

Abstract
Water oxidation catalysts are essential components of light-driven water splitting systems, which could convert water to H2 driven by solar radiation (H2O + hν → 1/2O2 + H2). The oxidation of water (H2O → 1/2O2 + 2H+ + 2e-) provides protons and electrons for the production of dihydrogen (2H+ + 2e- → H2), a clean-burning and high-capacity energy carrier. One of the obstacles now is the lack of effective and robust water oxidation catalysts. Aiming at developing robust molecular Ru-bda (H2bda = 2,2′-bipyridine-6,6′-dicarboxylic acid) water oxidation catalysts, we carried out density functional theory studies, correlated the robustness of catalysts against hydration with the highest occupied molecular orbital levels of a set of ligands, and successfully directed the synthesis of robust Ru-bda water oxidation catalysts. A series of mononuclear ruthenium complexes [Ru(bda)L2] (L = pyridazine, pyrimidine, and phthalazine) were subsequently synthesized and shown to effectively catalyze CeIV-driven [CeIV = Ce(NH4)2(NO3)6] water oxidation with high oxygen production rates up to 286 s-1 and high turnover numbers up to 55,400.
Keywords: catalysis, density function theory, seven coordination, photosystem II, solar fuels
In pursuit of sustainable energy systems such as solar fuels, much effort has been spent on water splitting to hydrogen and oxygen since hydrogen is a potential clean energy carrier and water is an abundant and environmentally benign resource (1–5). Water splitting consists of two half reactions: (i) water oxidation {H2O → 1/2O2 + 2H+ + 2e- [E = 1.23–0.059× pH V vs. normal hydrogen electrode (NHE)]} and (ii) proton reduction [2H+ + 2e- → H2 (E = -0.059× pH V vs. NHE)]. In practice, an overpotential is always present, leading to an even higher applied potential. The former half reaction requires strongly oxidizing conditions and is generally considered as the bottleneck of the whole water-splitting process due to the multiple proton–electron transfers and the formation of the O─O bond. Over the last few years, there has been an increasing development of water oxidation catalysts (WOCs) and many transition metal-based catalysts, including Ru (4, 5), Ir (6–8), Co (9–13), Fe (14, 15), and Mn (16–18), have been reported with oxygen production rates (OPRs: mole oxygen produced per mole catalyst per second) ≤ 5 s-1. Very recently, we reported a family of highly active Ru-based WOCs [Ru(bda)L2] (H2L = 2,2 -bipyridine-6,6′-dicarboxylic acid; L = 4-picoline, A; L = isoquinoline, B) (Fig. 1) with OPRs up to 300 s-1 (19, 20); a seven-coordinate dimeric RuIV intermediate (D7RuIV) (Fig. 1) is involved in the O─O bond formation step (20, 21).
Fig. 1.

Structures of complexes A, B, D7RuIV, and 1–3.
A general problem encountered in molecular WOCs is the decomposition of catalysts. Ligand dissociation and oxidative decomposition have been considered as the major deactivation pathways. The groups of Llobet, Meyer, and Sun have demonstrated an improved durability of their catalysts by immobilizing catalysts on the electrode/material surface and thereby dramatically suppressing the intermolecular oxidative decomposition pathway (22–25).
For our Ru-bda catalysts, the main deactivation pathway has been found to be the axial ligand dissociation (19). We believe our Ru-bda catalysts could be more robust if the binding affinity of the axial ligands could be increased. Therefore we carried out density function theory (DFT) calculations to predict which type of pyridyl ligands are more stable against ligand dissociation and thus to instruct us to design more rugged WOCs. Herein, we demonstrate the DFT-directed development of robust Ru-bda catalysts for water oxidation and report three Ru-bda WOCs [Ru(bda)L2] [L = pyridazine (pdz), 1; L = pyrimidine (pmd), 2; L = phthalazine (ptz), 3] (Fig. 1). The best case, complex 3, exhibits an impressively high catalytic activity toward water oxidation using Ce(NH4)2(NO3)6 (CeIV) as oxidant, with a OPR of 286 s-1 and a high turnover number (TON) of more than 55,400 (In this work, we reported the OPR and TON values as average of three runs with STDEV < 10%).
Results and Discussion
Theoretical Calculations.
As mentioned above, the main deactivation pathway of the Ru-bda catalysts is the axial ligand dissociation. For the water oxidation reaction catalyzed by the Ru-bda catalysts, the rate limiting step is the coupling step of 2RuV═O → RuIV─O─O─RuIV, and thus RuV is the dominant species at the catalytic steady state (19, 20). We thereby identified the ligand-exchange reaction at the RuV state, in which one of the ligands (L) is replaced by OH2 (Scheme 1). In addition, this reaction was found to be more favored at the RuV state compared to the RuIV and RuIII states, likely due to the higher hardness of the metal center at the high oxidation state. It is not clear why this hampers reactivity, but one hypothetical path of deactivation is that once another water molecule ligates, the complex is susceptible to further oxidation leading to inactive complexes. Another possibility is that hydrophilic ligands, in the current case an aqua ligand, can decrease the rate of the bimolecular O─O coupling. It was recently shown that large lipophilic groups like isoquinoline enhance the reaction rate (19). Our hypothesis is thus that increased stability and lifetime can be achieved by changing the ligand to one that is more difficult to replace with water.
Scheme 1.

Illustration of the ligand exchange reaction studied in DFT calculations.
One question arises now. What would be the underlying driving force for achieving more robust catalysts? To address this question, we have carried out calculations of the Gibbs free energy of the ligand exchange reactions (hydroxylation) for a number of ligands, including 4-picoline, isoquinoline, pyrimidine, pyrazine, pyridazine, cinnoline, phthalazine, and 4,5-dimethoxypyridazine (Fig. 2, Upper).
Fig. 2.

(Upper) Ligands screened in DFT calculations. The short names are given in the parentheses. (Lower) Gibbs free energy of reaction in pH 0 aqueous solution at 298 K as a function of HOMO energy of ligand in vacuum (in the inset are the HOMO orbital of 4-picoline and the HOMO - 1 orbital of phthalazine calculated with DFT).
After inspecting the electronic structure, structural properties, and pKa of the different systems, we have identified a correlation between the energy of the HOMO of the ligand in the gas phase and the stability of the corresponding Ru-bda complex. Fig. 2 Lower shows the plot of the Gibbs free energy of the reaction in pH 0 aqueous solution (Scheme 1) as a function of the HOMO energy of the dissociating ligand. To get better understanding of the relationship between these two variables we have run ordinary least square (OLS) estimation on the model presented in Eq. 1. It was found that β1 = 285.99 and β0 = 77.96 where both coefficients show statistical significance at 0.1% level and R2 is of 84.79%, indicating a monotonically increasing linear relationship between ΔG0 and HOMO. The estimated curve is shown as the red line in Fig. 2 Lower.
| [1] |
Very useful information obtained from Fig. 2 is that Ru-bda complexes with axial pyridazine, cinnoline, 4,5-dimethoxypyridazine, and phthalazine are significantly more stable against aquation compared to the complexes with axial 4-picoline, pyrimidine, and pyrazine. We expect that complex [Ru(bda)(ptz)2] would be a very robust WOC, which could be synthesized from readily available starting materials.
Synthesis and Characterization of Complexes 1–3.
We then selectively synthesized complexes [Ru(bda)(pdz)2] (1), [Ru(bda)(pmd)2] (2), and [Ru(bda)(ptz)2] (3) and evaluated their catalytic activities toward CeIV-driven water oxidation. Reaction of H2bda and [Ru(dmso)4Cl2] (dmso = dimethyl sulfoxide) in the presence of triethylamine, followed by addition of access axial ligands, gave the corresponding mononuclear RuII complexes 1–3 in moderate yields. All complexes were characterized by 1H NMR, mass spectrometry, elemental analysis, and cyclic voltammetry.
Cyclic voltammograms of complexes 1–3 recorded in the mixed CF3CH2OH/pH 1.0 aqueous solutions are shown in Fig. 3 (the oxidation potential of the solvent CF3CH2OH is approximately 1.85 V vs. NHE). Our previous study of Ru-bda complex A has shown that dissolution of this RuII-bda complex in aqueous solution results in the formation of the corresponding RuII-OH2 species. Its sequential electron-proton transfer upon oxidation at pH 1 conditions is as follows: RuII-OH2 → RuIII-OH2 → RuIV-OH → RuV═O (19). On the basis of the structural similarity between A and 1–3, the redox events at 0.43 V for 3, 0.49 V for 1, and 0.65 V for 2 were assigned to RuIII-OH2/RuII-OH2. Further oxidation of RuIV-OH/RuIII-OH2 was observed at 0.98 V, 0.99 V, and 1.00 V for 3, 1, and 2, respectively. Obviously, the axial ligand has a large influence on the redox potential of RuIII-OH2/RuII-OH2 but little on that of RuIV-OH/RuIII-OH2. The oxidation of RuIV-OH → RuV═O was observed at approximately 1.26 V for complex 2 as shown in the differential pulse voltammograms (Fig. S1), while this process was hardly seen for complexes 1 and 3.
Fig. 3.

Cyclic voltammograms of complexes 1–3 (0.5 mM) in the mixed CF3CH2OH/pH 1.0 (v∶v = 1∶2) aqueous solutions. Working electrode=pyrolytic graphite electroge (basal plane), scan rate = 100 mV s-1.
At E > 1.2 V, catalytic currents for all three complexes were observed, which were caused by the electrochemical oxidation of water (Fig. 3). Meanwhile, there was a reduction peak at ca. -0.40 V at the reverse scan of each complex, corresponding to the reduction of molecular O2 electrochemically generated at E > 1.2 V (Fig. 3). Accordingly, all three complexes are electrochemically active for water oxidation and can be potentially used for the modification of water oxidation electrodes.
CeIV-Driven Water Oxidation.
Longevity.
A first confirmation of our DFT prediction came from the comparison between 4-picoline- and pyridazine-containing complexes. The free energy for the ligand exchange reaction is clearly less favorable for the pyridazine complex 1 compared to the 4-picoline complex A, ΔG = 4.61 kcal/mol and -2.38 kcal/mol, respectively. This indicates that complex 1 has better resistance against the ligand exchange reaction than complex A. In other words, 1 could remain active for longer time. This is consistent with the experimental observations. Water oxidation by complexes 1–3 was evaluated using CeIV as oxidant in acidic aqueous solutions. The oxygen formation vs. time (Figs. 4 and 5) was recorded with a pressure transducer plus a data acquisition module, and the final amount of oxygen generated was calibrated by GC. Under the given catalytic conditions in Fig. 4, complex A deactivated in about 0.30 h while complex 1 lasted for more than 1.3 h; the corresponding TONs for complexes 1 and A were calculated to be 4,563 ± 172 and 935 ± 61, respectively. When complex 2 was evaluated under the same conditions, it deactivated even faster than A and resulted in a TON of 401 ± 30, probably due to the protonation/hydrogen bonding of the uncoordinated N atoms of the pyrimidine ligand of 2 under acidic aqueous conditions, which accelerates the ligand dissociation. When 2 was increased to 2.99 × 10-5 M, a TON of 1,702 ± 107 was obtained.
Fig. 4.

Oxygen evolution curves for complexes A (4 × 10-8 mol), 1 (4 × 10-8 mol), and 2 (4 × 10-8 mol) in acidic aqueous solutions. Conditions: volume of solutions = 3.22 mL, [cat.] = 1.242 × 10-5 M, [CeIV] = 0.365 M.
Fig. 5.

Oxygen evolution curve for complex 3 (5 × 10-9 mol) in acidic aqueous solutions. Conditions: volume of solutions = 3.225 mL, [cat.] = 1.550 × 10-6 M, [CeIV] = 0.365 M.
As we expected, complex 3 with phthalazine as axial ligands is the most robust one. Under the same conditions as used for other three complexes, CeIV was totally consumed in 150 seconds while the catalyst was still active. We thereby decreased the amount of 3 to 5 nmol. This time, the catalytic system lasted for more than three hours and a very large turnover number of 55,419 ± 959 (average of three runs) was achieved (Fig. 5). This large TON number is about 100 times higher than [Ru(terpy)(bpy)(OH2)]2+ (terpy = 2,2′∶6′,2′′-terpyridine; bpy = 2,2′-bipyridine) type complexes (26–28), the Ru-Hbpp [Hbpp = 2,6-bis(pyridyl)pyrazole] WOC (29), and our Ru-pdc (pdc = 2,6-pyridinedicarboxylate) complexes (30), and five times higher than the robust Ir-Cp* (Cp∗ = C5Me5) WOCs (8) and our dinuclear Ru complex (31). The specific lifetime of selected Ru-bda complexes under the given conditions as well as the free energy values of the ligand exchange reaction are collected in Table 1 and plotted in Fig. S2. Higher delta G values correlate longer lifetime, with the exception of complex A.
Table 1.
Longevity of selected Ru-bda catalysts under the given conditions and the calculated ligand exchange free energy of each complex
| complex | A | B | 2 | 1 | 3 |
| ΔG (kcal/mol) | −2.38 | −1.75 | 0.33 | 4.61 | 6.59 |
| Longevity (h)* | 0.29 | 0.064 | 0.332 | 1.07 | 1.37 |
Reactivity.
Since the rate of water oxidation by our Ru-bda complexes displays a second-order kinetic dependence in catalyst concentration (see below), a higher concentration of catalyst gives much higher reaction rates. We therefore investigated the catalytic activities of our catalysts at a relatively high concentration, [cat.] = 2.222 × 10-4 M. In Fig. 6 are the plots of oxygen evolution versus time for complexes 1–3 after mixing them with excess CeIV aqueous solutions. The data were recorded with a frequency of 9 Hz with a pressure transducer. Fast water oxidation was observed with 1–3 at varying rates. Complexes 1 and 2 gave OPRs of 31 ± 3 s-1 and 70 ± 12 s-1, respectively. Complex 3 with phthalazine exhibited a high OPR of 286 ± 21 s-1. These values, including those of complexes A and B (19), are significantly higher than those of other type WOCs known to date. Notably, complex 3 is far more active than complexes 1 and 2. An increased reactivity of complex B, similar to 3, was studied in ref. 19. There, the pi–pi stacking was concluded to contribute to the increased reactivity. In general, any effect that favors the radical coupling step (2RuV═O → RuIV─O─O─RuIV) would enhance the catalytic activity according to the second order reaction nature. Now we are systematically carrying out experiments to investigate the factors (electronic effects, noncovalent interactions, and steric effects) that influence the reactivity of the Ru-bda catalysts.
Fig. 6.

Oxygen evolution curves for complexes 1–3 (8 × 10-7 mol) in acidic aqueous solutions. Conditions: volume of solutions = 3.60 mL, [cat.] = 2.222 × 10-4 M, [CeIV] = 0.327 M.
Kinetic study.
The kinetic measurements were carried out by monitoring the decay of the absorbance of CeIV at 360 nm upon addition of catalysts. Similar to A, loss of CeIV is second order in catalysts for all three complexes, with the second-order rate constants being 2.64 × 106 M-1 s-1 for 1, 3.96 × 106 M-1 s-1 for 2, and 4.16 × 108 M-1 s-1 for 3 (Fig. S3). These observations imply that the rate-determining step is associated with the dimerization of two Ru species. Based on our previous study, we propose that the dimerization step involves the coupling of two seven-coordinate RuV═O units, which is the O─O bond formation step, forming a dimeric Ru peroxide RuIV─O─O─RuIV. Once this peroxide complex is formed, it is rapidly oxidized to a superoxyl complex 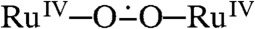 , which then releases dioxygen via a reductive elimination step and goes back to RuIII and RuIV states. See the proposed catalytic mechanism for complexes 1–3 in Scheme 2.
, which then releases dioxygen via a reductive elimination step and goes back to RuIII and RuIV states. See the proposed catalytic mechanism for complexes 1–3 in Scheme 2.
Scheme 2.

Proposed catalytic mechanism for O─O bond formation catalyzed by complexes 1–3. Axial ligands are omitted for clearity.
Conclusions
We herein have demonstrated the structure–longevity relationship of the Ru-bda WOCs. On the basis of DFT prediction, we have synthesized several Ru-bda WOCs (1–3) that exhibit promising catalytic activities toward CeIV-driven water oxidation. The proposal that an axial ligand with higher HOMO energy could form a Ru-bda complex with higher durability is in good agreement with the experimental observations. Notably, complex 3 with phthalazine axial ligands displayed an extraordinary TON of 55,400 and a very high initial OPR of 286 s-1. We have here a route to design more robust Ru-bda WOCs just by investigating the electronic structure of ligands in the gas phase, which will facilitate the discovery of more robust Ru-bda WOCs in a more efficient way. This may also have influence on the development of other types of WOCs, especially those based on the first-row transition metals such as Fe, Mn, etc.
Materials and Methods
Complex A was prepared according to the literature methods (20). Electrochemistry measurements were carried out with Autolab potentiostat, with pyrolytic graphite electrode (basal plane) as working electrode, Pt wire as auxiliary, and Ag/AgCl as reference electrode. All potentials reported herein are converted to their corresponding values versus NHE using an internal reference of [Ru(bpy)3]2+ [E1/2(Ru2+/3+) = 1.26 V vs. NHE]. The oxygen evolution was recorded with a pressure transducer (Omega PX138-030A5V) driven at 8.00 V using a power supply (TTi-PL601) plus a data acquisition module (Omega OMB-DAQ-2416), and the amount of oxygen was calibrated by GC (GC-2014 Shimadzu; equipped with a thermal conductive detector, a 5 Å Molecular sieve column, and He as carrier gas). The pressure transducer was connected with a two-neck round flask (space = 41 mL). A CF3SO3H aqueous solution (pH 1.0, 3 mL) was added to the flask containing 0.658 g of Ce(NH4)2(NO3)6 (98%) under stirring. Once all CeIV was dissolved, an acetonitrile/water (1/9) solution of catalyst was immediately injected to the above solution under vigorous stirring. The pressure change was recorded vs. time. After oxygen evolution ceased, the resulting gas phase was analyzed by GC.
All density functional theory calculations were performed using the Jaguar 7.6 program package by Scrödinger LLC. Geometry optimizations, single-point solvation, and frequency calculations were performed with the B3LYP hybrid functional (32) with the 6-31G** basis set for all atoms except for Ru, for which the LACVP** core potential and basis set was used. Single-point energy calculations were performed using the M06 functional (33) and LACV3P∗∗ ++ basis augmented by two f polarization functions, as suggested by Martin (34).
The free energy of each species was calculated by the formula G = E(M06/LACV3P∗∗ ++2fonRu) + Gsolv + ZPE + H298 - TS298 + 1.9 [concentration correction to the free energy of solvation from 1 M(g) → 1 M(aq) to 1 atm(g) → 1 M(aq)].
Other experimental procedures and details of DFT calculations are given in the SI Text. Calculated solvation energies of the ligands are listed in Table S1.
Supplementary Material
ACKNOWLEDGMENTS.
This work is supported by the Swedish Research Council, the Knut and Alice Wallenberg Foundation, the Swedish Energy Agency, the China Scholarship Council (CSC), the Basic Research Program of China (2009CB220009), and the Natural Science Foundation of China (21120102036). Computational resources were provided by the PDC supercomputer center at Kungliga Tekniska Högskolan.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1118347109/-/DCSupplemental.
References
- 1.Youngblood WJ, Lee S-HA, Maeda K, Mallouk TE. Visible light water splitting using dye-sensitized oxide semiconductors. Acc Chem Res. 2009;42:1966–1973. doi: 10.1021/ar9002398. [DOI] [PubMed] [Google Scholar]
- 2.Balzani V, Credi A, Venturi M. Photochemical conversion of solar energy. ChemSusChem. 2008;1:26–58. doi: 10.1002/cssc.200700087. [DOI] [PubMed] [Google Scholar]
- 3.Duan L, Tong L, Xu Y, Sun L. Visible light-driven water oxidation from molecular catalysts to photoelectrochemical cells. Energy Environ Sci. 2011;4:3296–3313. [Google Scholar]
- 4.Concepcion JJ, et al. Making oxygen with ruthenium complexes. Acc Chem Res. 2009;42:1954–1965. doi: 10.1021/ar9001526. [DOI] [PubMed] [Google Scholar]
- 5.Sala X, Romero I, Rodríguez M, Escriche L, Llobet A. Molecular catalysts that oxidize water to dioxygen. Angew Chem Int Ed Engl. 2009;48:2842–2852. doi: 10.1002/anie.200802659. [DOI] [PubMed] [Google Scholar]
- 6.McDaniel ND, Coughlin FJ, Tinker LL, Bernhard S. Cyclometalated iridium(III) aquo complexes: Efficient and tunable catalysts for the homogeneous oxidation of water. J Am Chem Soc. 2008;130:210–217. doi: 10.1021/ja074478f. [DOI] [PubMed] [Google Scholar]
- 7.Hull JF, et al. Highly active and robust Cp* iridium complexes for catalytic water oxidation. J Am Chem Soc. 2009;131:8730–8731. doi: 10.1021/ja901270f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lalrempuia R, McDaniel ND, Müller-Bunz H, Bernhard S, Albrecht M. Water oxidation catalyzed by strong carbene-type donor-ligand complexes of iridium. Angew Chem Int Ed Engl. 2010;49:9765–9768. doi: 10.1002/anie.201005260. [DOI] [PubMed] [Google Scholar]
- 9.Yin Q, et al. A fast soluble carbon-free molecular water oxidation catalyst based on abundant metals. Science. 2010;328:342–345. doi: 10.1126/science.1185372. [DOI] [PubMed] [Google Scholar]
- 10.Dogutan DK, McGuire R, Nocera DG. Electocatalytic water oxidation by cobalt(III) hangman b-octafluoro corroles. J Am Chem Soc. 2011;133:9178–9180. doi: 10.1021/ja202138m. [DOI] [PubMed] [Google Scholar]
- 11.Wasylenko DJ, Ganesamoorthy C, Borau-Garcia J, Berlinguette CP. Electrochemical evidence for catalytic water oxidation mediated by a high-valent cobalt complex. Chem Commun. 2011;47:4249–4251. doi: 10.1039/c0cc05522k. [DOI] [PubMed] [Google Scholar]
- 12.Shevchenko D, Anderlund MF, Thapper A, Styring S. Photochemical water oxidation with visible light using a cobalt containing catalyst. Energy Environ Sci. 2011;4:1284–1287. [Google Scholar]
- 13.Jiao F, Frei H. Nanostructured cobalt oxide clusters in mesoporous silica as efficient oxygen-evolving catalysts. Angew Chem Int Ed Engl. 2009;48:1841–1844. doi: 10.1002/anie.200805534. [DOI] [PubMed] [Google Scholar]
- 14.Ellis WC, McDaniel ND, Bernhard S, Collins TJ. Fast water oxidation using iron. J Am Chem Soc. 2010;132:10990–10991. doi: 10.1021/ja104766z. [DOI] [PubMed] [Google Scholar]
- 15.Fillol JL, et al. Efficient water oxidation catalysts based on readily available iron coordination complexes. Nat Chem. 2011;3:807–813. doi: 10.1038/nchem.1140. [DOI] [PubMed] [Google Scholar]
- 16.Najafpour M, Ehrenberg T, Wiechen M, Kurz P. Calcium manganese(III) oxides (CaMn2O4·xH2O) as biomimetic oxygen-evolving catalysts. Angew Chem Int Ed Engl. 2010;49:2233–2237. doi: 10.1002/anie.200906745. [DOI] [PubMed] [Google Scholar]
- 17.Hocking RK, et al. Water-oxidation catalysis by manganese in a geochemical-like cycle. Nat Chem. 2011;3:461–466. doi: 10.1038/nchem.1049. [DOI] [PubMed] [Google Scholar]
- 18.Yagi M, Narita K. Catalytic O2 evolution from water induced by adsorption of [(OH2)(Terpy)Mn(μ-O)2Mn(Terpy)(OH2)]3+ complex onto clay compounds. J Am Chem Soc. 2004;126:8084–8085. doi: 10.1021/ja039780c. [DOI] [PubMed] [Google Scholar]
- 19.Duan L, et al. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat Chem. 2012;4:418–423. doi: 10.1038/nchem.1301. [DOI] [PubMed] [Google Scholar]
- 20.Duan L, Fischer A, Xu Y, Sun L. Isolated seven-coordinate Ru(IV) dimer complex with [HOHOH]- bridging ligand as an intermediate for catalytic water oxidation. J Am Chem Soc. 2009;131:10397–10399. doi: 10.1021/ja9034686. [DOI] [PubMed] [Google Scholar]
- 21.Nyhlén J, Duan L, Åkermark B, Sun L, Privalov T. Evolution of O2 in a seven-coordinate RuIV dimer complex with a [HOHOH]-1 bridge: A computational study. Angew Chem Int Ed Engl. 2010;49:1773–1777. doi: 10.1002/anie.200906439. [DOI] [PubMed] [Google Scholar]
- 22.Mola J, et al. Ru-Hbpp-based water-oxidation catalysts anchored on conducting solid supports. Angew Chem Int Ed Engl. 2008;47:5830–5832. doi: 10.1002/anie.200704912. [DOI] [PubMed] [Google Scholar]
- 23.Chen Z, Concepcion JJ, Jurss JW, Meyer TJ. Single-site, catalytic water oxidation on oxide surfaces. J Am Chem Soc. 2009;131:15580–15581. doi: 10.1021/ja906391w. [DOI] [PubMed] [Google Scholar]
- 24.Concepcion JJ, Jurss JW, Hoertz PG, Meyer TJ. Catalytic and surface-electrocatalytic water oxidation by redox mediator-catalyst assemblies. Angew Chem Int Ed Engl. 2009;48:9473–9476. doi: 10.1002/anie.200901279. [DOI] [PubMed] [Google Scholar]
- 25.Li F, et al. Highly efficient oxidation of water by a molecular catalyst immobilized on carbon nanotubes. Angew Chem Int Ed Engl. 2011;50:12276–12279. doi: 10.1002/anie.201105044. [DOI] [PubMed] [Google Scholar]
- 26.Wasylenko DJ, et al. Electronic modification of the [RuII(tpy)(bpy)(OH2)]2+ scaffold: Effects on catalytic water oxidation. J Am Chem Soc. 2010;132:16094–16106. doi: 10.1021/ja106108y. [DOI] [PubMed] [Google Scholar]
- 27.Yoshida M, Masaoka S, Abe J, Sakai K. Catalysis of mononuclear aquaruthenium complexes in oxygen evolution from water: A new radical coupling path using hydroxocerium(IV) species. Chem Asian J. 2010;5:2369–2378. doi: 10.1002/asia.201000323. [DOI] [PubMed] [Google Scholar]
- 28.Yagi M, Tajima S, Komi M, Yamazaki H. Highly active and tunable catalysts for O2 evolution from water based on mononuclear ruthenium(II) monoaquo complexes. Dalton Trans. 2011;40:3802–3804. doi: 10.1039/c0dt01826k. [DOI] [PubMed] [Google Scholar]
- 29.Bozoglian F, et al. The Ru-Hbpp water oxidation catalyst. J Am Chem Soc. 2009;131:15176–15187. doi: 10.1021/ja9036127. [DOI] [PubMed] [Google Scholar]
- 30.Duan L, et al. Chemical and photochemical water oxidation catalyzed by mononuclear ruthenium complexes with a negatively charged tridentate ligand. Chem Eur J. 2010;16:4659–4668. doi: 10.1002/chem.200902603. [DOI] [PubMed] [Google Scholar]
- 31.Xu Y, et al. Chemical and light-driven oxidation of water catalyzed by an efficient dinuclear ruthenium complex. Angew Chem Int Ed Engl. 2010;49:8934–8937. doi: 10.1002/anie.201004278. [DOI] [PubMed] [Google Scholar]
- 32.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc. 2006;120:215–241. [Google Scholar]
- 34.Martin JML, Sundermann A. Correlation consistent valence basis sets for use with the Stuttgart–Dresden–Bonn relativistic effective core potentials: The atoms Ga–Kr and In–Xe. J Chem Phys. 2001;114:3408–3420. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.