

Abstract
In the photosynthetic photosystem II, electrons are transferred from the manganese-containing oxygen evolving complex (OEC) to the oxidized primary electron-donor chlorophyll P680•+ by a proton-coupled electron transfer process involving a tyrosine-histidine pair. Proton transfer from the tyrosine phenolic group to a histidine nitrogen positions the redox potential of the tyrosine between those of P680•+ and the OEC. We report the synthesis and time-resolved spectroscopic study of a molecular triad that models this electron transfer. The triad consists of a high-potential porphyrin bearing two pentafluorophenyl groups (PF10), a tetracyanoporphyrin electron acceptor (TCNP), and a benzimidazole-phenol secondary electron-donor (Bi-PhOH). Excitation of PF10 in benzonitrile is followed by singlet energy transfer to TCNP (τ = 41 ps), whose excited state decays by photoinduced electron transfer (τ = 830 ps) to yield 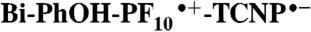 . A second electron transfer reaction follows (τ < 12 ps), giving a final state postulated as BiH+-PhO•-PF10-TCNP•-, in which the phenolic proton now resides on benzimidazole. This final state decays with a time constant of 3.8 μs. The triad thus functionally mimics the electron transfers involving the tyrosine-histidine pair in PSII. The final charge-separated state is thermodynamically capable of water oxidation, and its long lifetime suggests the possibility of coupling systems such as this system to water oxidation catalysts for use in artificial photosynthetic fuel production.
. A second electron transfer reaction follows (τ < 12 ps), giving a final state postulated as BiH+-PhO•-PF10-TCNP•-, in which the phenolic proton now resides on benzimidazole. This final state decays with a time constant of 3.8 μs. The triad thus functionally mimics the electron transfers involving the tyrosine-histidine pair in PSII. The final charge-separated state is thermodynamically capable of water oxidation, and its long lifetime suggests the possibility of coupling systems such as this system to water oxidation catalysts for use in artificial photosynthetic fuel production.
Keywords: photochemistry, biomimicry
The machinery of photosynthesis is an elaborate nanosystem composed of organic and inorganic cofactors assembled in large organized protein arrays that cooperatively perform the specific tasks needed to convert sunlight into biofuel. Through eons of evolution, nature has developed specialized enzymes able to oxidize water molecules and use the resulting electrons to reduce carbon dioxide to carbohydrates. The central component in the water oxidation process is the protein complex known as photosystem II (PSII), which uses sunlight to split water into molecular oxygen, protons, and reducing equivalents (1).
Essential to the function of PSII is its ability to combine single-photon excitation with the multielectron process of water oxidation. To perform this thermodynamically challenging reaction, PSII is equipped with the oxygen evolving complex (OEC), an inorganic complex featuring one calcium and four manganese ions (2–4). Functionally, the OEC is a charge accumulator able to store the four oxidizing equivalents needed for water oxidation. The OEC is electronically coupled to the chlorophyll complex P680 through a redox-active tyrosine-hystidine pair (TyrZ-D1His190). This pair acts as an electron relay between P680 and the OEC components through proton-coupled electron transfer (PCET) (5–8).
In PSII, the oxidizing equivalents are produced through photo-excitation of the chlorophyll molecules in the P680 complex, which decay by electron transfer to the quinone cofactors QA and QB via a pheophytin primary electron acceptor. The highly oxidizing P680•+ is reduced by the TyrZ residue on the nanosecond timescale. In this process, TyrZ loses its phenolic proton, likely to the nearby D1His190 group, to generate the neutral tyrosine radical TyrZ-O• (E0 = 0.9–1.0 V vs. SCE). The radical TyrZ-O• then removes one electron from the OEC on the microsecond timescale, increasing its oxidation state (6, 9–11). After four light-induced charge separations, the OEC oxidizes two water molecules, releases molecular oxygen, and returns to its initial oxidation state.
The functional principles of PSII can be used to guide the design of artificial photosynthetic devices. The highly efficient initial light-induced charge separation is a particularly attractive biomimetic target because the resulting electron-hole pair can in principle be used for conversion to electrical power or to generate a redox gradient to drive chemical reactions for fuel production. Therefore, development of synthetic systems able to produce useful forms of energy from abundant sunlight and water requires a better understanding of the thermodynamic and kinetic parameters of the primary processes of PSII.
In this context, we report the preparation and photophysical investigation of a bioinspired molecular triad engineered to functionally mimic the initial charge-separation events of PSII. The three units are designed and covalently assembled to simulate the specific interactions between P680, pheophytin, and the TyrZ-D1His190 pair of PSII. The energetics of the model are designed so that the final charge-separated state is thermodynamically capable of oxidizing water. A detailed spectroscopic investigation highlights the importance of electron transfer reactions associated with proton translocation in stabilizing the photoinduced charge-separated state and provides unique insight into how electron and proton motion can be used to bridge the gap between light and chemistry.
Results
Design and Synthesis.
Porphyrins are useful artificial alternatives to synthetically demanding and relatively unstable chlorophylls found in natural systems. In the case of PSII, the photogenerated P680•+ must be an extremely strong oxidant to provide the overpotential required for efficient water oxidation. Chlorophyll a in solution is incapable of carrying out such photo-oxidations, and the potential of chlorophyll in PSII is tuned by the protein environment to achieve the necessary redox power (12–14). Molecular triad 1 (Fig. 1) was engineered with the redox potentials of the porphyrin components designed to fulfill thermodynamic requirements similar to those found in P680 by adjusting the substituents on the macrocycles. In the design of 1, the tetracyanoporphyrin (TCNP) unit (Fig. 1, blue) acts as a powerful electron acceptor due to the electron withdrawing effects of the four cyano groups at the β-positions of the tetrapyrrolic core. The two pentafluorophenyl groups provide the primary electron-donor porphyrin (PF10) (Fig. 1, red) with the right redox balance to transfer one electron to the TCNP acceptor after irradiation of the complex, resulting in the  cation, which is thermodynamically competent to oxidize the benzimidazole-phenol (Bi-PhOH) secondary electron-donor component (Fig. 1, green). The Bi-PhOH unit features an intramolecular hydrogen bond between the phenolic proton and the lone pair of the nitrogen atom in the benzimidazole moiety. In the course of oxidation of Bi-PhOH by the photo-generated
cation, which is thermodynamically competent to oxidize the benzimidazole-phenol (Bi-PhOH) secondary electron-donor component (Fig. 1, green). The Bi-PhOH unit features an intramolecular hydrogen bond between the phenolic proton and the lone pair of the nitrogen atom in the benzimidazole moiety. In the course of oxidation of Bi-PhOH by the photo-generated 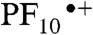 , the phenol is designed to transfer its proton to the benzimidazole group by a PCET mechanism to produce a neutral phenoxyl radical (E = 1.06 V vs. SCE in the case of 1) suitably poised for water oxidation (E0 = +0.58 V vs. SCE, pH 7), thus mimicking the role of the TyrZ-D1His190 in PSII.
, the phenol is designed to transfer its proton to the benzimidazole group by a PCET mechanism to produce a neutral phenoxyl radical (E = 1.06 V vs. SCE in the case of 1) suitably poised for water oxidation (E0 = +0.58 V vs. SCE, pH 7), thus mimicking the role of the TyrZ-D1His190 in PSII.
Fig. 1.

Molecular structure of triad 1, which is composed of three covalently linked subunits that mimic the redox processes in PS II. Upon irradiation, sequential electron transfer (ET) and PCET reactions eventually yield the final charge-separated state BiH+-PhO•-PF10-TCNP•-.
To assemble the three active units of 1, we developed the synthetic strategy depicted in Scheme 1. The synthesis begins with the preparation of porphyrin 4, which relies on an approach based on the selective condensation of the para-formyl group of compound 3. This selectivity is grounded in the reduced chemical reactivity of the formyl group involved in the intramolecular O-H---O = C hydrogen bond, which efficiently discourages its activation by acid catalysts. The benzimidazole moiety is then formed upon cyclization of the formyl group in 4 with ortho-phenylenediamine to afford 5, which is in turn hydrolyzed under acid conditions to yield 6. The resulting carboxylic acid in 6 is transformed into an amine group by a Curtius reaction to afford dyad 7. The TCNP 10 is efficiently prepared from tetrabromo derivative 8 (15) using a cyanation procedure developed in our laboratory. Finally, triad 1 is afforded through an amide-coupling reaction between 7 and 10. The dyad PF10-TCNP model compound 2 (Fig. 1) is prepared from the required precursors following similar procedures (SI Appendix).
Scheme 1.

Synthetic strategy and building blocks used to construct triad 1 and dyad 2.
Photophysical Investigation.
Spectroscopic investigations were carried out with triad 1, PF10-TCNP dyad 2, and Bi-PhOH-PF10 model compound 5, as well as with porphyrin references TCNP 9 and PF10 11 to gain insight into the photophysical properties of the photoactive units.
Steady-State Spectroscopic Investigations and Electrochemical Studies.
The absorption spectrum of PF10 11 in cyclohexane solution (Fig. 2, black line) exhibits characteristic electronic transitions of free base porphyrins (Soret absorption at 419 nm and Q-bands at 512, 547, 592, and 648 nm). The TCNP 9 (Fig. 2, solid red line) features a split in the Soret region with maxima at 440 and 444 nm, and broader Q-bands that appear at 543, 588, 654, and 711 nm (Fig. 2). These spectroscopic features are characteristic of a nonplanar porphyrin (16–22). Steric repulsion between the β-cyano groups and the meso-phenyl substituents leads to deformation of the tetrapyrrolic ring from its original planar geometry. The red shift is explained by two different phenomena, which operate cooperatively to reduce the highest-occupied molecular orbital–lowest-unoccupied molecular orbital (HOMO–LUMO) gap. The strong electron withdrawing effect induced by the four cyano groups is known to stabilize the LUMO orbital, whereas distortion of the porphyrin macrocycle destabilizes the HOMO orbital (22).
Fig. 2.

Ground state absorption spectra in cyclohexane (solid lines) and benzonitrile (broken lines). In cyclohexane: PF10 reference 11 (black solid line), TCNP 9 (red solid line), Bi-PhOH-PF10 model compound 5 (blue solid line), and PF10-TCNP dyad 2 (green solid line). In benzonitrile: PF10-TCNP dyad 2 (green broken line) and triad 1 (magenta broken line).
The absorption spectrum of Bi-PhOH-PF10 model compound 5 (Fig. 2, solid blue line) shows a slight broadening of the Soret band compared to that of 11, suggesting some electronic interaction between the Bi-PhOH and PF10 components in the ground state. On the other hand, the spectrum of PF10-TCNP dyad 2 (solid green line) is similar to a linear combination of the absorption spectra of its components, indicating no strong electronic interactions between the photoactive units when covalently linked to each other. Triad 1 is not sufficiently soluble in cyclohexane to obtain a useful absorption spectrum, but the spectrum in benzonitrile (Fig. 2, magenta broken line) resembles that of 2 in the same solvent (green broken line).
Steady-state fluorescence investigations in cyclohexane with excitation in the corresponding Soret region (SI Appendix, Fig. S2) disclose that PF10 reference 11 emits at 657 and 723 nm, whereas TCNP 9 fluoresces at 727 and 815 nm. From the absorption and emission data, the energies of the first singlet excited states (E00) are estimated as 1.90 eV for 11 and 1.72 eV for 9.
Estimation of the energies of the charge-separated states was afforded from electrochemical studies (SI Appendix, Figs. S6 and S7; Table S1). The first redox potentials (vs. SCE) of a PF10 model (SI Appendix, Fig. S8) are at Eox = +1.29 V and Ered = -0.96 V, whereas the Bi-PhOH first oxidation in model compound 5 occurs at Eox = +1.04 V. The first reduction of TCNP in triad 1 takes place at Ered = -0.27 V. Therefore, the intermediate 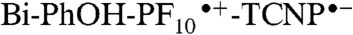 and final BiH+-PhO•-PF10-TCNP•- charge-separated states are roughly 1.56 eV and 1.34 eV above the ground state in triad 1.
and final BiH+-PhO•-PF10-TCNP•- charge-separated states are roughly 1.56 eV and 1.34 eV above the ground state in triad 1.
Time-Resolved Spectroscopic Investigations.
Time-resolved emission experiments in cyclohexane with excitation in the corresponding Soret regions yielded lifetimes of 8.89 ns (χ2 = 1.17) for the first singlet excited state of PF10 11 and 9.00 ns (χ2 = 1.03) for that of Bi-PhOH-PF10 model compound 5. The decay of reference TCNP 9 requires three components to satisfactorily fit the decay data (χ2 = 1.07): 102 ps, 2.47 ns, and 4.87 ns. A preliminary investigation of the photophysical properties of 9 revealed that the singlet excited state dynamics significantly depend on the solvent polarity and viscosity, which signals a complex excited state manifold and the possibility of different conformations in the excited state. We postulate that the 1TCNP state formed immediately after excitation undergoes a rather slow, solvent dependent (102 ps in cyclohexane) structural reorganization that yields a relaxed singlet excited state, which then decays via ordinary relaxation pathways with a fluorescence lifetime of 2.47 ns. The 4.87 ns component may represent the decay of an additional minor conformation, or in principle a minor impurity, although no other evidence for impurities was found.
Global analysis (χ2 = 1.07) of transient fluorescence experiments on dyad 2 in cyclohexane with excitation at 400 nm yielded five decay components, which are depicted in the form of decay-associated spectra (DAS) in Fig. 3. The 41 ps DAS spectrum shows positive amplitude at approximately 650 nm, where most of the fluorescence comes from the PF10 subunit, and negative amplitude between 700–800 nm, where the fluorescence is primarily due to the TCNP moiety (SI Appendix, Fig. S2). This spectrum reflects singlet-singlet energy transfer from PF10 to TCNP. Thus, the lifetime of 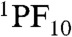 in dyad 2 is reduced from the 8.89 ns found for PF10 model 11 to 41 ps by the addition of the energy transfer decay pathway. From these numbers, we calculate a rate constant of kEnT = 2.4 × 1010 s-1 and a quantum yield of 98% for the energy transfer process in 2.
in dyad 2 is reduced from the 8.89 ns found for PF10 model 11 to 41 ps by the addition of the energy transfer decay pathway. From these numbers, we calculate a rate constant of kEnT = 2.4 × 1010 s-1 and a quantum yield of 98% for the energy transfer process in 2.
Fig. 3.

Fluorescence DAS in cyclohexane of dyad 2 following approximately 100 fs laser pulse excitation at 400 nm. Global analysis (χ2 = 1.07) yields five exponential components with lifetimes of 41 ps (black), 136 ps (red), 2.41 ns (green), 4.15 ns (blue), and 7.94 ns (turquoise).
The 136 ps, 2.4 ns, and 4.15 ns DAS spectra have very similar lifetimes to those obtained for the TCNP model 9 in cyclohexane. The 136 ps spectrum shows band-shift-like character with positive amplitude at the blue side and negative amplitude at the red side of the TCNP fluorescence emission bands and can be attributed to the relaxation of the singlet excited state in the complex excited state manifold to form a relaxed singlet excited state that lives for 2.4 ns (Fig. 3, red and green lines). The two minor components (4.15 and 7.94 ns) required in the global analysis may reflect conformational heterogeneity, minor impurities, and/or fitting artifacts. The fact that the decay time constants observed for 1TCNP in 2 are essentially identical to those observed for model 9 show that no charge-separated states are formed by decay of 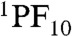 or 1TCNP in cyclohexane. Therefore, spectroscopic investigations of 2 were carried out in a more polar medium, benzonitrile, to facilitate charge transfer processes through driving force and reorganization energy effects.
or 1TCNP in cyclohexane. Therefore, spectroscopic investigations of 2 were carried out in a more polar medium, benzonitrile, to facilitate charge transfer processes through driving force and reorganization energy effects.
Time-resolved emission experiments with 2 in benzonitrile reveal a different scenario. Global analysis of the fluorescence decays (χ2 = 1.16) reveals time constants of 41 ps, 129 ps, 536 ps, 1.8 ns, and 9.3 ns. The first two decays represent energy transfer from 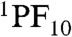 to TCNP and relaxation of 1TCNP, respectively, as was observed in cyclohexane. The 536 ps component indicates a new decay pathway for 1TCNP that was not present in 9 or in 2 in cyclohexane. The most likely mechanism is photoinduced electron transfer to form
to TCNP and relaxation of 1TCNP, respectively, as was observed in cyclohexane. The 536 ps component indicates a new decay pathway for 1TCNP that was not present in 9 or in 2 in cyclohexane. The most likely mechanism is photoinduced electron transfer to form 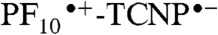 . This interpretation is consistent with the strong solvent dependence of the time constant and the spectroscopic and electrochemical results discussed above. Photoinduced electron transfer between porphyrins joined by similar linkages has been reported (23, 24). The fact that the shortest decay component in benzonitrile has a time constant that is essentially identical to the corresponding component in cyclohexane is consistent with an absence of significant photoinduced electron transfer from
. This interpretation is consistent with the strong solvent dependence of the time constant and the spectroscopic and electrochemical results discussed above. Photoinduced electron transfer between porphyrins joined by similar linkages has been reported (23, 24). The fact that the shortest decay component in benzonitrile has a time constant that is essentially identical to the corresponding component in cyclohexane is consistent with an absence of significant photoinduced electron transfer from 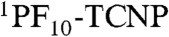 in both solvents. The 1.8 and 9.3 ns DAS are attributed to minor impurities or conformations, or fitting artifacts.
in both solvents. The 1.8 and 9.3 ns DAS are attributed to minor impurities or conformations, or fitting artifacts.
Transient absorption techniques were used to gain further insights into the spectral evolution of dyad 2. To identify the transient spectroscopic changes that are indicative of the formation of charge-separated states upon excitation, we first determined the spectroscopic absorption signatures of each possible cation and anion by spectroelectrochemical investigation of reference compounds 4 and 9. The results reveal that the 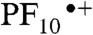 cation is characterized by a broad absorption in the 650–780 nm region, whereas the TCNP•- anion has maximum absorptions at 660 and 940 nm (SI Appendix). The redox processes are reversible in both cases, reflecting the redox stability of the porphyrins.
cation is characterized by a broad absorption in the 650–780 nm region, whereas the TCNP•- anion has maximum absorptions at 660 and 940 nm (SI Appendix). The redox processes are reversible in both cases, reflecting the redox stability of the porphyrins.
Excitation at 740 nm of an air-saturated benzonitrile solution of TCNP model porphyrin 9 yielded evolution associated difference spectra (EADS) with time constants of 116 ps, 1.9 ns, and a component with a lifetime too long to resolve on the timescale of the experiment (SI Appendix, Fig. S3). The 116 ps component is assigned to relaxation of the initially formed 1TCNP to a more stable species (spectral evolution shows band shift character), which decays in 1.9 ns. One of the products of this 1.9 ns decay is 3TCNP, which decays very slowly in the absence of oxygen (last, nondecaying EADS).
The results of a similar experiment with 2, excited at 740 nm in benzonitrile, where only TCNP absorbs, are presented in Fig. 4A. Five EADS with time constants of 12 ps, 120 ps, 590 ps, 1.8 ns (very minor EADS), and a component that did not decay on the < 10 ns timescale were obtained. Based on the EADS shapes, the time-resolved fluorescence results for 2 and 9, and the transient absorption data for 9, we interpret the 120 ps process as relaxation of the initially formed 1TCNP, which has a strong induced absorption with a maximum at approximately 520 nm. The 590 ps component, with strong induced absorption at approximately 570 nm, is due to decay of the resulting relaxed excited state by photoinduced electron transfer to yield 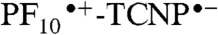 . The nondecaying component is assigned to porphyrin triplet states having induced absorption maxima at approximately 540 nm; consistent with this interpretation, the lifetime is reduced in the presence of oxygen. This triplet is formed in part by intersystem crossing of 1TCNP, and potentially in part by decay of the charge-separated state. Consistent with the fluorescence results discussed above, the 1.8 ns EADS is attributed to minor impurities, conformations, or fitting artifacts. The EADS with the 12 ps lifetime can be attributed to charge recombination of
. The nondecaying component is assigned to porphyrin triplet states having induced absorption maxima at approximately 540 nm; consistent with this interpretation, the lifetime is reduced in the presence of oxygen. This triplet is formed in part by intersystem crossing of 1TCNP, and potentially in part by decay of the charge-separated state. Consistent with the fluorescence results discussed above, the 1.8 ns EADS is attributed to minor impurities, conformations, or fitting artifacts. The EADS with the 12 ps lifetime can be attributed to charge recombination of 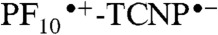 , at least in part to the ground state. The kinetic traces depicted in Fig. 4, Inset clearly show a rise/formation of stimulated emission at 780 nm and characteristic induced absorption due to the
, at least in part to the ground state. The kinetic traces depicted in Fig. 4, Inset clearly show a rise/formation of stimulated emission at 780 nm and characteristic induced absorption due to the 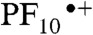 radical cation at 650 nm. The combined transient absorption and fluorescence results therefore reveal that the formation (590 ps) of the
radical cation at 650 nm. The combined transient absorption and fluorescence results therefore reveal that the formation (590 ps) of the 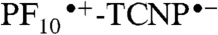 state is slower than its decay (12 ps). This order of rate constants is an example of inverted kinetics, where the rate constant for recombination is larger than that for charge separation, and transient signals can be inverted in amplitude relative to their usual appearance (24, 25). A quantum yield of 77% is calculated for formation of
state is slower than its decay (12 ps). This order of rate constants is an example of inverted kinetics, where the rate constant for recombination is larger than that for charge separation, and transient signals can be inverted in amplitude relative to their usual appearance (24, 25). A quantum yield of 77% is calculated for formation of 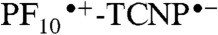 in 2. This yield is independent of which porphyrin is excited, as the quantum yield of PF10-1TCNP from
in 2. This yield is independent of which porphyrin is excited, as the quantum yield of PF10-1TCNP from 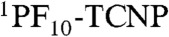 is essentially unity.
is essentially unity.
Fig. 4.

Transient absorption of dyad 2 (Fig. 3A) and triad 1 (Fig. 3B) in benzonitrile upon femtosecond excitation at 740 nm presented in the form of EADS. (A) For dyad 2, global analysis yielded five components: 12 ps (black), 120 ps (red), 590 ps (green), 1.8 ns (blue), and nondecaying (turquoise). (B) For triad 1, global analysis afforded four components: 120 ps (red), 610 ps (green), 2.1 ns (blue), and nondecaying (turquoise). (A, Inset) Transient absorption kinetic traces in benzonitrile for dyad 2 at 650 nm (red) and 780 nm (pink) and for triad 1 at 780 nm (blue).
Turning now to triad 1, incorporation of Bi-PhOH into the structure creates a redox gradient that provides sufficient driving force for secondary electron transfer from the Bi-PhOH to the 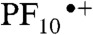 cation. The resulting oxidation of the Bi-PhOH unit dramatically enhances the acidity of the phenolic proton (Δ pKa ∼ 12) (25, 26), which creates the chemical potential required for proton transfer to the hydrogen-bonded benzimidazole moiety (27–30). Therefore, excitation of triad 1 is expected to lead to eventual formation of a charge-separated state characterized by cationic benzimidazole, a phenoxyl radical, a neutral PF10, and a reduced TCNP porphyrin (BiH+-PhO•-PF10-TCNP•-). Transient absorption techniques were employed to investigate this possibility. Based on the observation that excitation of the PF10 unit leads to a fast and nearly quantitative energy transfer to the TCNP moiety, the transient absorption experiments with 1 were carried out with exclusive excitation of the TCNP component at 740 nm.
cation. The resulting oxidation of the Bi-PhOH unit dramatically enhances the acidity of the phenolic proton (Δ pKa ∼ 12) (25, 26), which creates the chemical potential required for proton transfer to the hydrogen-bonded benzimidazole moiety (27–30). Therefore, excitation of triad 1 is expected to lead to eventual formation of a charge-separated state characterized by cationic benzimidazole, a phenoxyl radical, a neutral PF10, and a reduced TCNP porphyrin (BiH+-PhO•-PF10-TCNP•-). Transient absorption techniques were employed to investigate this possibility. Based on the observation that excitation of the PF10 unit leads to a fast and nearly quantitative energy transfer to the TCNP moiety, the transient absorption experiments with 1 were carried out with exclusive excitation of the TCNP component at 740 nm.
Global analysis of the femtosecond excitation data for 1 in the form of EADS (Fig. 4B) gives four time constants of 120 ps, 610 ps, 2.1 ns, and a long-lived nondecaying component. As with the model compounds, the 120 ps component is assigned to decay of the initially formed 1TCNP to a relaxed first excited singlet state. The 610 ps EADS is ascribed to decay mainly by photoinduced electron transfer to give 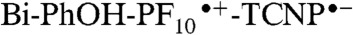 (kET = 1.2 × 109 s-1). An important observation from these experiments is the absence of the 12 ps component in the triad dynamics (Fig. 4A, Inset, blue line). According to our interpretation of the dyad spectral evolution, this 12 ps component represents charge recombination of the
(kET = 1.2 × 109 s-1). An important observation from these experiments is the absence of the 12 ps component in the triad dynamics (Fig. 4A, Inset, blue line). According to our interpretation of the dyad spectral evolution, this 12 ps component represents charge recombination of the 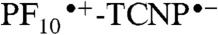 radical ion pair. Therefore, the
radical ion pair. Therefore, the 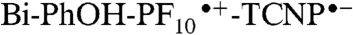 intermediate charge-separated state is rapidly quenched due to a new decay channel, which we interpret as charge shift to yield the BiH+-PhO•-PF10-TCNP•- state.
intermediate charge-separated state is rapidly quenched due to a new decay channel, which we interpret as charge shift to yield the BiH+-PhO•-PF10-TCNP•- state.
Complementary nanosecond transient absorption experiments on triad 1 dissolved in air-saturated benzonitrile solutions were carried out to investigate the relatively long-lived transient species. Global analysis of data for 1 in benzonitrile excited at 735 nm is presented in the form of DAS in Fig. 5. The first DAS (0.8 μs, black line) clearly shows TCNP ground state bleaching at 450 nm and 3TCNP excited state absorption at 540 nm. This DAS corresponds to the TCNP triplet excited state quenched by molecular oxygen. This lifetime increases when oxygen is removed from the sample. The 3.8 μs DAS (Fig. 5, red line), whose amplitude is also shown magnified ten times for better visualization (open symbols), shows ground state bleaching at 450 and 740 nm as well as induced absorptions in the 560–700 nm and > 800 nm regions. A closer inspection of the spectrum < 490 nm clearly reveals competition between the TCNP ground state bleaching at 450 nm and an induced absorption with a maximum at approximately 400 nm. Based on the spectroelectrochemistry results (SI Appendix, Figs. S9 and S10), the induced absorption at 400–500 nm and > 800 nm is assigned to formation of BiH+-PhO•, whereas those at 660 and 940 nm are due to TCNP•-. Therefore, the 3.8 μs DAS contains the spectroscopic signature of the BiH+-PhO•-PF10-TCNP•- final charge-separated state, providing compelling evidence that such a state is indeed produced upon excitation of triad 1 in benzonitrile. From comparison of triad and dyad kinetic traces at 940 nm, where the TCNP•- radical anion has a transient signature, the quantum yield of the final charge-separated state is estimated as approximately 52% (SI Appendix, Fig. S5).
Fig. 5.

Transient absorption of triad 1 in air-saturated benzonitrile upon nanosecond excitation at 735 nm presented in the form of DAS. The time constants associated with the spectra are 0.8 μs (black line and black circles) and 3.8 μs (red line and red circles). The 3.8 μs spectrum is also shown as a × 10 expansion (red line and open circles).
These spectroscopic observations together with the electrochemical data allow elaboration of the energy diagram depicted in Fig. 6, which highlights the photophysical decay pathways for 1 in benzonitrile. The first singlet excited state 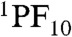 decays quantitatively through energy transfer to the TCNP unit with a time constant kEnT = 2.4 × 1010 s-1. The 1TCNP excited state undergoes an excited state relaxation/structural reorganization (k = 8.3 × 109 s-1) to yield a relaxed first excited singlet state that decays through electron transfer to form the
decays quantitatively through energy transfer to the TCNP unit with a time constant kEnT = 2.4 × 1010 s-1. The 1TCNP excited state undergoes an excited state relaxation/structural reorganization (k = 8.3 × 109 s-1) to yield a relaxed first excited singlet state that decays through electron transfer to form the 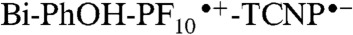 radical ion pair (kET = 1.2 × 109 s-1). Favorably competing with charge recombination of
radical ion pair (kET = 1.2 × 109 s-1). Favorably competing with charge recombination of 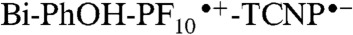 is the extremely fast charge shift reaction that oxidizes the phenol (kPCET > 8 × 1010 s-1). This proton-coupled electron transfer reaction involves both electron transfer from the phenol to
is the extremely fast charge shift reaction that oxidizes the phenol (kPCET > 8 × 1010 s-1). This proton-coupled electron transfer reaction involves both electron transfer from the phenol to 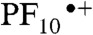 and transfer of the phenolic proton to the appended benzimidazole group to afford the BiH+-PhO•-PF10-TCNP•- final charge-separated state. The final state undergoes charge recombination with kCR = 2.6 × 105 s-1.
and transfer of the phenolic proton to the appended benzimidazole group to afford the BiH+-PhO•-PF10-TCNP•- final charge-separated state. The final state undergoes charge recombination with kCR = 2.6 × 105 s-1.
Fig. 6.

Energy level diagram, proposed decay pathways, and rate constants in s-1 for triad 1 following excitation of the PF10 unit in benzonitrile. The energy levels were derived from steady-state absorption, fluorescence, and electrochemical measurements. The rate constants were obtained from transient absorption and fluorescence measurements. kEnT = rate constant for energy transfer; kET = rate constant for photoinduced electron transfer; kPCET = rate constant for the proton-coupled electron transfer, and kCR = rate constant for charge recombination.
Previous studies of benzimidazole-phenol systems with internal hydrogen bonds have suggested that oxidation of the phenol is accompanied by transfer of a proton to the hydrogen-bonded imidazole, thereby lowering the oxidation potential of the phenol unit (31, 32). The ultrafast secondary charge shift (< 12 ps) observed for 1 is consistent with such a PCET mechanism. The PCET avoids formation of a Bi-PhOH•+-PF10-TCNP•- intermediate. Electrochemical studies of a model phenol that lacks the internal hydrogen bond, 2,4,6-tri-tert-butylphenol, show that the oxidation potential of the phenol is approximately 1.36 V vs. SCE (32, 33). Using this number, we estimate the energy of Bi-PhOH•+-PF10-TCNP•- to be approximately 1.63 eV. Thus, formation of this state from 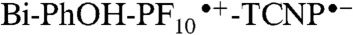 at 1.56 eV would be endergonic, and would be expected to occur slowly or not at all. As mentioned above, electron transfer in the photosynthetic OEC is thought to occur by a similar pathway.
at 1.56 eV would be endergonic, and would be expected to occur slowly or not at all. As mentioned above, electron transfer in the photosynthetic OEC is thought to occur by a similar pathway.
The final charge-separated state, proposed to be BiH+-PhO•-PF10-TCNP•-, is relatively long-lived (∼4 μs). In related triads of the carotenoid-porphyrin-porphyrin (C-P-P) type, where the carotenoid is the secondary electron-donor and no proton transfer is involved in the charge-separation or recombination processes, the final charge-separated states C•+-P-P•- typically live only hundreds of nanoseconds at room temperature (23, 24). The systems are not strictly comparable to 1, as the thermodynamic driving forces and solvents differ in the various systems. However, it is possible that nuclear rearrangements resulting from the PCET process may increase the energy barrier for charge recombination. Such a possibility has been suggested for the natural photosynthetic system (25) and we are in the process of investigating this hypothesis by femtosecond infrared spectroscopy using molecules related to the one described in this work.
Conclusion
The results for triad 1 demonstrate that structural and mechanistic motifs involving proton-coupled electron transfer in the TyrZ-D1His190 couple that are found in natural PSII can also be realized in a synthetic model system. Excitation of either porphyrin moiety of the triad powers a sequence of rapid electron transfer reactions that generate a long-lived charge-separated state in good yield. The results are consistent with the involvement of PCET in the second step of the charge-separation, a charge shift reaction. Furthermore, the design of the triad incorporates both a high-potential electron-donor porphyrin and a low-potential electron accepting porphyrin, leading to a final charge-separated state that is thermodynamically capable of water oxidation. Such constructs may be of value in the development of synthetic fuel production systems that draw their energy from sunlight in this issue of PNAS (34).
Materials and Methods
Synthesis.
All chemicals were purchased from Aldrich, Alfa Aesar, and Acros and were used without further purification. Solvents were obtained from EM Science and were used as received unless otherwise noted. Thin layer chromatography (TLC) was performed with silica gel coated glass plates from Analtech. Column chromatography was carried out using Silicycle silica gel 60 with 230–400 mesh. All new compounds were characterized by 1H-NMR spectroscopy and MALDI-TOF mass spectrometry. Details of the synthesis and characterization are given in SI Appendix.
Spectroscopy.
Steady-state absorption spectra were measured on a Shimadzu UV2100U UV-visible (UV-vis) and/or UV-3101PC UV-vis-near infrared spectrometer. Steady-state fluorescence spectra were measured using a Photon Technology International MP-1 spectrometer and corrected for detection system response. All time-resolved spectroscopic studies were carried out with laboratory-build instrumentation (SI Appendix).
Electrochemical and Spectroelectrochemical Studies.
All electrochemical and spectroelectrochemical studies were carried out using a CH Instruments 760D potentiostat. Spectroelectrochemical experiments were performed with an indium doped tin oxide (ITO) working electrode having a glass cover slip attached over the conductive face of the ITO with heat-shrink plastic. The spectroelectrochemical setup was contained in a 1 cm quartz cuvette under a constant Ar flow, and a Shimadzu UV-3101PC spectrophotometer was used for all experiments (SI Appendix).
Supplementary Material
Acknowledgments.
This work was supported as part of the Center for Bio-Inspired Solar Fuel Production, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0001016.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1118348109/-/DCSupplemental.
References
- 1.Blankenship RE. Molecular Mechanisms of Photosynthesis. Oxford: Blackwell Science; 2002. [Google Scholar]
- 2.Umena Y, Kawakami K, Shen JR, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 angstrom. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 3.Ferreira KN, et al. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 4.Loll B, et al. Towards complete cofactor arrangement in the 3.0 angstrom resolution structure of photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- 5.Limburg J, et al. A functional model for O-O bond formation by the O-2-evolving complex in photosystem II. Science. 1999;283:1524–1527. doi: 10.1126/science.283.5407.1524. [DOI] [PubMed] [Google Scholar]
- 6.Debus RJ, Barry BA, Babcock GT, Mcintosh L. Site-directed mutagenesis identifies a tyrosine radical involved in the photosynthetic oxygen-evolving system. Proc Natl Acad Sci USA. 1988;85:427–430. doi: 10.1073/pnas.85.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tommos C, Babcock GT. Oxygen production in nature: A light-driven metalloradical enzyme process. Acc Chem Res. 1998;31:18–25. [Google Scholar]
- 8.Hammarstrom L, Styring S. Proton-coupled electron transfer of tyrosinesin photosystem II and model systems for artificial photosynthesis: The role of a redox-active link between catalyst and photosensitizer. Energy Environ Sci. 2011;4:2379–2388. [Google Scholar]
- 9.Groot ML, et al. Initial electron donor and acceptor in isolated photosystem II reaction centers identified with femtosecond mid-IR spectroscopy. Proc Natl Acad Sci USA. 2005;102:13087–13092. doi: 10.1073/pnas.0503483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rappaport F, et al. Kinetics and pathways of charge recombination in photosystem II. Biochemistry. 2002;41:8518–8527. doi: 10.1021/bi025725p. [DOI] [PubMed] [Google Scholar]
- 11.Barry BA, Babcock GT. Tyrosine radicals are involved in the photosynthetic oxygen-evolving system. Proc Natl Acad Sci USA. 1987;84:7099–7103. doi: 10.1073/pnas.84.20.7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin X, et al. Specific alteration of the oxidation potential of the electron-donor in reaction centers from Rhodobacter sphaeroides. Proc Natl Acad Sci USA. 1994;91:10265–10269. doi: 10.1073/pnas.91.22.10265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalman L, LoBrutto R, Allen JP, Williams JC. Modified reaction centers oxidize tyrosine in reactions that mirror photosystem II. Nature. 1999;402:696–699. [Google Scholar]
- 14.Dutton PL, Mosser CC. Quantum biomechanics of long-range electron-transfer in protein-hydrogen bonds and reorganization energies. Proc Natl Acad Sci USA. 1994;91:10247–10250. doi: 10.1073/pnas.91.22.10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chumakov DE, Khoroshutin AV, Anisimov AV, Kobrakov KI. Bromination of porphyrins (Review) Chem Heterocycl Comp. 2009;45:259–283. [Google Scholar]
- 16.Birnbaum ER, et al. F-19 NMR-spectra and structures of halogenated porphyrins. Inorg Chem. 1995;34:3625–3632. [Google Scholar]
- 17.Drain CM, et al. Dynamic photophysical properties of conformationally distorted nickel porphyrins. 1. Nickel(II) dodecaphenylporphyrin. J Phys Chem. 1996;100:11984–11993. [Google Scholar]
- 18.Jentzen W, et al. Ruffling in a series of nickel(Ii) meso-tetrasubstituted porphyrins as a model for the conserved ruffling of the heme of cytochromes C. J Am Chem Soc. 1995;117:11085–11097. doi: 10.1021/ja00150a008. [DOI] [PubMed] [Google Scholar]
- 19.Kadish KM, et al. Synthesis and electrochemical studies of a series of fluorinated dodecaphenylporphyrins. Inorg Chem. 1999;38:2188–2198. doi: 10.1021/ic9811695. [DOI] [PubMed] [Google Scholar]
- 20.Nurco DJ, et al. Conformational flexibility in dodecasubstituted porphyrins. J Am Chem Soc. 1996;118:10918–10919. [Google Scholar]
- 21.Sazanovich IV, et al. Photophysical and structural properties of saddle-shaped free base porphyrins: Evidence for an “orthogonal” dipole moment. J Phys Chem B. 2001;105:7818–7829. [Google Scholar]
- 22.Shelnutt JA, et al. Nonplanar porphyrins and their significance in proteins. Chem Soc Rev. 1998;27:31–41. [Google Scholar]
- 23.Gust D, et al. Long-lived photoinitiated charge separation in carotene-diporphyrin triad molecules. J Am Chem Soc. 1991;113:3638–3649. [Google Scholar]
- 24.Gust D, Moore TA, Moore AL. Molecular mimicry of photosynthetic energy and electron transfer. Acc Chem Res. 1993;26:198–205. [Google Scholar]
- 25.Faller P, Goussias C, Rutherford AW, Un S. Resolving intermediates in biological proton-coupled electron transfer: A tyrosyl radical prior to proton movement. Proc Natl Acad Sci USA. 2003;100:8732–8735. doi: 10.1073/pnas.1530926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harriman A. Further comments on the redox potentials of tryptophan and tyrosine. J Phys Chem. 1987;91:6102–6104. [Google Scholar]
- 27.Costentin C, Robert M, Saveant JM. Concerted proton-electron transfers in the oxidation of phenols. Phys Chem Chem Phys. 2010;12:11179–11190. doi: 10.1039/c0cp00063a. [DOI] [PubMed] [Google Scholar]
- 28.Markle TF, Rhile IJ, DiPasquale AG, Mayer JM. Probing concerted proton-electron transfer in phenol-imidazoles. Proc Natl Acad Sci USA. 2008;105:8185–8190. doi: 10.1073/pnas.0708967105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westlake BC, et al. Concerted electron-proton transfer in the optical excitation of hydrogen-bonded dyes. Proc Natl Acad Sci USA. 2011;108:8554–8558. doi: 10.1073/pnas.1104811108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang MT, Irebo T, Johansson O, Hammarstrom L. Proton-coupled electron transfer from tyrosine: A strong rate dependence on intramolecular proton transfer distance. J Am Chem Soc. 2011;133:13224–13227. doi: 10.1021/ja203483j. [DOI] [PubMed] [Google Scholar]
- 31.Moore GF, et al. A bioinspired construct that mimics the proton coupled electron transfer between P680*+ and the Tyr(z)-His190 pair of photosystem II. J Am Chem Soc. 2008;130:10466–10467. doi: 10.1021/ja803015m. [DOI] [PubMed] [Google Scholar]
- 32.Moore GF, et al. Effects of protonation state on a tyrosine-histidine bioinspired redox mediator. J Phys Chem B. 2010;114:14450–14457. doi: 10.1021/jp101592m. [DOI] [PubMed] [Google Scholar]
- 33.Richards JA, Whitson PE, Evans DH. Electrochemical oxidation of 2,4,6-tri-tert-butylphenol. J Electroanal Chem. 1975;63:311–327. [Google Scholar]
- 34.Zhou Y, et al. Improving the efficiency of water splitting in dye-sensitized solar cells by using a biomimetic electron transfer mediator. Proc Natl Acad Sci USA. 2012 doi: 10.1073/pnas.1118339109. 10.1073/pnas.1118339109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.