLC analysis of monoamine tissue content.
Tissue was sonicated in 1.0 ml of 0.25 N perchloric acid and centrifuged at 14,000 × g for 20 min at 4°C. For reverse dialysis experiments, 300 μl of perchloric acid was used. Supernatant was then analyzed for 5-HT and dopamine using HPLC (Breier et al., 2006). Supernatant was injected on a C18 column (100 × 2 mm, 3 μm particle diameter, Phenomenex) and eluted with a mobile phase containing 32 mm citric acid, 54.3 mm sodium phosphate, 0.215 mm octyl sodium sulfate, and 11% methanol, pH 4.4. Concentration of monoamines was determined with a LC-4C amperometric detector (Bioanalytical Systems) and data were recorded using EZ Chrom software. Monoamine levels were normalized to protein content of samples. The pellet from the centrifugation was neutralized using 1.0 ml of 1 m NaOH and protein amount was determined using a Bradford assay. For reverse dialysis experiments, 100 μl of NaOH was used.
Western blot for dopamine transporter immunoreactivity.
To prepare synaptosomes, striatal tissue was homogenized in 0.32 m sucrose. Homogenate was then centrifuged at 800 × g for 24 min. The pellet was discarded (P1). Supernatant (S1) was decanted and centrifuged at 22,000 × g for 17 min. Supernatant was discarded (S2) and the pellet (P2) was resuspended in cold Millipore water. A Bradford assay was then used to determine protein concentration and samples were diluted (1:4) with lithium dodecylsulfate sample buffer (Invitrogen). Ten micrograms of each protein was loaded onto a NuPAGE 4–12%, 1.5 mm × 10 wells, Bis-Tris Gel (Invitrogen). Proteins were then run at 150 V for 80 min and transferred to polyvinylidine difluoride membranes at 28 V for 2 h. Membranes were then washed with TBS-T (20 mm Tris, 137 mm NaCl, and 0.5% Tween 20) and blocked for 1 h at room temperature using TBS-T with 5% powdered milk. Membranes were incubated overnight at 4°C with primary antibody to DAT (C-20, Santa Cruz Biotechnology). Membranes were then washed with TBS-T incubated with the appropriate horseradish peroxidase secondary antibodies (1:2500 dilution) at room temperature for 1 h, and developed using the HyGLO-enhanced chemiluminescence (Denville Scientific). All membranes were imaged using the Fuji LAS-4000 minisystem and software. The band at 70 kDa was quantified and all bands were normalized to an α-tubulin internal loading control.
Mass spectroscopy.
Mass spectroscopy was used to determine METH concentrations in striata. Tissue was prepared as in the 5-HT and dopamine tissue content assays. Supernatant was then injected on to a MetaChem MetaSil Basic column (3.0 × 100 mm; pore size, 3 μm; Varian) and measured using electrospray ionization as the ionization source.
Statistical analysis.
Analysis for AST and ALT levels in saline-treated and METH-treated rats and brain concentration of METH in rats treated with lactulose or vehicle and METH was performed using a t test. Analysis of plasma ammonia, hyperthermia, and tissue content of dopamine and 5-HT in the striatum was performed using a two-way ANOVA followed by post hoc Tukey's multiple-comparison tests or t tests. These tests were performed using SigmaPlot 11.0 software (SigmaPlot for Windows, Systat Software). Statistical analysis of brain ammonia levels was performed using a repeated-measures ANOVA test, with each time point as a within-subjects factor and lactulose and METH treatment as between-subjects factor. These tests were performed using SPSS Statistics 17.0 software (IBM). All data are presented as mean ± SEM. α Level in all studies is ≤0.05 and sample sizes were chosen to result in a power of ≥80%.
Results
METH and liver damage
Hematoxylin and eosin staining were used to examine the effects of METH (10 mg/kg, i.p., every 2 h × 4 injections) or saline (1 ml/kg, i.p., every 2 h × 4 injections) on the livers of treated rats. Livers were examined at 2 and 24 h after the last of the four injections. At 2 h, there is moderate vascular and sinusoidal congestion evident throughout the livers of all METH-treated rats (Fig. 1). This vascular and sinusoidal congestion was observed in the livers of all METH-treated rats examined at this time point and the image in Figure 1 is representative of the congestion seen in the livers of all rats. Similarly, at 24 h, extensive cytoplasmic damage is observed in the livers of 100% of METH-treated rats. When METH-induced hyperthermia was attenuated by cooling rats to the temperature of saline-treated rats, METH-induced changes in hepatocellular morphology were attenuated (data not shown).
Figure 1.

METH causes hepatocellular damage visible by hematoxylin and eosin staining. Hematoxylin and eosin staining was used to examine the effects of METH (10 mg/kg, i.p., every 2 h × 4) or saline (1 ml/kg, i.p., every 2 h × 4) on the livers of treated rats. Livers were examined at 2 and 24 h after the last injection of METH or saline. A representative image of a hepatic lobule near the central vein is shown (asterisks denote central vein). The images in the figure are representative of all rats examined for each treatment and at each time point. At 2 h, there is moderate vascular and sinusoidal congestion evident throughout the livers of METH-treated rats (white arrows). At 24 h, this congestion leads to extensive cytoplasmic disappearance in the livers of all METH-treated rats (black arrows) (magnification, 40×).
To confirm that the morphologic changes depicted in Figure 1 represent significant hepatocellular damage, the serum concentration of hepatocyte cytosolic enzymes was measured at 2 h after the last injection of METH (10 mg/kg, i.p., every 2 h × 4) or saline (1 ml/kg, i.p., every 2 h × 4) in the same rats examined in Figure 1. When hepatocytes are damaged, cytosolic contents, such as AST and ALT, are released into the blood and can be measured. At 2 h after the last injection of METH, serum AST concentration was 193.4 ± 15.7 IU/L in saline-treated rats and significantly elevated to 447.8 ± 74.8 IU/L in METH-treated rats (t = 215.00, p < 0.001). At this time point, ALT concentration was 70.6 ± 3.2 IU/L in saline-treated rats and significantly increased to 109.3 ± 9.8 IU/L in METH-treated rats (t = 95.0, p < 0.005). Accordingly, AST and ALT levels are significantly elevated by 141.0 ± 44.1% and 54.9 ± 15.2%, respectively, in METH-treated rats (Fig. 2).
Figure 2.
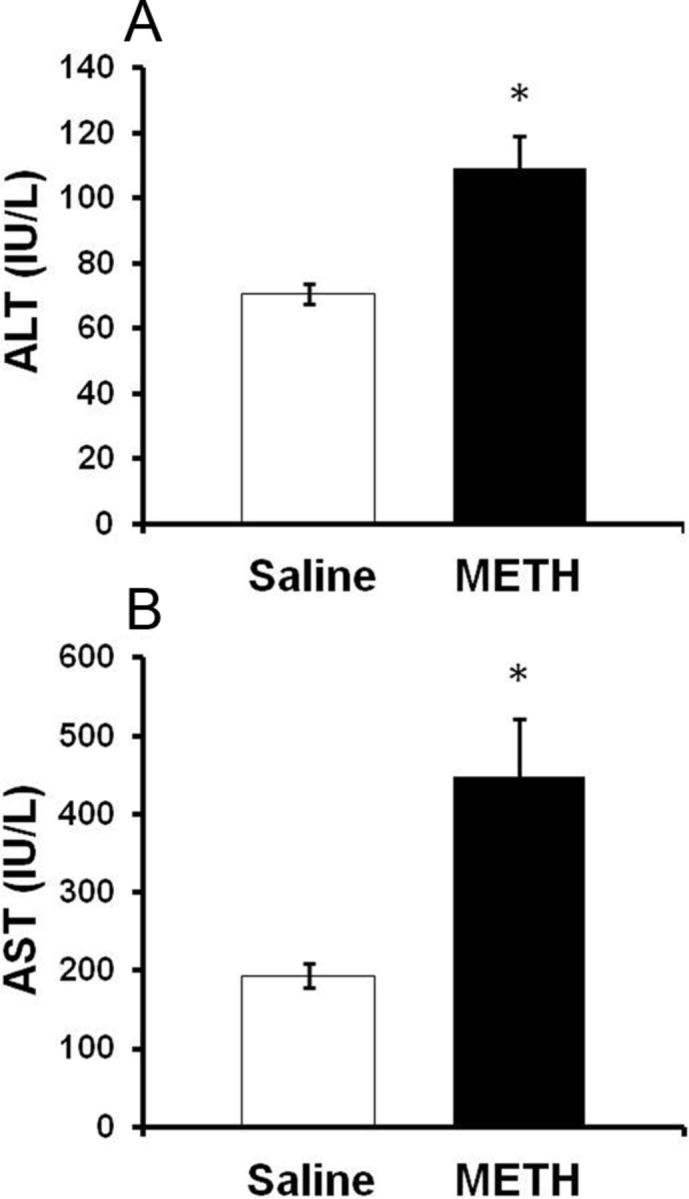
Serum AST and ALT after METH treatment. A, B, Rats were treated with METH (10 mg/kg, i.p., every 2 h × 4) or saline (1 ml/kg, i.p., every 2 h × 4). At 2 h after the last injection of METH or saline, (A) ALT and (B) AST levels were significantly elevated to 54.9 ± 15.2% and 141.0 ± 44.1% in METH-treated rats compared with saline controls, respectively. Values are mean ± SEM, *p < 0.05 (n = 12 rats per group).
METH and plasma and brain ammonia
To determine whether METH affected ammonia metabolism, plasma ammonia was measured at 2 h after the end of METH (10 mg/kg, i.p., every 2 h × 4) or saline (1 ml/kg, i.p., every 2 h × 4) treatment. A two-way ANOVA indicated a significant effect of METH treatment (F(1,31) = 18.25, p < 0.001) and of lactulose treatment (F(1,31) = 13.60, p < 0.001), and a significant interaction between METH and lactulose treatment (F(1,31) = 22.98, p < 0.001). At 2 h after the last injection of METH, plasma ammonia levels were significantly elevated by 64.04 ± 9.98%, and this increase was blocked with lactulose treatment (Fig. 3).
Figure 3.

METH and plasma ammonia. Rats were treated with METH (10 mg/kg, i.p., every 2 h × 4) or saline (1 ml/kg, i.p., every 2 h × 4) and lactulose (5.3 g/kg, p.o., every 12 h) or vehicle (8 ml/kg, p.o., every 12 h). At 2 h after the last injection of METH or saline, plasma ammonia levels were significantly elevated by 64.04 ± 9.98% (mean ± SEM) and this increase was blocked by lactulose treatment (*p < 0.001 compared with Lac Sal, &p < 0.001 compared with Veh METH) (n = 8 rats per group). Lac, lactulose; Sal, saline; Veh, vehicle.
To examine whether elevations in plasma ammonia translate to elevations in brain ammonia, the brain concentration of ammonia was measured using in vivo microdialysis during METH (10 mg/kg, i.p., every 2 h × 4) or saline (1 ml/kg, i.p., every 2 h × 4) treatment. Rats were also treated with lactulose (5.3 g/kg) or vehicle (8 ml/kg) every 12 h for 2 d before and on the morning of drug treatment. Baseline brain ammonia levels in all groups were 29.34 ± 5.33 μm. A repeated-measures ANOVA indicated a significant effect of METH treatment (F(7,279) = 2.43, p < 0.050) and a significant interaction between METH and lactulose treatment (F(7,279) = 2.79, p < 0.050) on brain ammonia, indicating that METH increased brain ammonia and that this increase was blocked by the peripheral administration of lactulose. Brain ammonia levels in METH-treated rats were significantly elevated beginning at the 5 h time point (F(3,31) = 4.22, p < 0.05). At the 8 h time point (Fig. 4), brain concentration of ammonia in saline-treated rats was 34.5 ± 8.0 μm and was 67.9 ± 8.7 μm in METH-treated rats, representing a 196.8 ± 18.0% increase in ammonia in the striata of METH-treated rats. Based on the observation that the in vivo recovery of ammonia by the microdialysis probe is 35% (data not shown), brain ammonia at 8 h after the first METH injection is ∼200 μm.
Figure 4.

METH and brain ammonia. Rats were treated with METH (10 mg/kg × 4, i.p., every 2 h) or saline (1 ml/kg × 4, i.p., every 2 h) and lactulose (5.3 g/kg, p.o., every 12 h) or vehicle (8 ml/kg, p.o., every 12 h). During drug treatment (arrows denote injections), striatal ammonia concentration was measured using in vivo microdialysis. Vehicle plus METH treatment significantly increased brain ammonia concentrations starting at time point 5 and lactulose pretreatment attenuated this increase (*p < 0.05). Baseline, 29.34 ± 5.33 μm. (n = 8–10 rats per group). Lac, lactulose; Sal, saline; Veh, vehicle.
METH and increased peripheral ammonia excretion
Rats were given lactulose (5.3 g/kg) or vehicle (8 ml/kg) every 12 h, by oral gavage, for 2 d before and 6 d after METH (10 mg/kg, i.p., every 2 h × 4) or saline treatment (1 ml/kg, i.p., every 2 h × 4) to ensure that ammonia excretion was consistently increased throughout the presumed time course over which METH causes neuronal damage.
During drug treatment, the effects of METH and lactulose on body temperature were measured 1 h after each METH injection. METH treatment caused significant hyperthermia in both vehicle plus METH-treated and lactulose plus METH-treated rats. Lactulose had no significant effect on hyperthermia (Fig. 5) indicating that the effects of lactulose are not dependent on alterations in hyperthermia.
Figure 5.

Hyperthermia after METH treatment. Rats were treated with METH (10 mg/kg, i.p., every 2 h × 4) or saline (1 ml/kg, i.p., every 2 h × 4) and lactulose (5.3 g/kg, p.o., every 12 h) or vehicle (8 ml/kg, p.o., every 12 h). During drug treatment, temperature was measured 1 h after each METH injection (denoted by arrows). METH treatment caused significant hyperthermia in both vehicle plus METH-treated and lactulose plus METH-treated rats, and lactulose had no significant effect on hyperthermia (*p < 0.001 compared with Veh Sal) (n = 6–9 per group). Lac, lactulose; Sal, saline; Veh, vehicle.
To examine whether lactulose affected the brain concentration of METH, the concentration of METH in the striatum was measured at 2 h after the last injection of METH using liquid chromatography-tandem mass spectrometry. Each of the four injections were within the half-life of METH and at this time point the drug presumably reached an approximate steady-state concentration in the brain, thus optimizing the detection of any effect that lactulose may have on the brain concentration of METH. Striatal concentration of METH in vehicle plus METH-treated rats was 2.11 × 10−2 ± 0.28 × 10−2 ppm/mg protein and 1.96 × 10−2 ± 0.13 × 10−2 ppm/mg protein in lactulose plus METH-treated rats. The concentrations of METH in the brain did not differ significantly between the vehicle plus METH and lactulose plus METH groups, supporting the view that lactulose does not alter the concentration of METH in the brain.
To examine the effects of METH and lactulose on striatal tissue content of dopamine and 5-HT, rats were killed at 7 d after METH or saline treatment and tissue content of dopamine and 5-HT was measured. A two-way ANOVA indicated a significant effect of METH treatment (F(1,23) = 30.51, p < 0.001) and a significant interaction between METH and lactulose treatment (F(1,23) = 11.03 p < 0.005) on dopamine tissue content, indicating that the effects of METH on dopamine content varied according to whether the rats were treated with lactulose. Tukey's post hoc analysis revealed that there was no effect of lactulose treatment within saline-treated groups (q = 2.5; p = 0.093); however, lactulose attenuated the dopamine depletion caused by METH (q = 8.08; p < 0.001). Treatment with lactulose significantly attenuated the striatal dopamine depletions caused by METH as METH depleted striatal dopamine by only 34.5 ± 7.9% in lactulose-treated rats (Fig. 6A), suggesting that ammonia may play a role in the dopaminergic toxicity produced by METH.
Figure 6.

Effect of lactulose on METH-induced monoamine depletions. Rats were treated with METH (10 mg/kg, i.p., every 2 h × 4) or saline (1 ml/kg, i.p., every 2 h × 4) and lactulose (5.3 g/kg, p.o., every 12 h) or vehicle (8 ml/kg, p.o., every 12 h). Tissue content of dopamine or 5-HT in the striatum was measured 7 d later. A, Treatment with lactulose significantly attenuated the striatal dopamine depletions caused by METH as METH depleted dopamine by only 34.5 ± 7.9% (mean ± SEM) in lactulose-treated rats. B, METH depleted 5-HT by 15.2 ± 8.2% (mean ± SEM) in lactulose-treated rats, which was not statistically significant compared with saline-treated rats (*p < 0.001 compared with Lac Sal; &p < 0.001 compared with Veh METH) (n = 6–9 rats per group). Lac, lactulose; Sal, saline; Veh, vehicle.
Likewise, a two-way ANOVA showed a significant effect of METH treatment on striatal 5-HT content (F(1,23) = 27.59, p < 0.0.001) and a significant interaction between METH treatment and lactulose treatment on striatal 5-HT (F(1,23) = 2.28 p < 0.050). Tukey's post hoc tests indicated that lactulose had no effect within saline-treated rats (q = 0.98; p = 0.34); however, lactulose attenuated the 5-HT depletion caused by METH (q = 3.80; p < 0.001). METH treatment produced a depletion of 5-HT by only 15.2 ± 8.2% in lactulose-treated rats, which was not significantly different compared with saline-treated rats (Fig. 6B), indicating that ammonia may play a key role in METH-induced serotonergic damage.
Both pretreatments and post-treatments with lactulose were required for attenuation of METH-induced neuronal terminal damage (data not shown), indicating that elevations in brain ammonia both during and in the days subsequent to METH treatment may play a role in monoaminergic terminal damage. Pretreatment with lactulose was required as the drug takes 24–48 h of dosing to be effective (Jia and Zhang, 2005; Nicaise et al., 2008; Al Sibae and McGuire, 2009).
To confirm the effects of lactulose on dopamine tissue content, DAT immunoreactivity was measured in the striatum at 7 d after METH treatment. A two-way ANOVA indicated a significant effect of METH treatment (F(1,23) = 4.708, p < 0.05) and a significant interaction between METH and lactulose treatment (F(1,23) = 4.708, p < 0.05). Post hoc tests reveal no effect of lactulose within saline treatment groups (q = 2.129, p = 0.311) and a significant effect of lactulose within METH treatment groups (q = 5.729, p < 0.05). METH treatment produced a 50.5 ± 19.5% depletion in vehicle plus METH-treated rats and a nonstatistically significant 19.5 ± 9.8% depletion in lactulose plus METH-treated rats (Fig. 7).
Figure 7.
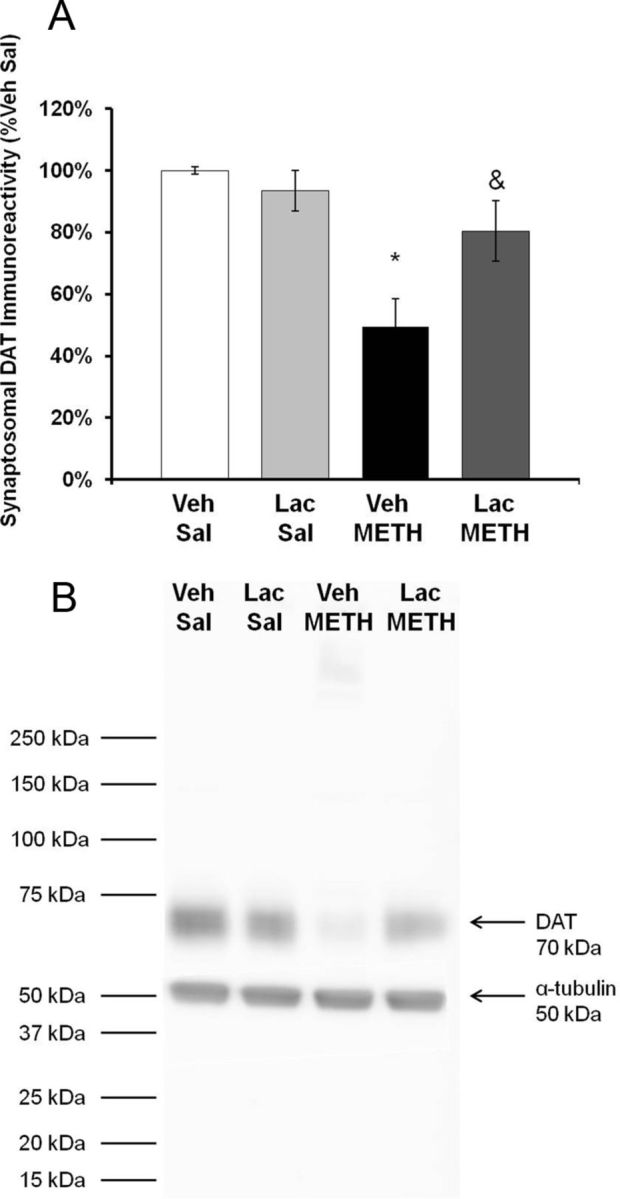
Effect of lactulose on METH-induced reductions in DAT immunoreactivity. Rats were treated with METH (10 mg/kg, i.p., every 2 h × 4) or saline (1 ml/kg, i.p., every 2 h × 4) and lactulose (5.3 g/kg, p.o., every 12 h) or vehicle (8 ml/kg, p.o., every 12 h). Tissue content of dopamine or 5-HT in the striatum were measured 7 d later. A, Treatment with lactulose significantly blocked the decrease in DAT immunoreactivity as METH produced only a 19.5 ± 9.8% depletion in lactulose plus METH-treated rats. B, Representative Western blot for DAT immunoreactivity and α-tubulin internal control (*p < 0.05 compared with Lac Sal. &p < 0.05 compared with Veh METH) (n = 6 rats per group). Lac, lactulose; Sal, saline; Veh, vehicle.
Local administration of METH and ammonia
To examine whether ammonia directly contributes to the neuronal damage caused by METH, METH (100 μm) and/or ammonia (570 μm) were reverse dialyzed into the striatum for 8 h. Dopamine and 5-HT tissue content of the tissue adjacent to the microdialysis probe was dissected 7 d later. Our previous studies showed that the local administration of 100 μm METH into the striatum increased striatal dopamine release to the same levels caused by the systemic administrations of 10 mg/kg, every 2 h × 4 injections (Burrows et al., 2000). Ammonia (570 μm) was reverse dialyzed to approximate the same brain concentration of ammonia previously measured after systemic METH treatment (Fig. 4), considering the in vivo recovery of ammonia by the microdialysis probe is 35%. The effects of reverse dialysis of METH and ammonia on dopamine and 5-HT tissue content in the striatum were determined (Fig. 8). A two-way ANOVA showed a significant interaction between METH and ammonia on dopamine tissue content (F(1,32) = 4.5, p < 0.050) and post hoc Tukey's tests revealed a significant difference between the combination of METH and ammonia compared with METH alone (q = 4.8, p < 0.005) or ammonia alone (q = 4.9, p < 0.005) (Fig. 8A). Similarly, a two-way ANOVA showed a significant interaction between METH and ammonia on 5-HT tissue content (F(1,32) = 5.6, p < 0.050). Tukey's post hoc tests revealed a significant difference between the combination of METH and ammonia compared with METH alone (q = 4.4, p < 0.005) or ammonia alone (q = 6.0, p < 0.001) (Fig. 8B). Specifically, the combination of ammonia and METH produced a 51.5 ± 12.5% depletion of dopamine and 41.8 ± 7.0% depletion of 5-HT, when compared with artificial CSF controls, demonstrating that ammonia synergizes with METH to directly contribute to the neuronal damage caused by METH.
Figure 8.

Effects of local administration of ammonia, METH, and GYKI 52466 into the striatum. Rats were reverse dialyzed with METH (100 μm), ammonia (570 μm), and/or GKYI 52466 (100 μm) in the striatum for 8 h. Dopamine and 5-HT tissue content of the striatal tissue around the probe membrane (1 mm) was measured 7 d later. A, Treatment with the combination of METH and ammonia depletes dopamine in the tissue around the probe by 51.5 ± 12.5% (mean ± SEM). B, Treatment with the combination of METH and ammonia depleted 5-HT in the tissue around the probe by 41.8 ± 7.0% (mean ± SEM) compared with aCSF. C, Treatment with GYKI 52466 significantly attenuated depletions in dopamine tissue content produced by METH and ammonia. D, Treatment with GKYI 52466 significantly attenuated depletions in 5-HT tissue content produced by the combination of METH and ammonia (*p < 0.05 compared with aCSF, &p < 0.05 compared with ammonia METH) (n = 10 rats per group). GYKI, GYKI 52466.
To identify a mechanism by which ammonia and METH may contribute to striatal damage, ammonia (570 μm) plus METH (100 μm) and/or GYKI 52466 (100 μm) were reverse dialyzed for 8 h and the tissue adjacent to the microdialysis probe was dissected 7 d later for measurements of dopamine and 5-HT tissue content. GYKI 52466 is an AMPA receptor antagonist that has previously been shown to prevent METH-induced increases in calpain-mediated spectrin proteolysis (Staszewski and Yamamoto, 2006). The effect GYKI 52466 on METH-induced and ammonia-induced decreases in dopamine and 5-HT tissue content was determined. A two-way ANOVA revealed a significant interaction between the combination of METH plus ammonia and GYKI 52466 treatment on dopamine tissue content (F(1,39) = 3.392, p < 0.05). A post hoc t test revealed that there is a significant effect of 52466 GYKI treatment on the METH plus ammonia-induced depletions in dopamine tissue content (t = 3.298, p < 0.05) (Fig. 8C). Similarly, when the effects of GYKI 52466 on METH plus ammonia-induced depletions in 5-HT were examined, a two-way ANOVA revealed a significant interaction between the combination of METH plus ammonia and GYKI 52466 treatment (F(1,39) = 3.624, p < 0.05). A post hoc Tukey's test revealed a significant effect of GYKI 52466 treatment on METH plus ammonia-induced depletions in 5-HT content (q = 5.541, p < 0.05) (Fig. 8D). Thus, GYKI 52466 blocked the decreases in dopamine and 5-HT content produced by ammonia plus METH.
Discussion
This study examined the relationship between changes in ammonia in the periphery and the long-term depletions of dopamine and 5-HT in the brain typically associated with a high-dose binge regimen of METH. METH caused an increase in plasma ammonia and acute liver damage that were paralleled by increases in brain ammonia. These increases were partially offset by the enhancement of peripheral ammonia excretion, which in turn attenuated the long-term depletions of brain dopamine and 5-HT produced by systemic exposure to METH. Furthermore, the local administration of ammonia with METH reproduced the depletions of striatal dopamine and 5-HT produced by the systemic administrations of METH alone and were blocked consequently by antagonism of the AMPA receptor.
Until now, the concurrence of peripheral organ toxicity and brain damage after exposure to METH had not been examined. While the effects of METH on the brain have been extensively studied, the possible damage to peripheral organs has received less attention. Consistent with clinical reports that METH causes peripheral organ damage (Smith and Fischer, 1970; Kamijo et al., 2002), the current study is the first to show evidence of hepatotoxicity in a rat model of binge METH exposure. Within 2 h after binge METH treatment, there is considerable venous and sinusoidal congestion in the liver (Fig. 1). This congestion is a typical early marker of hepatotoxicity and may be caused by cardiac dysfunction (Hong et al., 1991; Wijetunga et al., 2003), posthepatic vascular constriction (Wang et al., 1990; Chen, 2007), and/or direct drug-induced hepatotoxic damage to the liver. Similarly, serum AST and ALT are well established and sensitive markers for liver damage and were elevated at 2 h after binge METH treatment (Fig. 2). Although increases in AST or ALT could represent damage to other tissues, the elevations in both, along with the hepatic histopathological findings, support the view that METH alone does cause acute liver damage (Ozer et al., 2008). At 24 h after the last METH injection, the damage escalated to severe global, cytoplasmic disappearance (Fig. 1), suggestive of autophagy (Rautou et al., 2008). Although the mechanisms by which METH causes hepatotoxicity were not evaluated, the findings are consistent with reports that the metabolism of METH by the liver via the 2D6 p450 enzymes (Lin et al., 1997) may promote hepatocellular damage (Pourahmad et al., 2010; Letelier et al., 2011). Furthermore, amphetamines have been shown to cause hepatocellular mitochondrial damage (Moon et al., 2008). In addition, the well characterized hyperthermia produced by METH can exacerbate the oxidative and mitochondrial mechanisms that contribute to liver damage (Kew et al., 1970; Wills et al., 1976; Skibba et al., 1989) and most likely contribute to the liver damage observed after METH exposure.
Based on the fact that the liver is the primary organ for ammonia metabolism, we examined possible concurrent increases in plasma ammonia. In fact, compromised liver function resulting from inborn errors of metabolism or frank liver damage can increase plasma ammonia and contribute to compromised neurological symptoms, including altered mental status, peripheral neuropathy, and coma (Felipo and Butterworth, 2002). No previous studies have examined whether and how this peripheral damage and/or ammonia contribute to the long-term neuronal damage produced by METH. The current findings show that METH produced elevations of ammonia in the plasma and striatum that were temporally and physiologically related to the liver damage produced by the drug. In parallel with the liver pathology at 2 h after the last METH injection, brain ammonia increased to ∼70 μm in the striatal microdialysate (Fig. 4). This translates to a brain ammonia concentration of ∼200 μm, considering the recovery of ammonia from microdialysis is ∼35%. In cases of hepatic encephalopathy, ammonia concentrations in brain range from 300 to 1000 μm (Swain et al., 1992; Clemmesen et al., 1999). Although the concentrations of ammonia observed in the brain after binge METH treatment are less than those in hepatic encephalopathy, lower concentrations of ammonia in combination with METH exposure appear to cause neurotoxic effects. Thus, liver damage may be an unrecognized consequence of METH exposure and a cause of neuronal damage mediated by peripherally derived ammonia even in the absence of hepatic encephalopathy per se. Also worth noting is that METH-induced damage to other peripheral organ systems may contribute to increases in ammonia either directly, as in the case of renal damage and rhabdomyolysis, or indirectly via influence on hepatic function, as in the case cardiac dysfunction and, especially, right-sided heart failure (Smith and Fischer, 1970; Kamijo et al., 2002; Wijetunga et al. 2003; Ago et al., 2006). Thus, studies of the possible contributions by these organ systems and ammonia to METH-induced neurotoxicity are warranted.
We also investigated whether ammonia derived from the periphery contributes to the elevations in brain ammonia and the neuronal damage after METH exposure. Peripheral ammonia excretion was increased using lactulose. Lactulose is a synthetic disaccharide that is not absorbed systemically but remains in the gut. It is degraded by bacteria in the colon, thereby acidifying the colonic contents, converting NH3 to the ammonium ion (NH4+), thus trapping NH3 and preventing its absorption. Lactulose also functions as a laxative to reduce time for absorption and metabolism of protein, which is a source of ammonia. Lactulose prevented the acute increases in brain ammonia and the long-term decreases in dopamine and 5-HT content, without affecting METH-induced hyperthermia (Fig. 5) and the brain concentrations of METH. This supports the view that the effects of lactulose are likely due to its effects on peripheral ammonia and not alterations in the direct pharmacological actions of METH in the brain.
Excretion of ammonia partially protected against dopamine, but blocked the depletions of 5-HT tissue content produced by METH treatment. The partial protection of dopamine terminals can be explained by the fact that the established causes of dopaminergic terminal damage, such as the interaction of METH with DAT and vesicular monoamine transporter-2 (Fleckenstein et al., 1997; Hogan et al., 2000; Brown et al., 2002), might be relatively less affected by ammonia. Therefore, METH-induced increases in intracellular and extracellular dopamine can produce ROS and dopamine quinones and oxidatively damage dopamine terminals regardless of the presence of ammonia (Schmidt et al., 1985; Fornstedt et al., 1989; LaVoie and Hastings, 1999). In contrast, there may be causes of serotonergic terminal damage that are especially ammonia-dependent. Specifically, hyperammoniemia increases the uptake of l-tryptophan (Grippon et al., 1986), the expression and activity of monoamine oxidase (Mousseau et al., 1997), and production of 5-HIAA (Batshaw et al., 1986). Accordingly, ammonia-induced increases in 5-HT turnover may increase the production of oxidative intermediates. Furthermore, hyperammonemia increases the production of the excitotoxin and l-tryptophan metabolite, quinolinic acid (Moroni et al., 1986).
The central application of METH with ammonia recapitulated the neuronal damage produced by systemic METH exposure, even though administration of METH or ammonia alone did not produce neuronal damage (Fig. 8). The apparent synergy between ammonia and METH indicates both are necessary, but alone are not sufficient to produce toxicity. Ammonia could be toxic through excitotoxic and oxidative damage (Fan and Szerb, 1993; Kosenko et al., 1995, 1997; Chan and Butterworth, 1999), while METH confers selectivity to dopamine and 5-HT terminals through its effects on dopamine and 5-HT transporters and on the dopamine-dependent activation of the striatonigral pathway, resulting in increases in extracellular glutamate in the striatum (Mark et al., 2004).
The neuronal damage produced by the local administration of ammonia with METH into the striatum was blocked by the local administration of the AMPA antagonist, GYKI 52466. The combination of ammonia and METH could activate presynaptic AMPA receptors through elevations in extracellular glutamate. Systemic but not the central administration of METH treatment has been shown to increase extracellular glutamate (Tata and Yamamoto, 2007). Increased extracellular glutamate can then activate calcium-permeable AMPA receptors on striatal dopamine and 5-HT terminals (Hollmann et al., 1991; Lai et al., 2003), resulting in both increased dopamine and 5-HT release (Ohta et al., 1994), and in excitotoxic terminal damage (Staszewski and Yamamoto, 2006). Ammonia has been shown to increase glutamate release from astrocytes (Görg et al., 2010) and to increase extracellular glutamate by decreasing glutamate transporter expression (Chan et al., 2000) and glutamine synthetase activity (Kosenko et al., 2003), all of which can combine with the effects of METH on dopamine and 5-HT terminals to produce long-term 5-HT and dopamine depletions.
In conclusion, these studies identify a small molecule derived from the periphery that mediates the neuronal damage produced by METH and note the importance of considering the periphery in the context of METH-induced neurological dysfunction and damage. These findings suggest that comorbid conditions affecting liver function or ammonia metabolism may negatively contribute to the neurotoxic consequences of METH exposure. In particular, comorbid alcoholic cirrhosis or viral hepatitis with METH use may exacerbate acute METH-mediated liver damage and subsequent neurotoxicity. Furthermore, these findings highlight acute peripheral organ damage as a possible mediator of damage in other neurological disorders.
Footnotes
This work was supported by National Institutes of Health Grant DA07606. We thank Dr. Sadik A. Khuder for assistance with statistical analysis, Dr. John H. Irlam for assistance with examination of liver pathology, and Dr. Arunan Nadarajah and Lidia Rodriguez for assistance with the liquid chromatography-tandem mass spectrometry experiments.
The authors declare no competing financial interests.
References
- Ago M, Ago K, Hara K, Kashimura S, Ogata M. Toxicological and histopathological analysis of a patient who died nine days after a single intravenous dose of methamphetamine: a case report. Leg Med (Tokyo) 2006;8:235–239. doi: 10.1016/j.legalmed.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Al Sibae MR, McGuire BM. Current trends in the treatment of hepatic encephalopathy. Ther Clin Risk Manag. 2009;5:617–626. doi: 10.2147/tcrm.s4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batshaw ML, Hyman SL, Mellits ED, Thomas GH, DeMuro R, Coyle JT. Behavioral and neurotransmitter changes in the urease-infused rat: a model of congenital hyperammonemia. Pediatr Res. 1986;20:1310–1315. doi: 10.1203/00006450-198612000-00025. [DOI] [PubMed] [Google Scholar]
- Breier JM, Bankson MG, Yamamoto BK. l-tyrosine contributes to (+)-3,4-methylenedioxymethamphetamine-induced serotonin depletions. J Neurosci. 2006;26:290–299. doi: 10.1523/JNEUROSCI.3353-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Riddle EL, Sandoval V, Weston RK, Hanson JE, Crosby MJ, Ugarte YV, Gibb JW, Hanson GR, Fleckenstein AE. A single methamphetamine administration rapidly decreases vesicular dopamine uptake. J Pharmacol Exp Ther. 2002;302:497–501. doi: 10.1124/jpet.302.2.497. [DOI] [PubMed] [Google Scholar]
- Burrows KB, Nixdorf WL, Yamamoto BK. Central administration of methamphetamine synergizes with metabolic inhibition to deplete striatal monoamines. J Pharmacol Exp Ther. 2000;292:853–860. [PubMed] [Google Scholar]
- Cauli O, Rodrigo R, Piedrafita B, Boix J, Felipo V. Inflammation and hepatic encephalopathy: ibuprofen restores learning ability in rats with portacaval shunts. Hepatology. 2007;46:514–519. doi: 10.1002/hep.21734. [DOI] [PubMed] [Google Scholar]
- Chan H, Butterworth RF. Evidence for an astrocytic glutamate transporter deficit in hepatic encephalopathy. Neurochem Res. 1999;24:1397–1401. doi: 10.1023/a:1022532623281. [DOI] [PubMed] [Google Scholar]
- Chan H, Hazell AS, Desjardins P, Butterworth RF. Effects of ammonia on glutamate transporter (GLAST) protein and mRNA in cultured rat cortical astrocytes. Neurochem Int. 2000;37:243–248. doi: 10.1016/s0197-0186(00)00026-7. [DOI] [PubMed] [Google Scholar]
- Chen JP. Methamphetamine-associated acute myocardial infarction and cardiogenic shock with normal coronary arteries: refractory global coronary microvascular spasm. J Invasive Cardiol. 2007;19:E89–E92. [PubMed] [Google Scholar]
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology. 1999;29:648–653. doi: 10.1002/hep.510290309. [DOI] [PubMed] [Google Scholar]
- Fan P, Szerb JC. Effects of ammonium ions on synaptic transmission and on responses to quisqualate and N-methyl-D-aspartate in hippocampal CA1 pyramidal neurons in vitro. Brain Res. 1993;632:225–231. doi: 10.1016/0006-8993(93)91157-n. [DOI] [PubMed] [Google Scholar]
- Felipo V, Butterworth RF. Neurobiology of ammonia. Prog Neurobiol. 2002;67:259–279. doi: 10.1016/s0301-0082(02)00019-9. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Metzger RR, Wilkins DG, Gibb JW, Hanson GR. Rapid and reversible effects of methamphetamine on dopamine transporters. J Pharmacol Exp Ther. 1997;282:834–838. [PubMed] [Google Scholar]
- Fornstedt B, Brun A, Rosengren E, Carlsson A. The apparent autoxidation rate of catechols in dopamine-rich regions of human brains increases with the degree of depigmentation of substantia nigra. J Neural Transm Park Dis Dement Sect. 1989;1:279–295. doi: 10.1007/BF02263482. [DOI] [PubMed] [Google Scholar]
- Görg B, Morwinsky A, Keitel V, Qvartskhava N, Schrör K, Häussinger D. Ammonia triggers exocytotic release of l-glutamate from cultured rat astrocytes. Glia. 2010;58:691–705. doi: 10.1002/glia.20955. [DOI] [PubMed] [Google Scholar]
- Grippon P, Le Poncin Lafitte M, Boschat M, Wang S, Faure G, Dutertre D, Opolon P. Evidence for the role of ammonia in the intracerebral transfer and metabolism of tryptophan. Hepatology. 1986;6:682–686. doi: 10.1002/hep.1840060424. [DOI] [PubMed] [Google Scholar]
- Hawkins RA, Miller AL, Nielsen RC, Veech RL. The acute action of ammonia on rat brain metabolism in vivo. Biochem J. 1973;134:1001–1008. doi: 10.1042/bj1341001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan KA, Staal RG, Sonsalla PK. Analysis of VMAT2 binding after methamphetamine or MPTP treatment: disparity between homogenates and vesicle preparations. J Neurochem. 2000;74:2217–2220. doi: 10.1046/j.1471-4159.2000.0742217.x. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Hong R, Matsuyama E, Nur K. Cardiomyopathy associated with the smoking of crystal methamphetamine. JAMA. 1991;265:1152–1154. [PubMed] [Google Scholar]
- Jia L, Zhang MH. Comparison of probiotics and lactulose in the treatment of minimal hepatic encephalopathy in rats. World J Gastroenterol. 2005;11:908–911. doi: 10.3748/wjg.v11.i6.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo Y, Soma K, Nishida M, Namera A, Ohwada T. Acute liver failure following intravenous methamphetamine. Vet Hum Toxicol. 2002;44:216–217. [PubMed] [Google Scholar]
- Kew M, Bersohn I, Seftel H, Kent G. Liver damage in heatstroke. Am J Med. 1970;49:192–202. doi: 10.1016/s0002-9343(70)80075-4. [DOI] [PubMed] [Google Scholar]
- Kosenko E, Kaminsky Y, Grau E, Miñana MD, Grisolía S, Felipo V. Nitroarginine, an inhibitor of nitric oxide synthetase, attenuates ammonia toxicity and ammonia-induced alterations in brain metabolism. Neurochem Res. 1995;20:451–456. doi: 10.1007/BF00973101. [DOI] [PubMed] [Google Scholar]
- Kosenko E, Kaminsky Y, Kaminsky A, Valencia M, Lee L, Hermenegildo C, Felipo V. Superoxide production and antioxidant enzymes in ammonia intoxication in rats. Free Radic Res. 1997;27:637–644. doi: 10.3109/10715769709097867. [DOI] [PubMed] [Google Scholar]
- Kosenko E, Llansola M, Montoliu C, Monfort P, Rodrigo R, Hernandez-Viadel M, Erceg S, Sánchez-Perez AM, Felipo V. Glutamine synthetase activity and glutamine content in brain: modulation by NMDA receptors and nitric oxide. Neurochem Int. 2003;43:493–499. doi: 10.1016/s0197-0186(03)00039-1. [DOI] [PubMed] [Google Scholar]
- Lai SK, Tse YC, Yang MS, Wong CK, Chan YS, Yung KK. Gene expression of glutamate receptors GluR1 and NR1 is differentially modulated in striatal neurons in rats after 6-hydroxydopamine lesion. Neurochem Int. 2003;43:639–653. doi: 10.1016/s0197-0186(03)00080-9. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letelier ME, López-Valladares M, Peredo-Silva L, Rojas-Sepúlveda D, Aracena P. Microsomal oxidative damage promoted by acetaminophen metabolism. Toxicol In Vitro. 2011;25:1310–1313. doi: 10.1016/j.tiv.2011.04.022. [DOI] [PubMed] [Google Scholar]
- Lin LY, Di Stefano EW, Schmitz DA, Hsu L, Ellis SW, Lennard MS, Tucker GT, Cho AK. Oxidation of methamphetamine and methylenedioxymethamphetamine by CYP2D6. Drug Metab Dispos. 1997;25:1059–1064. [PubMed] [Google Scholar]
- Mark KA, Soghomonian JJ, Yamamoto BK. High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J Neurosci. 2004;24:11449–11456. doi: 10.1523/JNEUROSCI.3597-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCandless DW, Schenker S. Effect of acute ammonia intoxication on energy stores in the cerebral reticular activating system. Exp Brain Res. 1981;44:325–330. doi: 10.1007/BF00236570. [DOI] [PubMed] [Google Scholar]
- McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci. 1998;18:8417–8422. doi: 10.1523/JNEUROSCI.18-20-08417.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon KH, Upreti VV, Yu LR, Lee IJ, Ye X, Eddington ND, Veenstra TD, Song BJ. Mechanism of 3,4-methylenedioxymethamphetamine (MDMA, ecstasy)-mediated mitochondrial dysfunction in rat liver. Proteomics. 2008;8:3906–3918. doi: 10.1002/pmic.200800215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni F, Lombardi G, Carl à V, Lal S, Etienne P, Nair NP. Increase in the content of quinolinic acid in cerebrospinal fluid and frontal cortex of patients with hepatic failure. J Neurochem. 1986;47:1667–1671. doi: 10.1111/j.1471-4159.1986.tb13071.x. [DOI] [PubMed] [Google Scholar]
- Mousseau DD, Baker GB, Butterworth RF. Increased density of catalytic sites and expression of brain monoamine oxidase A in humans with hepatic encephalopathy. J Neurochem. 1997;68:1200–1208. doi: 10.1046/j.1471-4159.1997.68031200.x. [DOI] [PubMed] [Google Scholar]
- Nicaise C, Prozzi D, Viaene E, Moreno C, Gustot T, Quertinmont E, Demetter P, Suain V, Goffin P, Devière J, Hols P. Control of acute, chronic, and constitutive hyperammonemia by wild-type and genetically engineered Lactobacillus plantarum in rodents. Hepatology. 2008;48:1184–1192. doi: 10.1002/hep.22445. [DOI] [PubMed] [Google Scholar]
- Ohta K, Araki N, Shibata M, Komatsumoto S, Shimazu K, Fukuuchi Y. Presynaptic ionotropic glutamate receptors modulate in vivo release and metabolism of striatal dopamine, noradrenaline, and 5-hydroxytryptamine: involvement of both NMDA and AMPA/kainate subtypes. Neurosci Res. 1994;21:83–89. doi: 10.1016/0168-0102(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Ozer J, Ratner M, Shaw M, Bailey W, Schomaker S. The current state of serum biomarkers of hepatotoxicity. Toxicology. 2008;245:194–205. doi: 10.1016/j.tox.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Pourahmad J, Eskandari MR, Nosrati M, Kobarfard F, Khajeamiri AR. Involvement of mitochondrial/lysosomal toxic cross-talk in ecstasy induced liver toxicity under hyperthermic condition. Eur J Pharmacol. 2010;643:162–169. doi: 10.1016/j.ejphar.2010.06.019. [DOI] [PubMed] [Google Scholar]
- Rautou PE, Cazals-Hatem D, Moreau R, Francoz C, Feldmann G, Lebrec D, Ogier-Denis E, Bedossa P, Valla D, Durand F. Acute liver cell damage in patients with anorexia nervosa: a possible role of starvation-induced hepatocyte autophagy. Gastroenterology. 2008;135:840–848. 848.e1–848.e3. doi: 10.1053/j.gastro.2008.05.055. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- Sáez R, Llansola M, Felipo V. Chronic exposure to ammonia alters pathways modulating phosphorylation of microtubule-associated protein 2 in cerebellar neurons in culture. J Neurochem. 1999;73:2555–2562. doi: 10.1046/j.1471-4159.1999.0732555.x. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 1985;233:539–544. [PubMed] [Google Scholar]
- Seiden LS, Commins DL, Vosmer G, Axt K, Marek G. Neurotoxicity in dopamine and 5-hydroxytryptamine terminal fields: a regional analysis in nigrostriatal and mesolimbic projections. Ann N Y Acad Sci. 1988;537:161–172. doi: 10.1111/j.1749-6632.1988.tb42104.x. [DOI] [PubMed] [Google Scholar]
- Skibba JL, Stadnicka A, Kalbfleisch JH. Hyperthermic liver toxicity: a role for oxidative stress. J Surg Oncol. 1989;42:103–112. doi: 10.1002/jso.2930420208. [DOI] [PubMed] [Google Scholar]
- Smith DE, Fischer CM. An analysis of 310 cases of acute high-dose methamphetamine toxicity in Haight-Ashbury. Clin Toxicol. 1970;3:117–124. doi: 10.3109/15563657008990106. [DOI] [PubMed] [Google Scholar]
- Staszewski RD, Yamamoto BK. Methamphetamine-induced spectrin proteolysis in the rat striatum. J Neurochem. 2006;96:1267–1276. doi: 10.1111/j.1471-4159.2005.03618.x. [DOI] [PubMed] [Google Scholar]
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology. 1992;15:449–453. doi: 10.1002/hep.1840150316. [DOI] [PubMed] [Google Scholar]
- Tata DA, Yamamoto BK. Interactions between methamphetamine and environmental stress: role of oxidative stress, glutamate and mitochondrial dysfunction. Addiction. 2007;102(Suppl 1):49–60. doi: 10.1111/j.1360-0443.2007.01770.x. [DOI] [PubMed] [Google Scholar]
- Wagner GC, Ricaurte GA, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- Wang AM, Suojanen JN, Colucci VM, Rumbaugh CL, Hollenberg NK. Cocaine- and methamphetamine-induced acute cerebral vasospasm: an angiographic study in rabbits. AJNR Am J Neuroradiol. 1990;11:1141–1146. [PMC free article] [PubMed] [Google Scholar]
- Wijetunga M, Seto T, Lindsay J, Schatz I. Crystal methamphetamine-associated cardiomyopathy: tip of the iceberg? J Toxicol Clin Toxicol. 2003;41:981–986. doi: 10.1081/clt-120026521. [DOI] [PubMed] [Google Scholar]
- Wills EJ, Findlay JM, McManus JP. Effects of hyperthermia therapy on the liver. II. Morphological observations. J Clin Pathol. 1976;29:1–10. doi: 10.1136/jcp.29.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto BK, Pehek EA. A neurochemical heterogeneity of the rat striatum as measured by in vivo electrochemistry and microdialysis. Brain Res. 1990;506:236–242. doi: 10.1016/0006-8993(90)91256-g. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Raudensky J. The role of oxidative stress, metabolic compromise, and inflammation in neuronal injury produced by amphetamine-related drugs of abuse. J Neuroimmune Pharmacol. 2008;3:203–217. doi: 10.1007/s11481-008-9121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]