

Abstract
Introduction:
Levocetirizine dihydrochloride is an orally active, third-generation non-sedative antihistamine used in the symptomatic relief of seasonal and perennial allergic rhinitis. The present work aimed at preparing quick release films of levocetirizine with the purpose of developing a dosage form for a very quick onset of action, which is beneficial in managing severe conditions of allergies, aiding in the enhancement of bioavailability, and is very convenient for administration, without the problem of swallowing and using water.
Materials and Methods:
The films of levocetirizine dihydrochloride were prepared by using polymers such as hydroxypropyl methylcellulose (HPMC) and polyvinyl alcohol (PVA), as either single polymer or in combination of two, by a solvent casting method. They were evaluated for physical characteristics such as uniformity of weight, thickness, folding endurance, drug content uniformity, surface pH, percentage elongation, and tensile strength, and gave satisfactory results. The formulations were subjected to disintegration, in vitro drug release tests, and in vivo studies on rats.
Results:
A marked increase in the dissolution rate was exhibited by fast-dissolving films of levocetirizine dihydrochloride containing HPMC as a polymer, when compared to conventional tablets. The haloperidol-induced catalepsy, milk-induced leukocytosis, and nasal provocation in vivo studies in rats proved that the fast-dissolving films of levocetirizine dihydrochloride produced a faster onset of action compared to the conventional tablets.
Conclusions:
Fast dissolving films of levocetirizine dihydrochloride can be considered suitable for clinical use in the treatment of allergic rhinitis and other conditions of allergies, where a quicker onset of action for a dosage form is desirable along with the convenience of administration.
Keywords: Haloperidol-induced catalepsy, levocetirizine, milk-induced leucocytosis, nasal provocation, quick release films
INTRODUCTION
Recently, fast-dissolving drug delivery systems have started gaining popularity and acceptance as new drug delivery systems, which aim to enhance safety and efficacy of a drug molecule by formulating it into a convenient dosage form for administration and to achieve better patient compliance. Some companies introduced more robust forms of fast-dissolving drug delivery; for example Lavipharm Laboratories Inc. (Lavipharm), invented an ideal fast-dissolving drug delivery system, which satisfied the unmet needs of the market. This novel intraoral drug delivery system, trademarked Quick-Dis™, is Lavipharm's proprietary patented technology, and is a thin, flexible, and quick-dissolving film. The film is placed on the top or the floor of the tongue.[1] When put on the tongue, this film disintegrates instantaneously, releasing the drug which dissolves in the saliva. Some drugs are absorbed from the mouth, pharynx, and esophagus as the saliva passes down into the stomach. In such cases, the bioavailability of the drug is significantly greater than that observed for conventional tablets.[2]
Levocetirizine dihydrochloride is an orally active, third-generation non-sedative antihistamine used in the symptomatic relief of seasonal and perennial allergic rhinitis, hay fever, and for the treatment of chronic idiopathic urticaria. It has twice the affinity for H1 histamine receptors when compared to cetirizine hydrochlorid.[3] Levocetirizine dihydrochloride is rapidly and extensively absorbed following oral administration, with the peak plasma concentration usually attained in 0.9 h. Antihistaminic effects occur within 1 h. Symptomatic improvement is observed as early as 1 day after the initiation of therapy for allergic rhinitis or chronic idiopathic urticaria. The duration of antihistaminic effects persist for at least 24 h.[4–8] Hence, for an antihistamine drug like levocetirizine dihydrochloride, a quick-disintegrating dosage form is suitable, since the disintegration and dissolution of the dosage form occurs rapidly, thus providing a rapid onset of action. It was thought worth to formulate oro-dispersible formulations of the drug, so that the patient can ingest the dosage form anywhere and at anytime, without the aid of water which would be helpful especially in cases of unavailability of water, motion sickness, sudden episodes of allergic attacks, and deglutition problems. Mouth-dissolving tablets of levocetirizine dihydrochloride were prepared by a direct compression method using different concentrations of spray-dried mannitol (Perlitol SD 200), menthol, and camphor. The sublimation technique was used to increase the porosity of the tablets in which menthol and camphor were used as subliming agents which in turn forms the porous structure on the surface of tablets after sublimation. The formulated tablets were evaluated for different parameters like hardness, friability, and in vitro and in vivo disintegration time along with other physical parameters. The tablets were also evaluated for drug release for 30 min in 0.1 N HCL using the USP Type II dissolution apparatus. The in vitro drug release study revealed that menthol and camphor (1:1) at a concentration of 20% (Batch – LMD-6) of the total weight of the tablet offer a fast release of levocetirizine dihydrochloride within 5 min. These tablets also dissolved within 15-20 seconds in saliva with pleasant taste and smooth mouth feel.[9] The present investigation was aimed at the formulation of fast dissolving films of levocetirizine dihydrochloride and evaluate its physicochemical properties, in vitro dissolution profile, and in vivo antihistamine efficacy performance in rats.
MATERIALS AND METHODS
Materials
Levocetirizine dihydrochloride was obtained as a gift sample from FDC Ltd, Goa. Polyvinyl alcohol (PVA) and hydroxypropyl methylcellulose (HPMC 15 cps and 50 cps) were procured from CDH Laboratories, New Delhi. All other chemicals used were of analytical grade.
Methods
Formulation of fast-dissolving films[10]
In the present study, fast-dissolving films of levocetirizine dihydrochloride were prepared by a solvent casting technique. Flat, square-shaped, aluminum foil-coated glass molds having a surface area of 25 cm2 were fabricated for casting the films.
Preparation of casting solutions
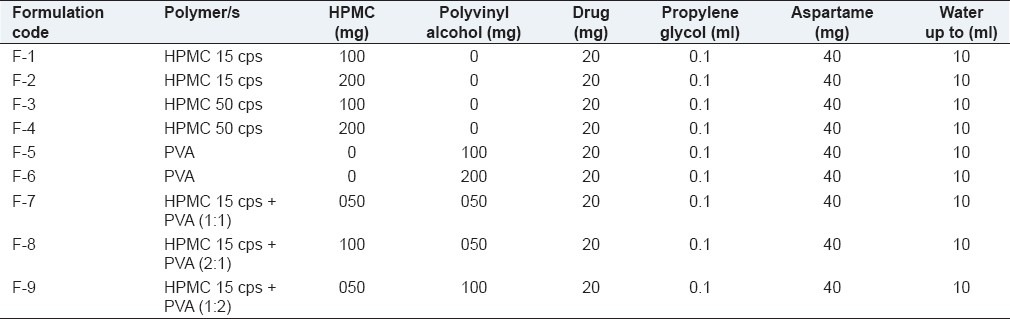
The weighed quantities of polymers [Table 1] were kept for swelling overnight in distilled water and dissolved (heated, if necessary). The drug and aspartame were dissolved in distilled water and added to the above mentioned polymer solution along with propylene glycol as a plasticizer, mixed thoroughly to form a homogenous mixture. The volume was made up to 10 ml with distilled water. Entrapped air bubbles were removed by applying vacuum.
Table 1.
Formulations of fast-dissolving films
Preparation of fast-dissolving films
The casting solution (10 ml) was poured into glass molds and dried at 40°C in a vacuum oven for 24 h for solvent evaporation. The patches were removed by peeling and cut into a square dimension of 5 cm × 5 cm (25 cm2). These patches were kept in a desiccator for 2 days for further drying and wrapped in aluminium foil, and packed in self-sealing covers. Fast-dissolving films were prepared with different polymers and ratios by maintaining the concentration of the plasticizer and sweetener constant [Table 1].
Evaluation of fast-dissolving films
Drug content uniformity
A fast-dissolving film (25 cm2) was transferred into a graduated flask containing 100 ml of distilled water. The flask was shaken for 4 h in a mechanical shaker. The solution was filtered and after suitable dilutions with distilled water, the absorbance value was measured at 230 nm using the placebo patch (patch without drug) solution as a blank, and the drug content was calculated.[11]
Folding endurance
The folding endurance is expressed as the number of folds (number of times the film is folded at the same place) required to break the specimen or to develop visible cracks. This also gives an indication of brittleness of the film. A strip of 2.5 cm × 2.5 cm (6.25 cm2) was subjected to folding endurance by folding the patch at the same place repeatedly several times until a visible crack was observed, and the values were reported.[11]
Surface pH
The film to be tested was placed in a Petri dish and was moistened with 0.5 ml of distilled water and kept for 30 s. The pH was noted after bringing the electrode of the pH meter in contact with the surface of the formulation and allowing equilibration for 1 min. The average of three determinations for each formulation was done.[12,13]
Elongation and tensile strength
This mechanical property was evaluated using the Instron universal testing instrument (Model F. 4026, Instron Ltd., Japan) with a 5 kg load cell. Film strips in a special dimension and free from air bubbles or physical imperfections were held between two clamps positioned at a distance of 3 cm. During measurement, the strips were pulled by the top clamps at a rate of 100 mm/min; the force and elongation were measured when the film broke. Results from film samples, which broke at and not between the clamps, were not included in the calculations. Measurements were run in triplicate for each film. Two mechanical properties, namely, tensile strength and percentage elongation were computed for the evaluation of the film. Tensile strength is the maximum stress applied to a point at which the film specimen breaks and can be computed from the applied load at rupture.[13] Disintegration test was performed to ensure the disintegration of the film in water. One film from each formulation was introduced into one tube of disintegration apparatus IP. A disc was added into the tube. The assembly was suspended in a beaker containing simulated saliva and the apparatus was operated until the film disintegrated.[14]
In vitro dissolution studies[9]
The simulated salivary fluid was taken as the dissolution medium to determine the drug release.
The dissolution profile of quick release films of levocetirizine dihydrochloride was carried out in a beaker containing 30 ml of the simulated salivary fluid (pH 6.8) as a dissolution medium, maintained at 37 ± 0.5°C. The medium was stirred at 100 rpm. Aliquots (5 ml) of the dissolution medium were withdrawn at 15, 30, 45, 60, 75, 90, 105 and 120 s time intervals and the same amount was replaced with the fresh medium.[8] Samples were assayed spectrophotometerically at 230 nm. Three trials were carried out for all the samples and the average value was taken. The percentage of the drug dissolved at various time intervals was calculated and plotted against time.
Stability studies
The stability study of the formulated fast-dissolving films was carried out under different environmental conditions. The film was packed in the aluminum foil and stored in a stability chamber for stability studies at 2-8°C (45% RH), 25-30°C (60% RH), and 45-50°C (75% RH) for a period of 45 days. The patches were characterized for the drug content and other parameters during the stability study period.[14]
In vivo studies
For the purpose of the in vivo study, separate patches were prepared in a similar way as described earlier, having similar physical properties; a film of 25 cm2 size (each film contains 2 mg of drug instead 20 mg) was subjected to the dissolution study (30 ml dissolution medium) for all the in vivo study methods so that each film could release 0.066 mg of drug/ml. Hence 1.3 ml of the dissolution medium contains 0.0858 mg of the drug, which is the suitable dose for the rats as per the literature.[15] Similarly, a conventional tablet formulation having similar hardness (3-4 kg/ cm2) as that of conventional tablet available in the market was prepared to have a similar dose during the administration of the drug from the dissolution study, as that of the film used for the study.
Haloperidol-induced catalepsy in rats[16]
The protocol of the experiment was approved by the Institutional Animal Ethics Committee (K.S Hegde Medical Academy, reg. no. 115/1999/CPCSEA). Male Wistar rats were divided into three groups (n= 6). Group-1 was the control group, and received saline; Group-2 was the test group, and received a single dose of the dissolution medium (1.3 ml) withdrawn at the 1 min. time interval during the dissolution study of the fast dissolving film. Group-3 was the reference group, and received a single dose (1.3 ml) of the dissolution medium withdrawn at the 1 min. time interval during the dissolution study of the conventional tablet preparation. All the groups were received haloperidol injection (1 mg/kg, i.p.). The duration of catalepsy was measured 10 min after the drug administration at 0, 15, 30, 45, and 60 min time intervals. The degree of catatonia was observed in four stages:
Stage I: the rat moves normally when placed on the table, score = 0.
Stage II: the rat moves only when touched/pushed, score = 0.5.
Stage III: the rat placed on the table with the front paws set alternatively on a 3 cm high block, fails to correct the posture in 10 s. score = 0.5 for each paw (total score 1).
Stage IV: the rat fails to move when the front paws were placed alternately on a 9-cm high block, score = 1 for each paw (total score 2).
Milk-induced leucocytosis in rats[16]
Male Wistar rats were divided into three groups (n= 6). Group-1 was the control group and received saline; Group-2 was the test group and received a single dose of the dissolution medium withdrawn at the 1 min. time interval during the dissolution study of the fast-dissolving film. Group-3 was the reference group and received a single dose of the dissolution medium withdrawn at the 1 min. time interval during the dissolution study of the conventional tablet preparation. All the groups received boiled and cooled milk injection (dose of 4 ml/kg, s.c). The total leucocyte count was done in each group before the induction of leucocytosis and 24 h after milk injection. The drug was administered 24 h. after the induction of leucocytosis, from the sample withdrawn at 1 min. of the dissolution study of the conventional tablet and fast-dissolving films (1 min. dissolution study). The leucocyte count was done using Neuber's chamber, after 30 and 60 min time intervals of drug administration, and drop in leucocytes was observed.
Nasal provocation method in rats[17]
Male Wistar rats (n = 6) weighing around 150-200 g were considered suitable for this study. Animals were divided into three groups (n = 6), control, test, and reference as specified above. All the animals were sensitized subcutaneously by injecting 20 mcg of ovalbumin in 0.2 ml of saline everyday for 10 consecutive days. After 10 days, nasal provocation was performed by spraying 1% ovalbumin in saline into nostrils. Nasal secretion was measured by using filter paper strips to measure the amount of nasal secretion by weight. The drug was administered as specified before in the earlier methods. The amount of nasal secretion secreted before and after giving the drug preparations was measured at the10 min. time intervals for 60 min. The difference in the amount of secretions secreted between two groups, i.e., test (film) and reference (tablet) groups, was measured-:

RESULTS AND DISCUSSION
Fast-dissolving films of levocetirizine dihydrochloride were prepared by the solvent casting method on glass molds, using HPMC 15 cps, HPMC 50 cps, and PVA as polymers. Propylene glycol was used as a plasticizer and aspartame as a sweetener. Distilled water was used as a solvent for HPMC and PVA. The effect of the concentration ratio of polymers and nature of polymers was studied by preparing various formulations of fast-dissolving films. In the preparation, the addition of ingredients, particularly propylene glycol and aspartame, was followed after the careful evaluation of films for physical characteristics. In all these formulations, a constant amount of drug (20 mg) was maintained. The casting solution (10 ml) was poured into 25 cm2 molds, so that each square centimeter contains approximately 0.8 mg of the drug. Polymers were used in different concentrations and the concentration of other ingredients such as plasticizer and sweetener were kept constant.
Fast-dissolving films of levocetirizine dihydrochloride were evaluated for various parameters. In the present study, nine formulations were prepared by varying the polymer concentration, and by using different polymers.
Effect of the polymer concentration
Different formulations (F1, F2, F3, F4, F5, F6, F7, F8, and F9) were prepared using HPMC 15 cps, HPMC 50 cps, PVA, and a combination of HPMC 15 cps and PVA in different concentrations [Tables 1 and 2] to study the effect of polymers/concentration on the physicochemical properties.
Table 2.
Drug content of the fast-dissolving films
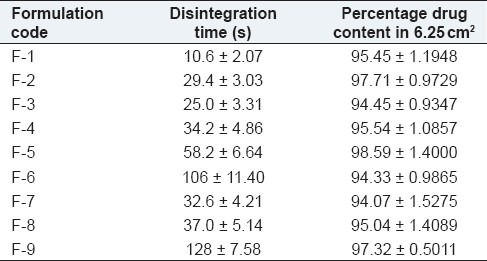
The physical appearance of the films was evaluated. All the films prepared with different polymer concentrations were found to be flexible, smooth, transparent, non-sticky, and homogeneous. The thickness of the films in each set was measured. The marginal difference in the thickness was observed among each group indicated that more the amount of polymer, higher the thickness values. All the films, except F1 showed good folding endurance (>250), indicated that the films have good flexibility. The effect of concentration of polymers was observed on the percentage elongation and tensile strength (1.2-1.6 kg/mm). It was found that as the concentration of PVA increased, the percentage elongation and tensile strength also increased. HPMC films showed a better tensile strength due to the hydrophobic nature of the polymers. No significant difference in the drug content among the films (approximately 100%), indicated good content uniformity. When placed over the tongue, the film dissolved instantly. Dissolution was also found to be improved due to salivary stimulation in the presence of the sweetener (aspartame).
It was observed that there was no significant difference in the thickness among the films, which indicated that the films were uniform, and the surface pH was found to be in the range of 6.2-7.08, which is close to the neutral pH, which indicated that films may have less potential to irritate the sublingual mucosa, and hence, more acceptable by the patients.
In vitro drug release study
From the in vitro drug release, it was observed that in formulations containing a single polymer, the drug release was found to be faster from films containing PVA as a polymer, and among the HPMC films, lower viscosity resulted in a faster release of drug. The release of the drug from HPMC 50 cps, was found to be slower compared to all other films containing a single polymer (F1-F6). Among the combination of polymers (F7, F8, and F9) it was found that the presence of PVA increased the drug release from films compared to films containing only HPMC 50 cps (F3 and F4). Further, as the concentration of the polymer increased, the drug release was found to be decreased due to the increase in the time required for wetting and dissolving the drug molecules present in the polymer matrices. The drug release was found to be in the following order: F5 > F1 > F3 > F6 > F2 > F4. For the group of formulations containing a combination of polymers, the drug release was found to be in the following order: F7 > F8 > F9 [Figure 1]. Among the nine formulations (F1, F2, F3, F4, F5, F6, F7, F8, and F9) prepared, formulations F7, F5, and F1 were found to be the best formulations in terms of drug release. The release profile of fast-dissolving films was compared with that of conventional marketed tablets and it was observed that the drug release from fast-issolving films was much faster than that from tablets.
Figure 1.
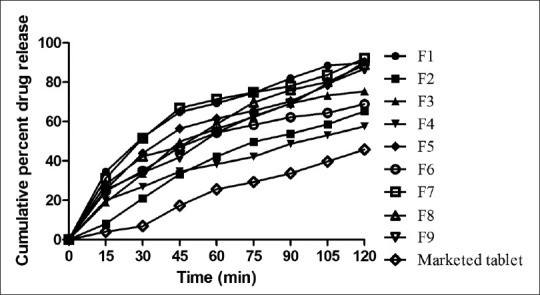
In vitro release profile of formulations
Stability studies
A stability study was carried out for 45 days at 2-8°C (45% RH), 2530°C (60% RH), and 45-50°C (75% RH). The films were observed for physical change, drug content, and % drug release. Fast-dissolving films of levocetirizine dihydrochloride were found to be physically and chemically stable as they showed no significant change in terms of physical characteristics and drug content at a lower temperature and room temperature. However, when stored at 45-50°C for 45 days, films became brittle.
In vivo study
Haloperidol-induced catalepsy in rats
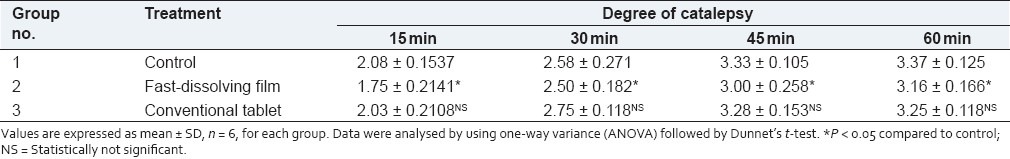
The comparison between fast-dissolving films and conventional tablets in terms of drug release and onset of action was done by haloperidol-induced catalepsy in a rat model. Haloperidol induces catalepsy by inhibiting dopamine D2 receptors and inhibits dopamine secretion. Dopamine is an agonist for adrenaline. Adrenaline is a physiological antagonist of histamine. Hence, whenever there is a fall in the level of dopamine, it leads to a decrease in the level of neurotransmitter adrenaline. This leads to a rise in the level of histamine. In this study, significant (P < 0.05) protection against haloperidol-induced catalepsy was observed more in fast-dissolving films as compared to the conventional tablet at the 10 min time interval. The results obtained were found to be statistically significant. However, the result for the conventional tablet- treated group at the 60-min time interval was not found to be statistically significant [Table 3].
Table 3.
Results of haloperidol-induced catalepsy in rats
Milk-induced leucocytosis in rats
The comparison between fast-dissolving films and conventional tablets was done by milk-induced leucocytosis in rats. Leucocytosis was observed after the parenteral administration of milk. The control group showed a significant (P < 0.05) increase in the leucocyte count [Table 4]. As expected, the fast-dissolving film-treated group showed a significant decrease in the number of leucocytes after 30 min. (time interval between 30 and 60 min) as compared to conventional tablets. There was no drop in leucocytes in the group treated with conventional tablets, as the dissolution medium expected to contain either very little drug or no drug. This is because; the conventional tablet can not release the drug within a minute. The value obtained was found to be statistically significant (P < 0.05) in animals treated with films. The normal leucocyte count ranged from 8000 ± 100 to 9100 ± 50. After injecting milk, the total leucocyte count was found to range from 10,325 ± 198 to 10,950 ± 323 showing the signs of leucocytosis.
Table 4.
Leucocyte count

Nasal provocation method
An objective of the present study was to provide a method for evaluating an anti-allergic substance characterized by measuring the amount of nasal secretion quantitatively, using rats with nasal mucosal hypersensitivity. The comparison between fast-dissolving films and conventional tablets was done by this method. The amount of nasal secretion before and after administering the drug preparations was measured. Also the difference in the amount of nasal secretion between the fast-dissolving film group and conventional tablet group was determined [Table 5].
Table 5.
Results showing the amount of nasal secretions secreted in rats
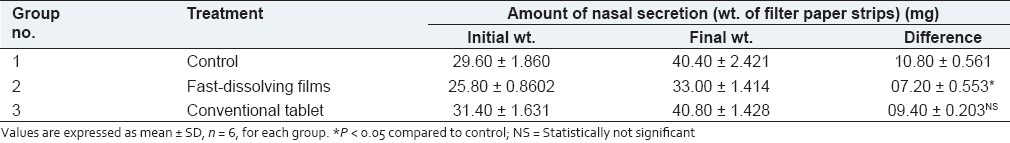
The control group showed a significant (P < 0.05) increase in nasal secretion. The fast-dissolving film group showed a decrease in nasal secretion as compared to the conventional tablet group at the 40 min. time interval. Also, the percentage decrease in the amount of nasal secretion for the films was found to be more than 50.0% compared to conventional tablets. Thus, the fast dissolving films showed a higher decrease in the amount of nasal secretion as compared to the conventional tablets.
CONCLUSIONS
Based on the encouraging results, the fast-dissolving films of levocetirizine dihydrochloride can be considered suitable for clinical use in the treatment of allergic rhinitis and other conditions of allergies, where a quicker onset of action for a dosage form is desirable along with the convenience of administration. The method of preparation was found to be simple and requires minimum excipients, thus making the product cost-effective. A further in vivo study proved that the fast-dissolving films of levocetirizine dihydrochloride produced a faster onset of action as compared to the conventional tablets.
ACKNOWLEDGEMENTS
The authors are grateful to Nitte University, Mangalore, and NITK, Surathkal, for providing the necessary facilities to carry out this study.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Suresh B, Osborne JL. Quick dissolving films- A novel approach to drug delivery. [Last accessed on 2011 Jan 10]. Available from: http://www.drugdeliverytechnology.com .
- 2.Revathi V. Fast Dissolving drug delivery system. Pharma Times. 2007;39:22–3. [Google Scholar]
- 3. [Last accessed on 2008 Sept 23]. Available from: http://en.wikipedia.org/wiki/Levocetirizine .
- 4.Tillement JP, Testa B, Brée F. Compared pharmacological characteristics in humans of racemic cetirizine and levocetirizine, two histamine H1-receptor antagonists. Biochem Pharmacol. 2003;66:1123–6. doi: 10.1016/s0006-2952(03)00558-6. [DOI] [PubMed] [Google Scholar]
- 5.Horak F, Zieglmayer PU, Zieglmayer R, Kavina A, Lemell P. Levocetirizine has a longer duration of action on improving total nasal symptoms score than fexofenadine after single administration. Br J Clin Pharmacol. 2005;60:24–31. doi: 10.1111/j.1365-2125.2005.02377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Potter PC Study Group. Levocetirizine is effective for symptom relief including nasal congestion in adolescent and adult (PAR) sensitized to house dust mites. Allergy. 2003;58:893–9. doi: 10.1034/j.1398-9995.2003.00171.x. [DOI] [PubMed] [Google Scholar]
- 7.Kapp A, Pichler WJ. Levocetirizine is an effective treatment in patients suffering from chronic idiopathic urticaria: A randomized, double-blind, placebo-controlled, parallel, multicenter study. Int J Dermatol. 2006;45:469–74. doi: 10.1111/j.1365-4632.2005.02609.x. [DOI] [PubMed] [Google Scholar]
- 8.Mashru RC, Sutariya VB, Sankalia MG, Parikh PP. Development and evaluation of fast-dissolving film of salbutamol sulphate. Drug Dev Ind Pharm. 2005;35:25–34. doi: 10.1081/ddc-43947. [DOI] [PubMed] [Google Scholar]
- 9.Uddhav Bagul, Kishore Gujar, Nancy Patel, Sanjeevani Aphale, Shalaka Dhat. Formulation and Evaluation of Sublimed Fast Melt Tablets of Levocetirizine Dihydrochloride. Int J Pharm Sci. 2010;2:76–80. [Google Scholar]
- 10.Raghuraman S, Velrajan G, Ravi R, Jeyabalan B, Benito Johnson D, Sankar V. Design and evaluation of propranolol hydrochloride buccal films. Indian J Pharm Sci. 2002;64:32–6. [Google Scholar]
- 11.Nafee NA, Boraie MA, Ismail FA, Mortada LM. Design and characterization of mucoadhesive buccal patches containing cetylpyridinium chloride. Acta Pharm. 2003;53:199–212. [PubMed] [Google Scholar]
- 12.Kumar GV, Krishna RV, William GJ, Konde A. Formulation and evaluation of buccal films of salbutamol sulphate. Indian J Pharm Sci. 2005;67:160–4. [Google Scholar]
- 13.Mashru RC, Sutariya VB, Sankalia MG, Parikh PP. Development and evaluation of fast dissolving film of salbutamol sulphate. Drug Dev Ind Pharm. 2005;31:25–34. doi: 10.1081/ddc-43947. [DOI] [PubMed] [Google Scholar]
- 14.Cilurzo F, Minghetti P, Como A, Montanari L. Maltodextrin fast dissolving film: A feasibility study. [Last accessed on 2011 Jan 10]. Available from: http://www.tecnova-srl.it .
- 15.Ghosh MN. Fundamentals of Experimental Pharmacology. 2nd ed. Culcutta: Scientific Book Agency; 1984. p. 155. [Google Scholar]
- 16.Pandit P, Singh A, Bafna AR, Kadam PV, Patil MJ. Evaluation of anti-asthamatic activity of Curculigo orchioides Gaertn rhizomes. Indian J Pharm Sci. 2008;70:330–44. doi: 10.4103/0250-474X.44590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Free patents online. [Last accessed on 2008 Oct 23]. Available from: http://www.freepatentsonline.com/5063045.html .