

Abstract
Current models of sleep/wake regulation posit that Hypocretin (Hcrt)-expressing neurons in the lateral hypothalamus promote and stabilize wakefulness by projecting to subcortical arousal centers. However, the critical downstream effectors of Hcrt neurons are unknown. Here we use optogenetic, pharmacological, and computational tools to investigate the functional connectivity between Hcrt neurons and downstream noradrenergic neurons in the locus coeruleus (LC) during nonrapid eye movement (NREM) sleep. We found that photoinhibiting LC neurons during Hcrt stimulation blocked Hcrt-mediated sleep-to-wake transitions. In contrast, when LC neurons were optically stimulated to increase membrane excitability, concomitant photostimulation of Hcrt neurons significantly increased the probability of sleep-to-wake transitions compared with Hcrt stimulation alone. We also built a conductance-based computational model of Hcrt-LC circuitry that recapitulates our behavioral results using LC neurons as the main effectors of Hcrt signaling. These results establish the Hcrt-LC connection as a critical integrator-effector circuit that regulates NREM sleep/wake behavior during the inactive period. This coupling of distinct neuronal systems can be generalized to other hypothalamic integrator nuclei with downstream effector/output populations in the brain.
Keywords: ChR2, norepinephrine, step function opsin
The neural basis of wakefulness and arousal is thought to depend on subcortical populations of “arousal-promoting” nuclei located in the hypothalamus and brainstem (1, 2). Neurons in the lateral hypothalamus that express hypocretins (Hcrts—also called “orexins”), a pair of neuropeptides produced from the same genetic precursor (3, 4), have been proposed to play a key role in stabilizing wake states by directly projecting throughout the brain to other arousal populations (1, 5, 6). Electrophysiological recordings of Hcrt neurons show that they are relatively silent during sleep compared with wakefulness, with phasic bursts of activity preceding transitions to wakefulness (7, 8). Loss-of-function perturbation of the Hcrts or their receptors causes a narcolepsy phenotype (9–11). When centrally administered, the Hcrts increase the time spent awake and decrease nonrapid eye movement (NREM) and rapid eye movement (REM) sleep (12, 13).
Although it is evident that Hcrts promote wakefulness and arousal, it is unknown which downstream structures are necessary and/or sufficient to mediate their effects. Hcrt neurons project diffusely throughout the brain (14), and it is possible that their effects on wakefulness are due to widespread, global postsynaptic targets. Alternatively, Hcrts may affect arousal primarily by projecting to key downstream arousal centers. An intriguing possibility is that Hcrt neurons affect wakefulness by projecting to the locus coeruleus (LC), a noradrenergic structure in the brainstem known to promote wakefulness and arousal (15, 16). Indeed, the LC receives the densest afferent projections from Hcrt neurons (14, 17). Application of Hcrts into the LC region depolarizes LC neurons (12, 18) and increases LC discharge rates (12, 17, 19–21). Interestingly, RNAi-mediated knockdown of Hcrt receptor signaling in the LC region increases REM sleep (22).
Although these studies support the hypothesis that Hcrts regulate arousal by directly exciting the LC, it is unknown whether the LC is necessary and/or sufficient to mediate Hcrt-mediated sleep-to-wake transitions. We previously used viral approaches to deliver genetically encoded optogenetic transgenes to Hcrt (23) and LC (24) neurons to study the effects of stimulation or inhibition on wakefulness with high spatial and temporal precision. Because Hcrt and LC neural populations are located in distinct brain regions (Fig. 1A), it is physically possible to access both structures simultaneously in the same animal. We therefore took a dual optogenetic approach to stimulate Hcrt neurons while concomitantly inhibiting or stimulating noradrenergic LC neurons during NREM sleep in the inactive period. We found that photoinhibiting LC neurons during Hcrt stimulation blocked Hcrt-mediated sleep-to-wake transitions. In contrast, we found that increasing the excitability of LC neurons during Hcrt stimulation enhanced Hcrt-mediated sleep-to-wake transitions. We also built a computational model of Hcrt-LC dynamics that elucidates the temporal effects of Hcrt stimulation on LC membrane potential. This model is sufficient to reproduce our behavioral data, using only LC neurons as downstream effectors of Hcrt neurons. Taken together, our results show that the LC serves as a necessary and sufficient downstream effector for Hcrt-mediated NREM sleep-to-wake transitions during the inactive period.
Fig. 1.

Hcrt and LC neuroanatomy and functional connectivity. (A) Diagram depicting the sagittal mouse brain with relative locations of Hcrt and LC neurons. Hcrt neurons send projections throughout the brain but project most strongly to LC neurons in the brainstem. (B and C) Spatial distribution of tyrosine hydroxylase-immunoreactive soma in the brainstem (B) closely parallels the density of Hcrt-immunoreactive fibers in an adjacent section (C). (Scale bar, 50 μm.) (D) In vivo unilateral photostimulation of Hcrt neurons increases c-Fos immunoreactivity in the LC. Shown are representative images of the ipsilateral and contralateral LC costained for TH (red) and c-Fos (white). (Scale bar, 25 μm.) (E) Quantification of the percentage of neurons showing TH immunoreactivity that also express c-Fos. Data represent the mean ± SEM from animals transduced with ChR2-mCherry (n = 4) and mCherry (n = 4). **P < 0.001, two-way ANOVA followed by Tukey’s posthoc test.
Results
Functional Connectivity Between Hcrt and LC Neurons.
To gain initial insight into the anatomical and functional connectivity between Hcrt and LC neurons, we collected coronal brain sections that spanned the anteroposterior length of the LC and compared expression of Hcrt with expression of tyrosine hydroxylase, a marker of LC neurons. Hcrt-containing fibers densely innervated the LC compared with surrounding brain regions (Fig. 1 B and C), confirming the relatively dense projections from Hcrt neurons to the LC (14).
To determine the effect of stimulating Hcrt neurons on LC activity, we unilaterally photostimulated Hcrt neurons with ChR2 and measured the expression of c-Fos, a biomarker of neural activity, within the LC (Fig. 1 D and E). We genetically targeted Hcrt neurons by injecting lentiviral vectors expressing either Hcrt::ChR2-mCherry or Hcrt::mCherry into the Hcrt field. These viruses were previously shown to deliver optogenetic transgenes specifically and efficiently to Hcrt neurons (23, 25). We applied a chronic stimulation paradigm (15-ms light pulses at 10 Hz for 10 s each minute for 1 h) to all animals and then immediately perfused the animals to examine expression of c-Fos in tyrosine hydroxylase-positive neurons. Unilateral stimulation in Hcrt::ChR2-mCherry animals significantly increased the percentage of c-Fos–expressing cells in both the ipsilateral and the contralateral LC compared with that in Hcrt::mCherry control animals (P < 0.001, n = 4 animals for each condition). These results demonstrate that unilateral stimulation of Hcrt neurons increases activity in both the ipsilateral and the contralateral sides of the LC.
Inhibition of LC Neurons Blocks Hcrt-Mediated Sleep-to-Wake Transitions.
To determine the effect of inhibiting LC neurons on Hcrt-mediated sleep-to-wake transitions, we unilaterally photostimulated Hcrt neurons while simultaneously photoinhibiting LC neurons during the inactive period (Fig. 2A). We genetically targeted optogenetic transgenes to LC neurons by stereotaxically injecting a Cre recombinase-dependent adeno-associated virus (AAV) into knock-in mice selectively expressing Cre in tyrosine hydroxylase neurons (TH::IRES-Cre mice) (26). AAV vectors contained either eNpHR3.0-eYFP or eYFP and were previously shown to specifically deliver transgenes to >98% of tyrosine hydroxylase-positive neurons in the LC (24). We simultaneously delivered non–Cre-dependent lentiviral vectors carrying either Hcrt::ChR2-mCherry or Hcrt::mCherry sequences into the Hcrt field. After viral injections, we implanted two bilateral cannulae, one above Hcrt neurons and the other above the LC, for subsequent light delivery (Fig. S1). We also implanted electroencephalographic (EEG) and electromyographic (EMG) electrodes into the skull and neck musculature, respectively, for sleep/wake analysis.
Fig. 2.

Simultaneous bilateral photoinhibition of LC neurons blocks Hcrt-mediated sleep-to-wake transitions. (A) Diagram depicting experimental conditions. Hcrt neurons were transduced with either ChR2-mCherry or mCherry and LC neurons were transduced with either eNpHR-eYFP or eYFP. (B) Latencies to wake during NREM sleep following photostimulation in Hcrt neurons with simultaneous photoinhibition in LC neurons. Data represent the mean ± SEM sleep-to-wake latencies (15 trials per mouse per condition, six mice per condition). **P < 0.001, Student’s t test between eNpHR-eYFP and eYFP conditions. (C) Latencies to wake during NREM sleep following photostimulation in Hcrt neurons and subsequent photoinhibition in LC neurons. Data represent the mean ± SEM sleep-to-wake latencies (15 trials per mouse per condition, six mice per condition).
To ensure that yellow light illumination of the LC decreased neuronal activity as assessed by c-Fos expression, we repeated unilateral Hcrt-stimulation experiments (15-ms light pulses at 10 Hz for 10 s each minute for 1 h) while simultaneously photoinhibiting the LC with yellow light (constant light for 10 s each minute concurrent with Hcrt stimulation). As expected, stimulation of ChR2-mCherry–transduced Hcrt neurons caused an increase in c-Fos expression in LC neurons compared with mCherry control animals (P < 0.001, n = 4 animals per condition) (Fig. S2). However, this effect was blocked with concurrent photoinhibition of the LC (Fig. S2), demonstrating that eNpHR-mediated inhibition blocks the increase in activity of LC neurons caused by stimulation of Hcrt neurons.
To determine the necessity of LC activity on Hcrt-mediated sleep-to-wake transitions, we unilaterally stimulated either ChR2-mCherry or mCherry-transduced Hcrt neurons with 10 Hz blue light, using a stimulation protocol of 15-ms light pulses for 10 s starting 15 s after the onset of NREM sleep. As expected, we found that 10 Hz stimulation significantly reduced the sleep-to-wake latency between ChR2 and control animals (P < 0.001, n = 6 animals per condition) (Fig. 2B). However, in animals in which the LC was transduced with eNpHR, 10 Hz photostimulation of Hcrt neurons had no effect on sleep-to-wake latency when the LC was simultaneously illuminated bilaterally with yellow light (Fig. 2B and Movie S1). Yellow light did not diminish Hcrt-mediated sleep-to-wake transitions in control animals in which the LC was transduced with eYFP. Interestingly, there was no difference between Hcrt-mediated sleep-to-wake latencies in eNpHR compared with eYFP animals (P > 0.05) when the LC was inhibited unilaterally on the ipsilateral or contralateral side (Fig. 2B). These results show that bilateral inhibition, but not unilateral inhibition, of LC neurons is sufficient to block Hcrt-mediated sleep-to-wake transitions.
To determine whether LC inhibition must occur simultaneously with Hcrt stimulation to block Hcrt-mediated effects on wakefulness, we repeated the prior experiments but illuminated LC neurons with yellow light immediately following Hcrt stimulation (Fig. 2C). We found no significant difference in Hcrt-mediated sleep-to-wake latency in eNpHR compared with eYFP animals (P > 0.05, n = 6 animals per condition) when the LC was inhibited bilaterally or unilaterally in the 10 s following Hcrt stimulation (Fig. 2C). These results demonstrate that inhibition of LC neurons must occur simultaneously with Hcrt stimulation to block Hcrt-mediated sleep-to-wake transitions.
To further investigate whether Hcrt signaling to LC neurons is necessary for Hcrt-mediated sleep-to-wake transitions, we repeated photostimulation experiments in Hcrt::ChR2-mCherry and Hcrt::mCherry control animals (15-ms pulses at 1 or 10 Hz for 10 s) 15 min after bilateral injection of a Hcrt receptor antagonist, SB-334867 (27), directly into the LC region. Stimulation of ChR2-mCherry animals at 10 Hz significantly reduced the sleep-to-wake latency compared with that in animals stimulated at 1 Hz or in mCherry control animals (P < 0.0001, n = 4 animals per condition), but this effect was blocked following microinjection of 100 μM SB-334867 (Fig. 3), demonstrating the necessity of Hcrt signaling in the LC region in eliciting sleep-to-wake transitions.
Fig. 3.

Pharmacological antagonism of Hcrt signaling in the LC region blocks Hcrt-mediated sleep-to-wake transitions. Photostimulation of Hcrt neurons at 10 Hz causes a significant decrease in sleep-to-wake latency between animals expressing ChR2-mCherry and control animals expressing mCherry; these effects are blocked by local injection of the Hcrt antagonist SB-334867 directly into the LC field. ***P < 0.0001, ANOVA between genotype and stimulation frequency, followed by Tukey’s posthoc test.
Taken together, these optogenetic and pharmacological experiments demonstrate that Hcrt-mediated NREM sleep-to-wake transitions in the inactive period depend on LC activity and Hcrt signaling to LC neurons.
Optogenetic Excitation of LC Neurons Enhances Hcrt-Mediated Sleep-to-Wake Transitions.
We next asked whether increasing the membrane excitability in LC neurons would enhance the effect of Hcrt stimulation on reducing the sleep-to-wake latency. To increase the excitability of LC neurons, we transduced the LC with a mutated form of ChR2, ChR2(C128S), a “step-function” opsin (SFO) that can convert a single, brief pulse of blue light into a stable step in membrane potential (28). The channel can be closed with a single pulse of yellow light. This SFO allows for the possibility of using a single light pulse to stimulate transduced neurons, but not necessarily so that the neuron reaches a threshold potential.
To test the functional expression of ChR2(C128S)-eYFP in LC neurons, we recorded from eYFP-positive neurons in acute brainstem slices using the whole-cell patch-clamp technique. In voltage-clamp mode, we found that transduced neurons exhibited inward photocurrents upon a single 10-ms pulse of blue light (Fig. 4A), with a peak level of 47 ± 4.2 pA (mean ± SEM; n = 6 cells). A single 10-ms pulse of yellow light immediately closed the channel (Fig. 4B). In current-clamp experiments, a single 10-ms pulse of blue light caused depolarizations of 4.7 ± 1.1 mV (mean ± SEM; n = 5 cells) that could be returned to baseline with a single 10-ms pulse of yellow light (Fig. 4C). To investigate the effects of light-stimulation duration on LC firing, we used steady depolarizing intracellular current injection to bring the membrane potential near the action potential threshold (∼45 mV) and stimulated neurons with single pulses of blue light of different durations. We found that increasing the duration of the light pulse from 1 ms to 5–10 ms significantly increased the number of action potentials (n = 5) (Fig. 4 D and E), demonstrating that increasing pulse duration increases the membrane excitability of LC neurons.
Fig. 4.

Functional expression of ChR2(C128S)-eYFP in LC neurons. (A) Voltage-clamp recording of an LC neuron expressing ChR2(C128S)-eYFP in an acute brainstem slice showing inward current in response to a single 10-ms pulse of blue light. (B) Voltage-clamp recording of a neuron showing inward current in response to a single 10-ms pulse of blue light and a return to baseline in response to a single 10-ms pulse of yellow light. (C) Current-clamp recording of a neuron showing a depolarization in response to a single 10-ms pulse of blue light and a return to baseline in response to a single 10-ms pulse of yellow light. (D) Action potential trains generated in current clamp in response to single 1-, 5-, and 10-ms pulses of blue light. (E) Quantification of the fold-increase number of spikes relative to 1-ms pulses of light. Data represent mean ± SD fold increase from n = 5 neurons. **P < 0.001, Student’s t test between conditions. (F) Probability of a sleep-to-wake transition within 30 s of stimulation with single pulses of light of different durations between ChR2(C128S)-eYFP and eYFP control animals. Data represent the mean ± SEM probability values for n = 6 mice, 10 trials per mouse. ***P < 0.0001, Student’s t test between transduced mice.
To test the behavioral effects of stimulating ChR2(C128S)-eYFP–transduced LC neurons in vivo, we stimulated animals with single pulses of light at different pulse durations and determined the probability of causing a sleep-to-wake event within 30 s of stimulation. There was no difference in the probability of awakening between animals in which the LC was transduced with ChR2(C128S)-eYFP and eYFP following a single light pulse of 1, 5, or 10 ms in duration (Fig. 4F). However, there was a significant increase (P < 0.0001) in the probability of awakening for pulses of 15, 20, and 25 ms. Therefore, single pulses of blue light increase the probability of sleep-to-wake transitions in ChR2(C128S)-eYFP transduced animals at pulse widths of 15 ms and above.
To test the effects of stimulating LC neurons during Hcrt-mediated sleep-to-wake transitions, we decided to stimulate ChR2(C128S)-eYFP–transduced LC neurons with a single 10-ms pulse, the longest pulse duration for which we observed no increase in the probability of sleep-to-wake transitions. We performed surgeries in which Hcrt neurons were transduced with either ChR2-mCherry or mCherry, and LC neurons were transduced with either ChR2(C128S)-eYFP or eYFP (Fig. 5A).
Fig. 5.

Stimulation of LC neurons with ChR2(C128S)-eYFP enhances Hcrt-mediated sleep-to-wake transitions. (A) Diagram depicting experimental conditions. Hcrt neurons were transduced with either ChR2-mCherry or mCherry and LC neurons were transduced with either ChR2(C128S)-eYFP or eYFP. (B–E) (Upper) Experimental stimulation paradigms. (Lower) Latencies to wakefulness during NREM sleep following photostimulation in Hcrt neurons and simultaneous photostimulation in LC neurons. Data represent the mean ± SEM sleep-to-wake latencies (15 trials per mouse per condition, six mice per condition). In B, LC stimulation precedes Hcrt stimulation by 5 s. In C, LC stimulation precedes Hcrt stimulation but is followed with a 10-ms pulse of yellow light before Hcrt stimulation. In D, LC stimulation occurs at the offset of Hcrt stimulation. In E, LC stimulation precedes 1-Hz Hcrt stimulation by 5 s. *P < 0.05, **P < 0.001, Student’s t test between ChR2(C128S)-eYFP and eYFP conditions.
As in previous experiments, we stimulated Hcrt neurons starting 15 s after the onset of NREM sleep with 15-ms pulses at 10 Hz for 10 s. Importantly, stimulating LC neurons with a single 10-ms pulse of blue light did not reduce the sleep-to-wake latency in Hcrt::mCherry animals (P > 0.05), demonstrating that this method of LC stimulation, by itself, has no effect on sleep-to-wake transitions (Fig. 5B). However, stimulating LC neurons with a single 10-ms pulse 5 s before stimulating ChR2-transduced Hcrt neurons caused a significant decrease in sleep-to-wake latency for ChR2(C128S)-eYFP animals compared with eYFP control animals (P < 0.05, n = 6 mice per condition, 15 trials per mouse) regardless of whether the stimulation was bilateral, ipsilateral, or contralateral (Fig. 5B and Movie S1). This effect was blocked when LC stimulation with a blue-light pulse was immediately followed with a single pulse of yellow light before Hcrt stimulation (P > 0.05, n = 6 mice per condition) (Fig. 5C). We also did not observe any significant difference in sleep-to-wake latencies in ChR2(C128S)-eYFP animals compared with eYFP control animals when the 10-ms pulse was delivered after Hcrt stimulation (P > 0.05, n = 6 mice per condition) (Fig. 5D). Finally, we also asked whether stimulating the LC with ChR2(C128S)-eYFP caused Hcrt-mediated sleep-to-wake transitions when Hcrt neurons were stimulated at 1 Hz, a frequency previously shown to be ineffective in decreasing the mean sleep-to-wake latency (23). We found no significant difference between ChR2(C128S)-eYFP and eYFP animals when Hcrt neurons were stimulated at 1 Hz (P > 0.05, n = 6 mice per condition), demonstrating that 1 Hz remains insufficient to affect sleep-to-wake transitions even during LC stimulation (Fig. 5E). Taken together, these experiments demonstrate that priming the LC with a single pulse of blue-light stimulation facilitates the effect of Hcrt stimulation at 10 Hz, but only if the pulse immediately precedes Hcrt stimulation.
Computational Model of Hcrt-LC Circuitry.
To better understand how Hcrt stimulation may affect LC neurons and ultimately sleep/wake behavior, we constructed a conductance-based model of Hcrt-LC circuitry on the basis of previous electrophysiological recordings. We reproduced the behavior of Hcrt neurons (29) under various ChR2-stimulation paradigms (30). Response properties of LC neurons are based on a model similar to those of Hcrt neurons but without endogenous activity and rebound potentials endowed by a low-threshold calcium current. We modeled Hcrt activity on LC neurons with AMPA synapses and slowly reacting Hcrt receptors on LC neurons (18, 31).
Our model describes the effects of Hcrt stimulation on LC neurons, with and without stimulation of ChR2(C128S)-eYFP–transduced LC neurons. During sleep, LC neurons are relatively silent (32–34). Our model suggests that stimulation of LC neurons transduced with ChR2(C128S)-eYFP with a single 10-ms pulse of blue light causes an increase in spike frequency of ∼0.5–0.6 Hz (Fig. 6A), a frequency previously shown to be inadequate to cause sleep-to-wake transitions (24). Stimulation of Hcrt neurons elevates the membrane potential of LC neurons with a delay of several seconds (Fig. 6B and Movie S2), mimicking the slow dynamics of calcium mobilization upon binding of Hcrt ligand to the receptor. Our model suggests that the AMPA component of the membrane conductance of LC neurons may not be sufficient to induce depolarization because the LC neurons are more responsive to AMPA current when they remain closer to the activation threshold. When LC neurons are stimulated with a single light pulse of 10 ms before Hcrt stimulation with ChR2, there is acceleration in the depolarization of LC neurons compared with the effect of Hcrt stimulation alone (Fig. 6C). This acceleration causes an increase in LC spike frequency that would be sufficient to cause immediate sleep-to-wake transitions (24), as observed in our behavioral experiments (Fig. 5B). Furthermore, our model demonstrates that Hcrt stimulation at 1 Hz, with or without LC stimulation with ChR2(C128S), is insufficient to increase LC firing to a degree necessary to cause sleep-to-wake transitions.
Fig. 6.

Conductance-based model of Hcrt-LC circuitry. (A) Model of the effect of a single pulse of blue light (1, 5, or 10 ms) in ChR2(C128S)-eYFP-transduced LC neurons. (B) Model of the effect of Hcrt stimulation on LC neurons. We executed our model with a paradigm of 10 s prestimulation baseline activity followed with 10 s ChR2-mediated stimulation of Hcrt neurons at 10 Hz. We then let the model run an additional 70 s to monitor poststimulation effects. (Top) Spike trains in Hcrt neurons throughout a run of the model. (Middle) Corresponding spike trains in LC neurons. (Bottom) Average number of spikes in LC neurons per 10-s bins during the run of the model. Dashed green line corresponds to the threshold of LC activity previously shown (24) to be sufficient to cause a sleep-to-wake transition. See also Movie S2. (C) Same as B but with an additional 10-ms blue-light pulse in ChR2(C128S)-eYFP–transduced LC neurons occurring 5 s before the onset of Hcrt stimulation.
Discussion
This study examined the effects of modulating LC activity on Hcrt-mediated NREM sleep-to-wake transitions during the inactive period (Movie S1). We found that photoinhibition of the LC blocked the effects of Hcrt stimulation on wakefulness (Fig. 2) whereas photostimulation of the LC intensified the effects (Fig. 5). These results build upon previous in vivo electrophysiological experiments demonstrating that Hcrts depolarize LC neurons (12, 18, 31) and increase LC discharge rates (12, 17, 19–21). We conclude that the LC gates Hcrt-mediated sleep-to-wake transitions during NREM sleep and serves as a necessary downstream effector for Hcrt function in promoting arousal.
Our results are consistent with the known role of the LC-norepinephrine system in desynchronizing cortical activity and promoting states of wakefulness (15, 16). Central administration of norepinephrine into the ventricles or forebrain desynchronizes the cortical EEG and promotes wakefulness (35), and central injections of pharmacological antagonists of noradrenergic receptors (36) or agonists of inhibitory autoreceptors (37) have substantial sedative effects. Recently, the LC-norepinephrine system was shown to be critical for maintaining the increased membrane potential of cortical neurons in awake compared with sleep states (38). These studies suggest that the LC-norepinephrine system has a crucial role in desynchronizing cortical activity and increasing cortical excitability during wakefulness. Because Hcrt neurons lie upstream of LC-norepinephrine circuitry, our current data suggest that Hcrts may facilitate sleep-to-wake transitions by increasing the probability of LC-norepinephrine discharge in the cortex and other brain regions.
An experiment that would strengthen our conclusions that the LC is sufficient to mediate Hcrt-mediated sleep-to-wake transitions would be local photostimulation of ChR2-transduced Hcrt terminals in the LC region. However, a technical limitation prevents us from performing this experiment in a way that yields meaningful conclusions: The endogenous Hcrt promoter is not efficient enough to drive expression of optogenetic transgenes in Hcrt terminals. We have three lines of evidence to back up our assertions: First, we can see mCherry fluorescence in Hcrt neurons transduced with Hcrt::ChR2-mCherry, but never see mCherry fluorescence in projections (including the LC region). Second, we tried stimulating the projections of Hcrt neurons transduced with Hcrt::ChR2-mCherry in acute brain slices containing the LC and measuring the responses of LC neurons, using whole-cell patch clamp. We never found any effect on LC depolarization due to stimulation of ChR2 fibers, even though the brain slices were taken from animals that positively responded to ChR2 stimulation in the Hcrt neurons themselves (i.e., stimulation decreased the sleep-to-wake latency in vivo) and even though we detected mCherry fluorescence in the Hcrt cell bodies. Finally, we tried stimulating the projections of Hcrt neurons in the LC region in vivo and measuring the induction of c-Fos. We found no statistically significant effect of stimulating Hcrt terminals on c-Fos expression in the LC. This result is in stark contrast to stimulating Hcrt neurons themselves (Fig. 1D) and to previous studies demonstrating that adding Hcrt peptides into the LC region causes a strong induction of c-Fos (19). Taken together, we believe that these experiments demonstrate that stimulating the fiber projections of Hcrt neurons transduced with Hcrt::ChR2-mCherry is insufficient to cause excitatory discharge from the terminals due to the relatively weak expression of ChR2 driven by the Hcrt promoter. Indeed, in all published optogenetic studies that stimulate fiber projections, a relatively strong promoter was used to drive transgene expression, such as the EF1α promoter.
To gain insight into the effects of Hcrt stimulation on LC membrane potential, we built a minimal realistic model of Hcrt-LC circuitry, using previous biophysical studies (18, 29, 31). Our model focuses on the effect of Hcrt activity on downstream LC neurons, showing that Hcrt stimulation slowly (over several seconds) increases the membrane potential of LC neurons from baseline (<1 Hz) to tonic discharge (1–3 Hz) (Fig. 6B). These patterns of tonic discharge are consistent with our previous behavioral results showing that stimulation of LC neurons at these frequencies was sufficient to increase the probability of wakefulness (24). Our model also shows that increasing the excitability of LC neurons with ChR2(C128S)-eYFP during Hcrt stimulation is sufficient to increase the probability of sleep-to-wake transitions. A 10-ms light pulse without Hcrt stimulation presumably increases LC membrane potential, but not to an extent that causes the tonic discharge rates necessary to cause awakenings. However, a 10-ms light pulse combined with subsequent Hcrt stimulation can increase LC tonic discharge in a shorter duration compared with Hcrt stimulation alone (Fig. 6C), increasing the probability of LC discharge leading to a sleep-to-wake transition.
Importantly, our mathematical model recapitulates our behavioral results using only two compartments with Hcrt neurons serving as regulators and LC neurons serving as effectors. On the basis of this model, the LC provides enough excitatory drive to the neocortex such that other arousal systems such as the histaminergic and cholinergic systems are not required to predict reliable sleep-to-wake transitions, although each of these structures may encode different components of arousal. This result is consistent with our previous results showing that Hcrt-mediated sleep-to-wake transitions occur in the absence of histaminergic signaling (25).
Our results also inform the functional anatomy between Hcrt and LC neurons. For example, we found that ipsilateral or contralateral inhibition of LC neurons did not block the effects of unilateral Hcrt stimulation. This may be due to the fact that efferent projections from Hcrt neurons typically project both ipsilaterally and contralaterally to downstream brain regions (39). Therefore, inhibiting the LC only on the ipsilateral or the contralateral side would still allow Hcrt neurons to excite the noninhibited LC. The divergent projections from Hcrt neurons to both hemispheres also potentially explain why unilateral stimulation of Hcrt neurons increases c-Fos expression in both the ipsilateral and the contralateral LC and why stimulating the LC with ChR2(C128S) unilaterally on either side enhances unilateral stimulation of Hcrt neurons. We therefore conclude that the LC gates Hcrt-mediated promotion of wakefulness, even if the phasic discharge of Hcrt neurons occurs only unilaterally.
If the LC is an important regulator of Hcrt-mediated sleep-to-wake transitions, then what is the role of the diffuse Hcrt projections throughout the brain? The fact that Hcrt dysfunction leads to narcolepsy indicates that there are no alternative or redundant neuronal circuits that could compensate its function. However, the neuronal effector systems are likely redundant and activated by distinct sets of inputs. Therefore, we cannot rule out that blocking other arousal systems, such as the central histaminergic and cholinergic systems, would also severely affect Hcrt-induced behavioral state transitions. It is also important to note that Hcrt neurons have been shown to play a role in other behaviors, such as feeding (4) and stress (40). This study specifically focused on the effects of Hcrt stimulation on acute sleep-to-wake transitions. Therefore, it is possible that LC activity is not required for other Hcrt-mediated behaviors. For example, Hcrt regulation of stress and anxiety may depend on other downstream regions, such as the corticotropin-releasing factor (CRF)-containing cells in the paraventricular hypothalamus (40). The effect of Hcrt activity on downstream nuclei is likely to be context specific and determined by specific environmental stimuli and the homeostatic states of the animal.
Taken together, our analysis of Hcrt-LC circuitry suggests a framework for understanding how brain arousal systems regulate the probability of sleep-to-wake transitions. According to global models of sleep/wake regulation, a balance of activity in arousal-promoting systems and sleep-promoting systems in the brainstem, hypothalamus, and basal forebrain regulates the arousal state of an animal (1, 2). During sleep, the membrane potential and degree of neuronal activity in each of these populations presumably determine the probability of a sleep-to-wake event (41). In our study, we modulated the activity of LC neurons to mimic relative periods of hyperpolarization or depolarization in these neurons, a manipulation that by itself had no effect on sleep-to-wake transitions. However, manipulating LC neurons had a profound effect on Hcrt-mediated sleep-to-wake transitions, demonstrating that these systems functionally interact on the basis of distinct biophysical properties and membrane dynamics. Future studies should investigate the activity and modulation of other arousal populations, both in isolation and in combination as performed here, with the ultimate goal of building a conductance-based model of brain sleep/wake circuitry that incorporates the connectivity and dynamics of several brain systems.
Methods
Animals.
Tyrosine hydroxylase (TH)::IRES-Cre knock-in mice (EM:00254) were obtained from the European Mouse Mutant Archive and mated with c57Bl/6 wild-type mice. We used only male heterozygous mice, aged 8–10 wk at the start of experimental procedures and no more than 18 wk at the end of experimental procedures. Mice were housed in individual Plexiglas recording chambers in custom-designed stainless-steel cabinets at constant temperature (22 ± 1 °C), humidity (40–60%), and circadian cycle (12-h light–dark cycle, starting at 9:00 AM). Food and water were available ad libitum. All experiments were performed in accordance with the guidelines described in the US National Institutes of Health Guide for the Care and Use of Laboratory Animals (42) and were approved by the Institutional Animal Care and Use Committee of Stanford University.
Virus Preparation.
Optogenetic transgenes were delivered using both lentiviral (for targeting of Hcrt neurons) and AAV (for targeting of LC neurons) delivery systems. Lentiviral vectors contained either Hcrt::ChR2-mCherry or Hcrt::mCherry sequences and were prepared as previously described (23). Cre-inducible recombinant AAV vectors carrying optogenetic transgenes were serotyped with AAV5 coat proteins and packaged by the viral vector core at the University of North Carolina. The final viral concentration was 2 × 1012 genome copies (gc) per milliliter. Aliquots of virus were stored at –80 °C before stereotaxic injection.
Surgery.
At the start of surgical procedures, mice were anesthetized with ketamine/xylazine anesthesia (80 and 16 mg/kg, i.p., respectively) and placed on a small animal stereotaxic frame (David Kopf Instruments). All stereotaxic injections were performed on the basis of the coordinates of Paxinos and Franklin (43). Recombinant AAV5 carrying Ef1α::eNpHR(3.0)-eYFP, Ef1α::ChR2(C128S)-eYFP, or Ef1α::eYFP was bilaterally injected adjacent to the LC [anteroposterior (AP), –5.45 mm; mediolateral (ML), ±1.28 mm; dorsoventral (DV), 3.65 mm] through an internal cannula (Plastics One) at a rate of 0.1 μL/min for 10 min (1 μL total volume). Next, lentivirus carrying either Hcrt::ChR2-mCherry or control Hcrt::mCherry was injected unilaterally above the lateral hypothalamus (AP, –1.5 mm; ML, ± 0.75 mm; DV, 4.5 mm) at a rate of 0.1 μL/min for 10 min (1 μL total volume).
Following viral injection, animals received surgical implantation of two cannulae (Plastics One). A 22-G bilateral cannula with 2 mm between individual cannulae and 3 mm shaft length was placed above the LC region (AP, 5.45 mm; ML, ± 1.0 mm; DV, 3.0 mm). A 22-G bilateral cannula with 1.5 mm between individual cannulae and 4.5 mm shaft length was placed above the Hcrt field (AP, –1.5 mm; ML, ± 0.75 mm; DV, 4.5 mm). Both cannulae were affixed to the skull with C&B Metabond (Parkell) and dental acrylic. Finally, animals were also implanted with a custom-made EEG/EMG implant placed anterior to the cannulae. EEG signals were recorded from electrodes placed on the frontal (AP, –2 mm; ML, ± 2.5 mm) cortices. EMG signals were recorded from two electrodes inserted in the neck musculature to record postural tone.
After surgical procedures, animals were allowed to recover in individual housing for at least 2 wk. Animals used for sleep recordings were then acclimated to a flexible EEG/EMG connection cable for an additional 7 d within individual recording chambers. Each cable was flexible so that mice could freely move about their cages. Fiber-optic cables were implanted at least 2 d before experiments and ran alongside the EEG/EMG connection cables.
Polysomnographic Recordings and Analysis.
All sleep recordings took place between 12:00 PM (noon) and 6:00 PM (light onset at 9:00 AM). EEG and EMG signals derived from the surgically implanted electrodes were amplified (Grass Instruments) and digitized at 256 Hz, using sleep-recording software (Vital Recorder; Kissei Comtec America). The signals were digitally filtered and spectrally analyzed by fast Fourier transformation, and polysomnographic recordings were scored using sleep analysis software (SleepSign for Animal; Kissei Comtec America). All scoring was performed manually on the basis of the visual signature of the EEG and EMG waveforms, as well as the power spectra of 5-s epochs.
We defined wakefulness as desynchronized low-amplitude EEG and heightened tonic EMG activity with phasic bursts. We defined NREM sleep as synchronized, high-amplitude, low-frequency (0.5–4 Hz) EEG with highly reduced EMG activity compared with wakefulness and no phasic bursts. We defined REM sleep as having a pronounced theta rhythm (4–9 Hz) and flat EMG. All sleep scoring was done by an investigator (M.E.C.) blind to the viral transgenes delivered to the animals.
Photostimulation and Photoinhibition.
All Hcrt photostimulation experiments were conducted unilaterally on the left Hcrt field. Fiber-optic cables (2 m long, 200 μm diameter; ThorLabs) were placed inside the implanted cannulae 2 d before stimulation/inhibition experiments. For photostimulation experiments, light pulse trains (15-ms pulses for 10 s at either 1 or 10 Hz) were programmed using a waveform generator (33220A Waveform Generator; Agilent Technologies) that provided input to a blue-light laser (473 nm; LaserGlow). Each stimulation epoch was applied 15 s after the onset of NREM sleep as detected by real-time online EEG/EMG analysis.
LC photostimulation or photoinhibition experiments were conducted unilaterally or bilaterally as described in Results. For photoinhibition experiments, light delivery was provided by a yellow-light laser (593.5 nm; LaserGlow) split into two fiber-optic cables. For photostimulation experiments, single light pulses (10 ms) were programmed using a Master-8 pulse stimulator (A.M.P.I.) that provided simultaneous input into two blue-light lasers. For experiments using ChR2(C128S), each trial was followed by a 15-ms pulse of yellow light before the start of future trials to ensure inactivation of the channel.
Pharmacology.
Hcrt antagonist SB-334867 (100 μM; Tocris) was dissolved in artificial cerebrospinal fluid consisting of 112 mM NaCl, 3.1 mM KCl, 1.2 mM MgSO4, 0.4 mM NaH2PO4, and 25 mM NaHCO3. Aliquots were stored at –20 °C before use. Microinfusions were performed bilaterally using a minipump (Harvard Apparatus; 70-2212) at a rate of 0.1 μL/min for 3 min (0.3 μL total volume) in freely moving animals 15 min before behavioral experiments. Injection coordinates were the same as those used to deliver AAV5 into the LC region.
Electrophysiology.
Acute brainstem coronal slices (250 μm) were collected on a vibratome. Transduced LC neurons were identified by eYFP fluorescence and recorded with whole-cell methods. Signals were amplified with a Multiclamp 700A amplifier, sampled at 10 kHz, and acquired using a Digidata 1320A digitizer (all Molecular Devices). Slices were maintained at 32 °C in the recording chamber and constantly superfused with oxygenated artificial cerebrospinal fluid (ACSF) (126 mM NaCl, 26 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH3PO4, 2 mM CaCl2, 2 mM MgCl2, 10 mM glucose, equilibrated with 95% (vol/vol) O2/5% CO2, pH 7.4) at a rate of 2 mL/min. The recording pipette contained 120 mM K-gluconate, 11 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM Hepes, 10 mM EGTA, pH 7.3, adjusted with KOH. Optogenetic stimulation was performed using a xenon arc lamp (Oligochrome; TILL Photonics) with a 455 ± 35-nm excitation filter for blue illumination (Chroma; D455/70x) and a 593 ± 25-nm excitation filter for yellow illumination (Semrock; FF01-593/40-25). Light was directed onto the slice through the microscope objectives for full-field illumination. To ensure maximal inactivation of the step-function opsin, all experiments were performed in the dark and we provided yellow inactivating pulses (10 ms) before each trial.
Histology.
After completion of experiments, mice were anesthetized with ketamine and xylazine anesthesia (80 and 16 mg/kg, respectively, i.p. injection) and perfused transcardially with PBS, pH 7.4, followed by 4% paraformaldehyde in PBS. The brains were extracted, allowed to postfix overnight in the same fixative at 4 °C, and cryoprotected in 30% sucrose dissolved in PBS for an additional 24 h at 4 °C. Each brain was sectioned at 30 μm on a cryostat (Leica Microsystems) and collected in cold PBS. Immunohistochemical procedures were performed using standard methods (described in SI Methods).
Statistics.
All data were analyzed using Prism 5.0 (GraphPad Software) as described in the text. Data were exported into Adobe Illustrator CS5 (Adobe Systems) for preparation of figures.
Computational Modeling.
Hcrt model neuron.
We built a conductance-based model using two compartments (44, 45). The model is publicly available for download from ModelDB at: https://senselab.med.yale.edu/ModelDB/showmodel.asp?model=145162. Both compartments are given by the following set of ordinary differential equations,
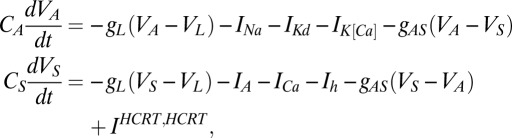 |
with 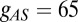 nS,
nS, 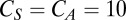 μF,
μF, 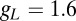 nS, and
nS, and  mV. The synaptic currents come from the neighboring Hcrt cells. The
mV. The synaptic currents come from the neighboring Hcrt cells. The 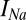 current is based on Traub and Miles (46); i.e.,
current is based on Traub and Miles (46); i.e., 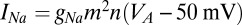 ,
,
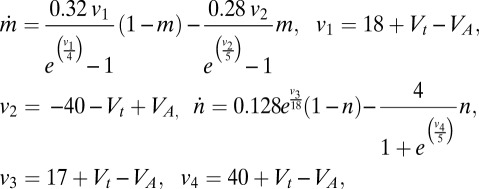 |
where 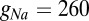 nS and
nS and 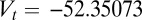 mV to replicate firing rates. The delayed rectifier potassium current is
mV to replicate firing rates. The delayed rectifier potassium current is 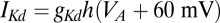 with
with 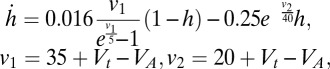 and
and 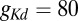 nS. The potassium dependent on calcium is
nS. The potassium dependent on calcium is 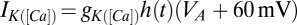 with
with 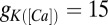 nS,
nS, 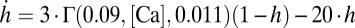 , and
, and 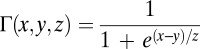 , which is used in the rest of the equations. The calcium dynamics are computed as
, which is used in the rest of the equations. The calcium dynamics are computed as 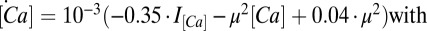
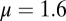 . The low-threshold current is
. The low-threshold current is 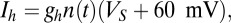 where
where 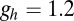 nS and
nS and
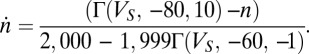 |
The calcium current is modeled by the Goldman–Hodgkin–Katz (GHK) formalism (47) as 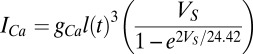 with
with 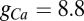 nS. The time evolution of the activation variable is
nS. The time evolution of the activation variable is 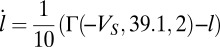 . The last current is
. The last current is 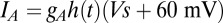 with
with 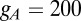 nS and
nS and 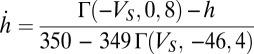 .
.
LC model neuron.
We also used two compartments to model the LC. The dynamics of both compartments are governed by
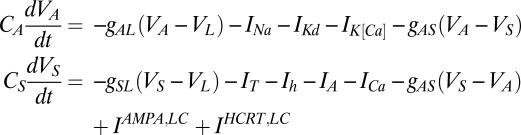 |
with 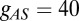 nS,
nS, 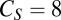 μF,
μF, 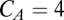 μF,
μF, 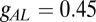 nS,
nS, 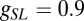 nS, and a resting value of
nS, and a resting value of 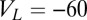 mV. The synaptic currents from the Hcrt neurons and LC neurons,
mV. The synaptic currents from the Hcrt neurons and LC neurons, 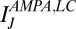 and
and 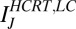 , have units of picoamperes. As in the Hcrt neuron, the
, have units of picoamperes. As in the Hcrt neuron, the 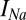 and
and 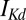 currents are modeled following ref. 46 but using
currents are modeled following ref. 46 but using 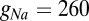 nS,
nS,  nS, and
nS, and 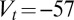 mV. The Goldman–Hodgkin–Katz calcium current is again used but with
mV. The Goldman–Hodgkin–Katz calcium current is again used but with 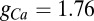 nS and the dynamics of the activation variable are written as
nS and the dynamics of the activation variable are written as 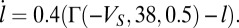 The calcium dynamics are modeled with
The calcium dynamics are modeled with 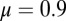 . The form of the
. The form of the 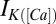 remains unchanged with a maximal conductance,
remains unchanged with a maximal conductance, 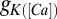 , of 40 nS. The timescale of the activation variable for this current is modified with respect to the rest as
, of 40 nS. The timescale of the activation variable for this current is modified with respect to the rest as
The 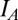 current has the same form as in both previous models with a high
current has the same form as in both previous models with a high 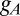 of 40 nS with activation as
of 40 nS with activation as
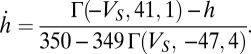 |
The low-threshold 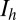 is exactly the same as for the Hcrt neurons, whereas the low-threshold calcium current,
is exactly the same as for the Hcrt neurons, whereas the low-threshold calcium current, 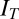 , is responsible for the after depolarization (48). The current is expressed using GHK formalism, because it is a calcium current, using
, is responsible for the after depolarization (48). The current is expressed using GHK formalism, because it is a calcium current, using 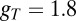 nS as
nS as 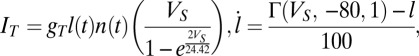 and
and 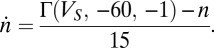
Network connections.
We model AMPA synaptic currents (48) as the release of the neurotransmitter in the form 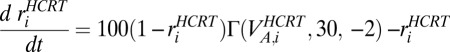 , where the AMPA current into the LC neurons is modeled as
, where the AMPA current into the LC neurons is modeled as 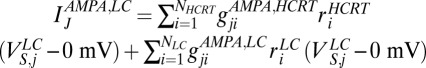 .
. 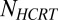 is the number of Hcrt neurons and
is the number of Hcrt neurons and 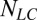 is the number of LC neurons. The release of Hcrt leads to a depolarization of the target cell (49). Then the release of neurotransmitter is represented by a first-order kinetic equation as
is the number of LC neurons. The release of Hcrt leads to a depolarization of the target cell (49). Then the release of neurotransmitter is represented by a first-order kinetic equation as 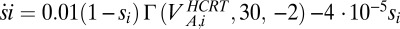 . Because it is unclear which ions are involved in the membrane exchange, we do not use reverse potential at this point. Thus,
. Because it is unclear which ions are involved in the membrane exchange, we do not use reverse potential at this point. Thus, 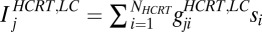 . There are AMPA connections from the Hcrt neurons to the LC neurons and from the LC neurons to the LC neurons themselves. The Hcrt neurons release the neurotransmitter globally and the strength,
. There are AMPA connections from the Hcrt neurons to the LC neurons and from the LC neurons to the LC neurons themselves. The Hcrt neurons release the neurotransmitter globally and the strength, 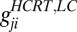 , is drawn from a uniform distribution from
, is drawn from a uniform distribution from 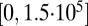 pA. The same applies for the synaptic conductances
pA. The same applies for the synaptic conductances 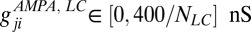 ,
, 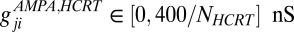 , and
, and 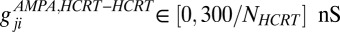 but in this case there is a 50% probability of making an AMPA connection from the Hcrt neurons to the LC neurons, a 25% probability of making connections between the LC neurons, and a 20% connection probability between the Hcrt neurons. Note that the strength of the connections is scaled by the number of neurons that send the afferents. The ChR2 depolarization is modeled by only two states (open and closed) because we are analyzing the slow dynamics of the system (30). Therefore, ChR2 is modeled using
but in this case there is a 50% probability of making an AMPA connection from the Hcrt neurons to the LC neurons, a 25% probability of making connections between the LC neurons, and a 20% connection probability between the Hcrt neurons. Note that the strength of the connections is scaled by the number of neurons that send the afferents. The ChR2 depolarization is modeled by only two states (open and closed) because we are analyzing the slow dynamics of the system (30). Therefore, ChR2 is modeled using 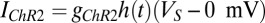 , where the reverse potential is set to 0 mV as suggested by Talathi et al. (50),
, where the reverse potential is set to 0 mV as suggested by Talathi et al. (50), 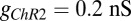 , and
, and 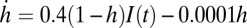 , where
, where 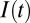 is the light stimulation that takes 0 values when there is no light and 1 when blue light is present. Pulses of 10-ms duration during 5 s are introduced on the Hcrt neurons. The ChR2(C128S) channel on the LC neurons is similarly modeled using
is the light stimulation that takes 0 values when there is no light and 1 when blue light is present. Pulses of 10-ms duration during 5 s are introduced on the Hcrt neurons. The ChR2(C128S) channel on the LC neurons is similarly modeled using 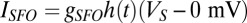 , with
, with 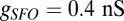 and
and  .
.
Supplementary Material
Acknowledgments
We thank Karl Deisseroth for optogenetic reagents and Ofer Yizhar for technical advice. We also thank Antoine Adamantidis, Alison Cross, Simal Ozen, Clara Tourino, and Kelly Zalocusky for critical feedback in the preparation of this manuscript. M.E.C. was supported by a National Science Foundation Graduate Research Fellowship and by National Institutes of Health National Research Service Award F31MH83439. M.E.C. and P.B. are both supported by postdoctoral fellowships from the Hilda and Preston Davis Foundation. J.B. is supported by a postdoctoral fellowship from the American Epilepsy Society. J.R.H. is supported by National Institute of Neurological Disorders and Stroke Grant NS12151. R.H. is supported by National Institute on Deafness and Other Communication Disorders Grant R01DC11422 and Office of Naval Research Grant N00014-07-01. L.d.L. is supported by National Institutes of Health Grants MH83702, MH87592, and DA21880 and the Klarman Family Foundation.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
See Author Summary on page 15551 (volume 109, number 39).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1202526109/-/DCSupplemental.
References
- 1.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 2.Pace-Schott EF, Hobson JA. The neurobiology of sleep: Genetics, cellular physiology and subcortical networks. Nat Rev Neurosci. 2002;3:591–605. doi: 10.1038/nrn895. [DOI] [PubMed] [Google Scholar]
- 3.de Lecea L, et al. The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sakurai T, et al. Orexins and orexin receptors: A family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 5.Sutcliffe JG, de Lecea L. The hypocretins: Setting the arousal threshold. Nat Rev Neurosci. 2002;3:339–349. doi: 10.1038/nrn808. [DOI] [PubMed] [Google Scholar]
- 6.Sakurai T. The neural circuit of orexin (hypocretin): Maintaining sleep and wakefulness. Nat Rev Neurosci. 2007;8:171–181. doi: 10.1038/nrn2092. [DOI] [PubMed] [Google Scholar]
- 7.Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–798. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005;25:6716–6720. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355:39–40. doi: 10.1016/S0140-6736(99)05582-8. [DOI] [PubMed] [Google Scholar]
- 10.Lin L, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- 11.Chemelli RM, et al. Narcolepsy in orexin knockout mice: Molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 12.Hagan JJ, et al. Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc Natl Acad Sci USA. 1999;96:10911–10916. doi: 10.1073/pnas.96.19.10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.España RA, Plahn S, Berridge CW. Circadian-dependent and circadian-independent behavioral actions of hypocretin/orexin. Brain Res. 2002;943:224–236. doi: 10.1016/s0006-8993(02)02653-7. [DOI] [PubMed] [Google Scholar]
- 14.Peyron C, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: Adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–450. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- 16.Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: Modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- 17.Horvath TL, et al. Hypocretin (orexin) activation and synaptic innervation of the locus coeruleus noradrenergic system. J Comp Neurol. 1999;415:145–159. [PubMed] [Google Scholar]
- 18.van den Pol AN, et al. Hypocretin (orexin) enhances neuron activity and cell synchrony in developing mouse GFP-expressing locus coeruleus. J Physiol. 2002;541:169–185. doi: 10.1113/jphysiol.2002.017426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bourgin P, et al. Hypocretin-1 modulates rapid eye movement sleep through activation of locus coeruleus neurons. J Neurosci. 2000;20:7760–7765. doi: 10.1523/JNEUROSCI.20-20-07760.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gompf HS, Aston-Jones G. Role of orexin input in the diurnal rhythm of locus coeruleus impulse activity. Brain Res. 2008;1224:43–52. doi: 10.1016/j.brainres.2008.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soffin EM, et al. SB-334867-A antagonises orexin mediated excitation in the locus coeruleus. Neuropharmacology. 2002;42:127–133. doi: 10.1016/s0028-3908(01)00156-3. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, et al. Knockdown of orexin type 1 receptor in rat locus coeruleus increases REM sleep during the dark period. Eur J Neurosci. 2010;32:1528–1536. doi: 10.1111/j.1460-9568.2010.07401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter ME, et al. Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat Neurosci. 2010;13:1526–1533. doi: 10.1038/nn.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carter ME, Adamantidis A, Ohtsu H, Deisseroth K, de Lecea L. Sleep homeostasis modulates hypocretin-mediated sleep-to-wake transitions. J Neurosci. 2009;29:10939–10949. doi: 10.1523/JNEUROSCI.1205-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindeberg J, et al. Transgenic expression of Cre recombinase from the tyrosine hydroxylase locus. Genesis. 2004;40:67–73. doi: 10.1002/gene.20065. [DOI] [PubMed] [Google Scholar]
- 27.Smart D, et al. SB-334867-A: The first selective orexin-1 receptor antagonist. Br J Pharmacol. 2001;132:1179–1182. doi: 10.1038/sj.bjp.0703953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berndt A, Yizhar O, Gunaydin LA, Hegemann P, Deisseroth K. Bi-stable neural state switches. Nat Neurosci. 2009;12:229–234. doi: 10.1038/nn.2247. [DOI] [PubMed] [Google Scholar]
- 29.Eggermann E, et al. The wake-promoting hypocretin-orexin neurons are in an intrinsic state of membrane depolarization. J Neurosci. 2003;23:1557–1562. doi: 10.1523/JNEUROSCI.23-05-01557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nikolic K, et al. Photocycles of channelrhodopsin-2. Photochem Photobiol. 2009;85:400–411. doi: 10.1111/j.1751-1097.2008.00460.x. [DOI] [PubMed] [Google Scholar]
- 31.Murai Y, Akaike T. Orexins cause depolarization via nonselective cationic and K+ channels in isolated locus coeruleus neurons. Neurosci Res. 2005;51:55–65. doi: 10.1016/j.neures.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 32.Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: Reciprocal discharge by two brainstem neuronal groups. Science. 1975;189:55–58. doi: 10.1126/science.1094539. [DOI] [PubMed] [Google Scholar]
- 34.McCarley RW, Hobson JA. Neuronal excitability modulation over the sleep cycle: A structural and mathematical model. Science. 1975;189:58–60. doi: 10.1126/science.1135627. [DOI] [PubMed] [Google Scholar]
- 35.Segal DS, Mandell AJ. Behavioral activation of rats during intraventricular infusion of norepinephrine. Proc Natl Acad Sci USA. 1970;66:289–293. doi: 10.1073/pnas.66.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berridge CW, España RA. Synergistic sedative effects of noradrenergic alpha(1)- and beta-receptor blockade on forebrain electroencephalographic and behavioral indices. Neuroscience. 2000;99:495–505. doi: 10.1016/s0306-4522(00)00215-3. [DOI] [PubMed] [Google Scholar]
- 37.De Sarro GB, Ascioti C, Froio F, Libri V, Nisticò G. Evidence that locus coeruleus is the site where clonidine and drugs acting at alpha 1- and alpha 2-adrenoceptors affect sleep and arousal mechanisms. Br J Pharmacol. 1987;90:675–685. doi: 10.1111/j.1476-5381.1987.tb11220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Constantinople CM, Bruno RM. Effects and mechanisms of wakefulness on local cortical networks. Neuron. 2011;69:1061–1068. doi: 10.1016/j.neuron.2011.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.España RA, Reis KM, Valentino RJ, Berridge CW. Organization of hypocretin/orexin efferents to locus coeruleus and basal forebrain arousal-related structures. J Comp Neurol. 2005;481:160–178. doi: 10.1002/cne.20369. [DOI] [PubMed] [Google Scholar]
- 40.Winsky-Sommerer R, et al. Interaction between the corticotropin-releasing factor system and hypocretins (orexins): A novel circuit mediating stress response. J Neurosci. 2004;24:11439–11448. doi: 10.1523/JNEUROSCI.3459-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Postnova S, Voigt K, Braun HA. A mathematical model of homeostatic regulation of sleep-wake cycles by hypocretin/orexin. J Biol Rhythms. 2009;24:523–535. doi: 10.1177/0748730409346655. [DOI] [PubMed] [Google Scholar]
- 42. Committee on Care and Use of Laboratory Animals (1985) Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD), DHHS Publ No (NIH) 85–23.
- 43.Paxinos G, Franklin KBJ. 2004. The Mouse Brain in Stereotaxic Coordinates (Elsevier Academic, Amsterdam), Compact 2nd Ed.
- 44.Huerta R, Sánchez-Montañés MA, Corbacho F, Sigüenza JA. A central pattern generator to control a pyloric-based system. Biol Cybern. 2000;82:85–94. doi: 10.1007/PL00007963. [DOI] [PubMed] [Google Scholar]
- 45.Szücs A, Huerta R, Rabinovich MI, Selverston AI. Robust microcircuit synchronization by inhibitory connections. Neuron. 2009;61:439–453. doi: 10.1016/j.neuron.2008.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Traub RD, Miles R. Neuronal Networks of the Hippocampus. Cambridge: Cambridge Univ Press; 1991. p. xviii. [Google Scholar]
- 47.Goldman DE. Potential, impedance, and rectification in membranes. J Gen Physiol. 1943;27:37–60. doi: 10.1085/jgp.27.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Cellular and network models for intrathalamic augmenting responses during 10-Hz stimulation. J Neurophysiol. 1998;79:2730–2748. doi: 10.1152/jn.1998.79.5.2730. [DOI] [PubMed] [Google Scholar]
- 49.Yamanaka A, Tabuchi S, Tsunematsu T, Fukazawa Y, Tominaga M. Orexin directly excites orexin neurons through orexin 2 receptor. J Neurosci. 2010;30:12642–12652. doi: 10.1523/JNEUROSCI.2120-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Talathi SS, Carney PR, Khargonekar PP. 2010. Control of neural synchrony using channelrhodopsin-2: A computational study. J Comput Neurosci 31:87–103. [DOI] [PMC free article] [PubMed]
