

Abstract
Photoelectrochemical water splitting directly converts solar energy to chemical energy stored in hydrogen, a high energy density fuel. Although water splitting using semiconductor photoelectrodes has been studied for more than 40 years, it has only recently been demonstrated using dye-sensitized electrodes. The quantum yield for water splitting in these dye-based systems has, so far, been very low because the charge recombination reaction is faster than the catalytic four-electron oxidation of water to oxygen. We show here that the quantum yield is more than doubled by incorporating an electron transfer mediator that is mimetic of the tyrosine-histidine mediator in Photosystem II. The mediator molecule is covalently bound to the water oxidation catalyst, a colloidal iridium oxide particle, and is coadsorbed onto a porous titanium dioxide electrode with a Ruthenium polypyridyl sensitizer. As in the natural photosynthetic system, this molecule mediates electron transfer between a relatively slow metal oxide catalyst that oxidizes water on the millisecond timescale and a dye molecule that is oxidized in a fast light-induced electron transfer reaction. The presence of the mediator molecule in the system results in photoelectrochemical water splitting with an internal quantum efficiency of approximately 2.3% using blue light.
Keywords: artificial photosynthesis, photoelectrochemistry
The design of biomimetic systems for artificial photosynthesis is of fundamental interest in the study of light-driven electron and proton transfer reactions. It also represents a potential route to the efficient conversion of solar energy to energy stored in fuel. System modeling has shown that it should be possible, using complementary dye molecules that absorb in the infrared and the visible, to construct artificial Z-schemes that split water with over 10% energy conversion efficiency (1, 2). It is simpler in many ways to design small molecules with the proper photoredox properties than it is to find a set of semiconductors that can be coupled for visible light water splitting. Nevertheless, the molecular approach has so far lagged behind the semiconductor-based approach where high efficiencies have been realized with expensive materials (3–7).
A ubiquitous problem in molecular artificial photosynthesis is back electron transfer, which rapidly thermalizes the energy stored by light-induced charge separation in donor-acceptor pairs. Recently, our group and several others have studied this problem in dye-sensitized solar cells where a molecular dye and a porous TiO2 electrode act as the donor-acceptor dyad (8–13). The dye is covalently coupled to a colloidal or molecular water oxidation catalyst. Fast back electron transfer, relative to the rate of water oxidation, results in low quantum yields for water splitting in these systems.
It is well known that charge-separation lifetimes in molecular donor-acceptor systems can be increased dramatically by adding secondary electron donors or acceptors to form triads, tetrads, and more complex supermolecules (14). This strategy spatially separates the ultimate donor and acceptor molecules in the chain, and the back electron transfer reaction is slowed because of the exponential dependence of outer-sphere electron transfer rates on distance. Multistep electron transfer is used in many biological systems, including photosynthesis, to control the kinetics of oxidation-reduction reactions. It has been noted that biology prefers to use distance rather than free energy differences to tune electron transfer rates because the former is sufficient and the latter is too expensive in terms of energy efficiency (15).
The four-electron oxidation of water is a kinetically demanding process that occurs on the millisecond timescale in the oxygen-evolving complex (OEC) of Photosystem II as well as in a number of synthetic catalysts (16–20). A key component of Photosystem II is the tyrosine-histidine pair that mediates electron transfer between the OEC and P680•+. Synthetic mimics of this biological redox relay have been studied in a number of molecular dyads and triads (21–25). We incorporated a redox mediator into a water-splitting dye-sensitized solar cell (see Scheme 1). Dramatically increased photocurrent is found relative to the simple TiO2-dye-catalyst system. Flash photolysis/transient absorbance measurements show that the mediator accelerates the reduction of the photo-oxidized dye; thus, its function in this system is similar to that of the tyrosine-histidine mediator in Photosystem II.
Scheme 1.

Electron transfer reactions in the mediator-based, dye-sensitized TiO2 photoanode showing molecular structures of the BIP mediator, 3P-Ru dye, and CEPA linking group.
Results and Discussion
The construction of the electron transport assembly shown in Scheme 1 begins with the synthesis of Iridium oxide nanoparticles that are bound to the benzimidazole-phenol (BIP) mediator, and the 2-carboxyethylphosphonic acid (CEPA) anchoring molecule. In earlier papers involving water oxidation using ligand-capped IrOx·nH2O nanoparticles, the nanoparticles were prepared by alkaline hydrolysis of 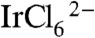 ions in the presence of a carboxylated ligand (11, 26–28). IrOx·nH2O nanoparticles prepared by that method cannot be easily linked to TiO2 photoelectrodes, possibly because relatively few capping ligands coordinate the particle surface under basic conditions. To address this problem, we combined the capping ligands and
ions in the presence of a carboxylated ligand (11, 26–28). IrOx·nH2O nanoparticles prepared by that method cannot be easily linked to TiO2 photoelectrodes, possibly because relatively few capping ligands coordinate the particle surface under basic conditions. To address this problem, we combined the capping ligands and 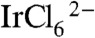 under slightly acidic conditions (pH 5–6 for 1 h at 80–90 °C to form the ligand-Ir(IV) complex. Then, we hydrolyzed these complexes to form IrOx·nH2O nanoparticles at pH 7–8. CEPA-capped IrOx·nH2O nanoparticles synthesized by this procedure persistently link onto TiO2 photoelectrodes at loadings that can be easily controlled by their concentration and adsorption time.
under slightly acidic conditions (pH 5–6 for 1 h at 80–90 °C to form the ligand-Ir(IV) complex. Then, we hydrolyzed these complexes to form IrOx·nH2O nanoparticles at pH 7–8. CEPA-capped IrOx·nH2O nanoparticles synthesized by this procedure persistently link onto TiO2 photoelectrodes at loadings that can be easily controlled by their concentration and adsorption time.
A second important problem in preparing photoelectrodes catalyzed by IrOx·nH2O nanoparticles is the elimination of the monomeric [Ir(OH)6]2- anion that is formed during hydrolysis of 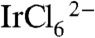 . This anion can be detected by its strong absorbance at 310 nm (29–31). Significant dark current and lower photocurrent were observed when TiO2 photoelectrodes or other mesoporous metal oxide photoelectrodes such as Fe2O3 and WO3 were soaked in IrOx·nH2O nanoparticle solutions that contained an [Ir(OH)6]2- impurity. We hypothesized that this electroactive anion can access the electrode back contact [a fluorine-doped tin oxide (FTO) film] and, thereby, mediate back electron transfer. The [Ir(OH)6]2- impurity can be removed by ion exchange on a DOWEX 1X8-50 resin, and excess free capping ligand in the solution can also be very efficiently removed. For example, free BIP molecules in a solution of BIP and IrOx·nH2O nanoparticles were completely removed after stirring with ion-exchange resin for 1 h. BIP- and CEPA-capped IrOx·nH2O colloids prepared by this method were a deep blue color and had negligible 310 nm absorbance in their UV-vis spectra, which indicates essentially complete conversion of
. This anion can be detected by its strong absorbance at 310 nm (29–31). Significant dark current and lower photocurrent were observed when TiO2 photoelectrodes or other mesoporous metal oxide photoelectrodes such as Fe2O3 and WO3 were soaked in IrOx·nH2O nanoparticle solutions that contained an [Ir(OH)6]2- impurity. We hypothesized that this electroactive anion can access the electrode back contact [a fluorine-doped tin oxide (FTO) film] and, thereby, mediate back electron transfer. The [Ir(OH)6]2- impurity can be removed by ion exchange on a DOWEX 1X8-50 resin, and excess free capping ligand in the solution can also be very efficiently removed. For example, free BIP molecules in a solution of BIP and IrOx·nH2O nanoparticles were completely removed after stirring with ion-exchange resin for 1 h. BIP- and CEPA-capped IrOx·nH2O colloids prepared by this method were a deep blue color and had negligible 310 nm absorbance in their UV-vis spectra, which indicates essentially complete conversion of 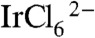 to IrOx·nH2O nanoparticles with negligible [Ir(OH)6]2- impurity.
to IrOx·nH2O nanoparticles with negligible [Ir(OH)6]2- impurity.
Fig. 1 compares spectra of purified solutions of IrOx·nH2O nanoparticles capped by CEPA, CEPA, and BIP designated 1-IrOx·nH2O and 2-IrOx·nH2O, respectively. There is a broad absorbance centered at 580 nm, which is typical of Iridium (IV) oxide nanoparticles; and, there is no impurity peak at 310 nm present in the 1-IrOx·nH2O spectrum. In addition to the 580 nm peak, the 2-IrOx·nH2O nanoparticles exhibited an absorbance attributed to BIP at 340 nm, which indicates that BIP is covalently bound. Using an extinction coefficient of 18,000 M-1 cm-1 for BIP, the concentration of BIP in the 2-IrOx·nH2O solution is approximately 0.1 mM, and the BIP:Ir mole ratio is approximately 1∶10, assuming complete conversion of 1 mM 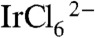 to IrOx·nH2O nanoparticles. In the following discussion, the ratios and concentrations refer to the number of Ir atoms rather than the number of IrOx·nH2O nanoparticles.
to IrOx·nH2O nanoparticles. In the following discussion, the ratios and concentrations refer to the number of Ir atoms rather than the number of IrOx·nH2O nanoparticles.
Fig. 1.

UV-vis of BIP, CEPA capped (1-IrOx·nH2O), and BIP and CEPA capped (2-IrOx·nH2O) iridium oxide nanoparticles.
TEM images (Fig. 2) show that 1-IrOx·nH2O and 2-IrOx·nH2O consist of irregularly shaped 2-nm nanoparticles. IrOx·nH2O nanoparticles synthesized using other ligands under the same conditions had similar particle sizes and UV-vis absorbance at 580 nm. In contrast, larger IrOx·nH2O aggregates were found if the solution was lower than pH 7 during the hydrolysis, regardless of the type of ligands used. Therefore, it is very important to keep the reaction pH constant at 7–8 during the hydrolysis step to obtain ligand-capped particles without aggregation. The pH of the solution decreases gradually during hydrolysis, and dilute NaOH must be added to maintain a constant pH.
Fig. 2.

TEM images of 1-IrOx·nH2O (Left) and 2-IrOx·nH2O (Right) nanoparticles.
An obvious shortcoming of the coadsorption strategy illustrated in Scheme 1 is that juxtaposition of the sensitizer and BIP-catalyst on the TiO2 surface is uncontrolled. If these components do intimately mix, then changing their ratio should alter the kinetics of the system. In a recent report of photoelectrodes containing a codeposited porphyrin sensitizer and water oxidation catalyst, the ratio of sensitizer to the catalyst, a Cp-Ir complex, was chosen to be 1∶1 (10). Previous studies on 2-nm ligand-capped IrOx·nH2O nanoparticles suggest that 20–30% of the Ir atoms should be on the surface (32–34). We experimented in preliminary studies with different molar ratios of the 3P-Ru dye to Ir ranging from 1∶1 to 1∶10, and we found the highest photocurrents at a 1∶5 ratio with 1-IrOx·nH2O or 2-IrOx·nH2O nanoparticles. For consistency, all subsequent electrochemical and transient spectroscopic data were obtained using TiO2 electrodes with 3P-Ru and IrOx·nH2O nanoparticles coadsorbed in molar ratio of 1∶5.
The electrocatalytic activity of the 1-IrOx·nH2O or 2-IrOx·nH2O nanoparticles was first assessed by cyclic voltammetry (CV) in the dark in Na2SiF6-NaHCO3 buffer solutions (pH 5.8) shown in Fig. 3. The porous TiO2 electrode itself gives negligible current under these conditions near the water oxidation potential, and an electrode derivatized with the 3P-Ru sensitizer is not catalytic, showing the reversible Ru(II/III) wave only. 1-IrOx·nH2O and 2-IrOx·nH2O nanoparticles deposited on TiO2 photoelectrodes showed significant catalytic current for water oxidation, which was slightly suppressed by coadsorption of 3P-Ru. Although these data establish that the nanoparticles are electrocatalytically active and bound to the electrode, we did not directly observe the oxidation/reduction of the BIP mediator. The (dark) anodic current was lower for electrodes catalyzed by 2-IrOx·nH2O than by 1-IrOx·nH2O. Electrochemical characterization of the diethyl ester of BIP (Supporting Information) showed a reversible one-electron oxidation at 1.06 V vs. SCE (+1.10 V vs Ag/AgCl) in acetonitrile.
Fig. 3.

Cyclic voltammetry of porous TiO2 photoelectrode, photoelectrodes with adsorbed 3P-Ru dye, 1-IrOx·nH2O nanoparticles, 3P-Ru dye +1-IrOx·nH2O nanoparticles, and 3P-Ru dye +2-IrOx·nH2O nanoparticles in 0.1 M silicate buffer solution at pH 5.8, scan rate = 50 mV/ sec.
The photoelectrochemical behavior of 3P-Ru-sensitized electrodes containing 1-IrOx·nH2O or 2-IrOx·nH2O under filtered white light illumination is shown in Fig. 4. The 3P-Ru/1-IrOx·nH2O electrode without the BIP mediator showed an initial photocurrent density of 130 μA/cm2, which rapidly decayed to a stable photocurrent of 25 μA/ cm2. This steady-state photocurrent is similar to that previously observed using TiO2 electrodes sensitized with IrOx·nH2O nanoparticles capped by a heteroleptic Ru Tris(bipyridyl) dye (9, 11). The rapid polarization of the electrode may arise from hole accumulation (as a consequence of the slow kinetics of water oxidation) or the local decrease in pH that arises from the four-electron oxidation of water. The initial photocurrent is recovered after switching the cell off for 1–2 min, which shows that the drop in photocurrent does not represent desorption or decomposition of the 3P-Ru dye. Electrodes with codeposited 3P-Ru dye and 2-IrOx·nH2O dyads showed less polarization and steady-state photocurrent densities up to 80 μA/ cm2-almost triple the value obtained without the BIP mediator. The significant increase in transient photocurrent and slower polarization of the electrode are consistent with faster electron transfer between the catalytic nanoparticles and the oxidized sensitizer when the IrOx·nH2O nanoparticles are capped by the BIP mediator. Using 450 nm light at 4.5 mW/cm2 intensity, we measured steady-state photocurrent densities of 10.1 and 29.1 μA/cm2 from electrodes containing 3P-Ru (with A464 = 0.75) and 1-IrOx·nH2O or 2-IrOx·nH2O, respectively, corresponding to internal quantum yields of approximately 0.8% and 2.3%. Measurements of the steady-state photocurrent as a function of applied bias (Fig. 4B) gave similar open circuit potentials of -380 and -400 mV vs. Ag/AgCl for electrodes with coadsorbed 3P-Ru and 1-IrOx·nH2O or 2-IrOx·nH2O nanoparticles, which corresponds to an open circuit photovoltage of 1.05 V. The 20 mV increase in photovoltage with the BIP mediator is consistent with faster forward electron transfer at the anode. Clark electrode measurements showed that the Faradaic efficiency for oxygen evolution was ≥85% in both cases, which is comparable to values reported previously for the TiO2/dye-IrOx·nH2O dyad system (11).
Fig. 4.

(A) Photocurrent density vs. time for TiCl4-treated porous TiO2 electrodes with coadsorbed 3P-Ru dye and 1-IrOx·nH2O or 2-IrOx·nH2O nanoparticles (Dye: Ir = 1∶5) in pH 5.8 silicate buffer at 0 V vs. Ag/AgCl. The initial sharp spike in current corresponds to switching from dark to light conditions (B) Steady-state photocurrent vs. anode potential.
Flash photolysis experiments were carried out on porous TiO2 electrodes derivatized with 3P-Ru and 1-IrOx·nH2O or 2-IrOx·nH2O in order to characterize the electron transfer kinetics of the system. In previous experiments on dye-IrOx·nH2O dyads adsorbed on TiO2, we observed photoinduced charge injection on a timescale faster (< 30 ns) than could be resolved by transient absorption measurements, followed by charge recombination on the timescale of hundreds of microseconds (9, 11). Transient absorbance decays recorded at the absorbance maximum of 3P-Ru, which correspond to charge recombination kinetics in the present system, are shown in Fig. 5. Under both open circuit conditions and at more positive potentials where the photocurrent reaches its maximum value, charge recombination occurs over a broad range of timescales. This is consistent with a trapping/detrapping process for photoinjected electrons. In both cases the kinetics were fit to a stretched exponential distribution, (Eq. 1), with β values in the range of 0.3–0.6.
| [1] |
Fig. 5.

(A) Normalized transient bleaching recovery curves monitored at 464 nm for TiCl4-treated TiO2 electrodes with adsorbed 3P-Ru dye, 3P-Ru dye +1-IrOx·nH2O, and 3P-Ru dye +2-IrOx·nH2O in pH 5.8 silicate buffer solution at open circuit. Kinetic fits are superimposed on the data. Calculated lifetimes (τ) were (i) 3P-Ru dye: 0.52 ms, (ii) 3P-Ru dye +1-IrOx·nH2O, 0.26 ms (iii) 3P-Ru dye +2-IrOx·nH2O, 0.15 ms. (B) Transient decays obtained at +100 mV vs. Ag/AgCl. Calculated lifetimes (τ) were (i) 3P-Ru dye: 0.72 ms, (ii) 3P-Ru dye +1-IrOx·nH2O, 0.44 ms (iii) 3P-Ru dye +2-IrOx·nH2O, 0.31 ms.
This long-lived component of the decay represented by the constant c is possibly an artifact of dye desorption at negative potentials under the conditions of the flash photolysis experiments. At open circuit, a very long lifetime component (c = 0.15–0.20) corresponding to 15–20% of the initial transient was needed to fit the data (Fig. 5A). At positive potentials, the data could be adequately fit with c = 0.10 (Fig. 5B).
At both potentials, coadsorption of ligand-capped IrOx·nH2O particles decreases the lifetime of the Ru(III) form of the sensitizer, which is consistent with electron transfer from Ir(IV) to Ru(III). The lifetime for 3P-Ru dye +1-IrOx·nH2O at open circuit (0.26 ms) is in good agreement with previous measurements in the TiO2-dye/IrOx·nH2O dyad system (9, 11). Interestingly, incorporation of the BIP mediator molecule decreases τ at open circuit and greater positive potentials. Because the distance between the BIP mediator molecules and 3P-Ru dye molecules is not controlled, there should be a distribution of rates; and, we cannot draw quantitative conclusions about the rate of electron transfer between the BIP mediator and Ru(III) from the data in Fig. 5. Nevertheless, the trend of faster bleaching recovery with the BIP mediator present is consistent with the increased photocurrent shown in Fig. 4.
In summary, porous TiO2 photoelectrodes with coadsorbed 3P-Ru sensitizer molecules and ligand-capped IrOx·nH2O nanoparticles exhibited similar performance to electrodes modified with dye-IrOx·nH2O dyads that were previously reported (9, 11). The synthesis of IrOx·nH2O from a ligand-Ir(IV) precursor solution under controlled pH conditions allows one to bind a mixture of capping ligands to the nanoparticles. This, in turn, allows one to make more complex electron transfer assemblies using electroactive ligands. Incorporation of the BIP tyrosine-histidine mimic into the system accelerates electron transfer between Ir(IV) and Ru(III) resulting in a more efficient water-splitting anode by almost a factor of three. In this study, the electron transfer rate between the BIP mediator and the 3P-Ru dye was not well controlled because the molecules were separately adsorbed onto the electrode surface. By modifying the molecular structure of the mediator and covalently linking it to the dye, as was done in previous studies (21–25), it should be possible to control the electron transfer distance in the dye-mediator dyad. This should, in turn, enable tuning of the electron transfer rate to improve the water-splitting efficiency of the system further.
Materials and Methods
Synthesis of 2-Carboxyethyl Phosphonic Acid (CEPA) Capped IrOx·nH2O Nanoparticles.
CEPA (0.3 mmol) was dissolved to 50 mL DI water and the pH was adjusted to 5.0 by the addition of 0.5 M NaOH. K2IrCl6 (0.05 mmol) was dissolved in this solution, which was then heated in a flask with a reflux condenser at 90 °C for 1 h. The solution was allowed to cool to 80 °C, and the pH was continuously adjusted by slow addition of dilute NaOH solution until the pH was stabilized at 7.5. The solution was then heated in a flask with a reflux condenser at 80 °C for 5 h. The resulting solution was deep blue with an absorbance peak at 580 nm after cooling to room temperature. The colloidal solution was stirred with 1 g ion-exchange resin [DOWEX 1X8-50(20-50 mesh)] for 1 h to remove a small impurity of monomeric [Ir(OH)6]2- and excess CEPA. The CEPA-capped IrOx·nH2O nanoparticles are denoted as 1-IrOx·nH2O.
Synthesis of BIP- and CEPA-Capped IrOx·nH2O Nanoparticles.
The synthesis of the benzimidazol-phenol mediator (2-(2-(3,5-di-tert-butyl-2-hydroxyphenyl)-1H-benzo[d]imidazol-5-yl-methylene) malonic acid, BIP) is described in the Supporting Information. 5.0 mg BIP was dissolved in 40 mL DI water at pH 6 (this is the maximum solubility for this compound). 0.05 mmol K2IrCl6 was then dissolved in the solution, and the resulting mixture was heated in a flask with a reflux condenser at 80 °C for 1 h. The solution was allowed to cool to room temperature, and 10 mL of 0.3 mmol CEPA at pH 5.5 was added. The solution was then heated at 80 °C for another 1 h. The pH was continuously adjusted by slow addition of dilute NaOH solution until the pH was stabilized at 7.5. The solution was then heated at 80 °C for 5 h, during which time it slowly turned deep blue. It was then allowed to cool to room temperature. The [Ir(OH)6]2- impurity and excess BIP and CEPA were removed by stirring with 1g ion-exchange resin [DOWEX 1X8-50 (20–50 mesh)] for 1 h. These CEPA- and BIP capped IrOx·nH2O nanoparticles are denoted as 2-IrOx·nH2O.
TiO2 Photoelectrode Preparation, TiCl4 Treatment, and Sensitization.
A TiO2 colloidal suspension was prepared by the method previously described (35). TiO2 films were coated onto fluorine-doped tin oxide-coated glass electrodes (FTO-glass, Hartford, USA, 8 and 15 Ω/cm) by the doctor-blade method using Scotch tape spacers to adjust the film thickness. The films were then sintered at 450 °C for 30 min. A TiCl4 treatment was performed following previous reports by soaking the electrodes in 0.05 M TiCl4 solution at 70 °C for 30 min followed by annealing at 450 °C for 30 min (36). The TiO2 photoelectrodes were then connected to insulated copper wires by using silver paste (DuPont 4922N), and the electrical contact was sealed with white epoxy (Hysol-C, McMaster-Carr). Electrodes with 7-μm thick TiO2 films deposited on the 8 Ω-cm FTO were used for photoelectrochemical measurement, whereas more transparent photoelectrodes with 4-μm thick TiO2 films deposited on 15 Ω-cm FTO were used for transient UV-vis spectroscopic measurements. The TiO2 photoanodes were sensitized using a solution of 3P-Ru dye and IrOx·nH2O nanoparticles in different ratios. A typical sensitizer solution contained 0.05 mM 3P-Ru dye and 0.25 mM 1- or 2-IrOx·nH2O in water. The TiO2 photoelectrodes with coadsorbed 3P-Ru dye and IrOx·nH2O nanoparticles that were used for photoelectrochemical and transient spectroscopic measurements had absorbance values of 0.8–1.0 and 0.4–0.6 at 464 nm, respectively, relative to a blank FTO reference sample.
Photoelectrochemical Measurements.
The TiO2 working electrode was placed in the quartz cuvette of an H-shaped cell with a Pt-wire counter electrode and Ag/AgCl reference electrode in the other compartment of the cell. The entire cell was filled with 37.5 mM Na2SiF6-NaHCO3 (1∶1.5) silicate buffer (pH 5.8), containing 0.1 M LiClO4 as the supporting electrolyte and kept under a blanket of Ar gas. The buffer solution was prepared the day prior to the measurements and was aged overnight. All buffer solutions were centrifuged at 4,000 rpm for 20 min to remove precipitates. A 150 W Xe lamp was used as the light source with a 410-nm, long-pass filter and a water filter. The photoelectrochemical data were recorded using a digital potentiostat (Bioanalytical Systems 100A).
Amperometric Detection of O2 Generated at the Photoanode.
Pt film electrodes were made by sputtering 1 μm thick Pt films onto glass slides with an adhesion layer of Ti and Au. The Pt film electrodes were connected to an insulated Cu wire with silver paste or by soldering and were sealed with white epoxy (Hysol-C, McMaster–Carr) covering the electrical contact. A 1.5-mm thick layer of epoxy was applied to the outer edge of the Pt electrodes as a spacer. Before each measurement, the Pt electrodes were electrochemically cleaned by the following procedure: the electrode was anodized at 1.5 V vs Ag/AgCl for 20 s in 0.5 M H2SO4, then the Pt electrode was continuously cycled between -0.3 V and 1.1 V vs. Ag/AgCl at 50 mV/s in 0.5 M H2SO4 until the peaks of hydrogen adsorption and stripping became pronounced. The cathodic current corresponding to reduction of dissolved O2 was measured on a Pine Bipotentiostat (model AFCBP1) using the setup described in our previous study (9).
Transient Spectroscopy.
Transient absorption measurements were carried out using TiCl4-treated, 4-μm thick TiO2 electrodes. Different components of the electron transfer system, 3P-Ru dye, 3P-Ru dye, 1-IrOx·nH2O 3P-Ru dye, and 2-IrOx·nH2O nanoparticles, were adsorbed as described above, and the working electrode was held at open circuit (by disconnecting the electrodes and purging with Ar for 15 min) and at +100 mV vs. AgCl/Ag by using a potentiostat as described above. The electrodes were irradiated with a 300 W Xe arc lamp as the analyzing light and photoexcited with a 532-nm Nd:YAG Spectra-Physics INDI laser (200 mJ/pulse, 7 ns). A mechanical shutter minimized exposure of the samples to the white analyzing light. A Spectral Products CM110 monochromator was placed between the Xe lamp and the sample. The analyzing light then passed through the sample and a monochromator (Spectral Products, CM110) and reached a photodiode detector (Thor Labs). Two laser line filters were placed in the beam path before the photodiode to eliminate scattered laser light. Each kinetic trace was recorded by using a Tektronix oscilloscope (TDS 540A) and approximately five shots were averaged to minimize photodegradation.
Supplementary Material
ACKNOWLEDGMENTS.
Work at Pennsylvania State University is supported by the Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Energy Biosciences, Department of Energy under contract DE-FG02-07ER15911, and work at Arizona State University is supported as part of the Center for Bio-Inspired Solar Fuel Production, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001016.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1118339109/-/DCSupplemental.
References
- 1.Bolton JR, Strickler SJ, Connolly JS. Limiting and realizable efficiencies of solar photolysis of water. Nature. 1985;316:495–500. [Google Scholar]
- 2.Blankenship RE, et al. Comparing photosynthetic and photovoltaic efficiencies and recognizing the potential for improvement. Science. 2011;332:805–809. doi: 10.1126/science.1200165. [DOI] [PubMed] [Google Scholar]
- 3.Khaselev O, Turner JA. A monolithic photovoltaic-photoelectrochemical device for hydrogen production via water splitting. Science. 1998;280:425–427. doi: 10.1126/science.280.5362.425. [DOI] [PubMed] [Google Scholar]
- 4.Khaselev O, Bansal A, Turner JA. High-efficiency integrated multijunction photovoltaic/electrolysis systems for hydrogen production. Int J Hydrogen Energy. 2001;26:127–132. [Google Scholar]
- 5.Licht S, et al. Over 18% solar energy conversion to generation of hydrogen fuel; theory and experiment for efficient solar water splitting. Int J Hydrogen Energy. 2001;26:653–659. [Google Scholar]
- 6.Kelly NA, Gibson TL. Design and characterization of a robust photoelectrochemical device to generate hydrogen using solar water splitting. Int J Hydrogen Energy. 2006;31:1658–1673. [Google Scholar]
- 7.Reece SY, et al. Wireless solar water splitting using silicon-based semiconductors and earth-abundant catalysts. Science. 2011;334:645–648. doi: 10.1126/science.1209816. [DOI] [PubMed] [Google Scholar]
- 8.Brimblecombe R, Koo A, Dismukes GC, Swiegers GF, Spiccia L. Solar driven water oxidation by a bioinspired manganese molecular catalyst. J Am Chem Soc. 2010;132:2892–2894. doi: 10.1021/ja910055a. [DOI] [PubMed] [Google Scholar]
- 9.Lee S-HA, et al. Electron transfer kinetics in water splitting dye-sensitized solar cells based on core-shell oxide electrodes. Faraday Discuss. 2012;155:165–176. doi: 10.1039/c1fd00083g. [DOI] [PubMed] [Google Scholar]
- 10.Moore GF, et al. A visible light water-splitting cell with a photoanode formed by codeposition of a high-potential porphyrin and an iridium water-oxidation catalyst. Energ Environ Sci. 2011;4:2389–2392. [Google Scholar]
- 11.Youngblood WJ, et al. Photoassisted overall water splitting in a visible light-absorbing dye-sensitized photoelectrochemical cell. J Am Chem Soc. 2009;131:926–927. doi: 10.1021/ja809108y. [DOI] [PubMed] [Google Scholar]
- 12.Sherman BD, et al. A porphyrin-stabilized iridium oxide water oxidation catalyst. Can J Chem. 2011;89:152–157. [Google Scholar]
- 13.Xu Y, Duan L, Tong L, Akermark B, Sun L. Visible light-driven water oxidation catalyzed by a highly efficient dinuclear ruthenium complex. Chem Commun. 2010;46:6506–6508. doi: 10.1039/c0cc01250e. [DOI] [PubMed] [Google Scholar]
- 14.Gust D, Moore TA, Moore AL. Solar fuels via artificial photosynthesis. Acc Chem Res. 2009;42:1890–1898. doi: 10.1021/ar900209b. [DOI] [PubMed] [Google Scholar]
- 15.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 16.Yin Q, et al. A fast soluble carbon-free molecular water oxidation catalyst based on abundant metals. Science. 2010;328:342–345. doi: 10.1126/science.1185372. [DOI] [PubMed] [Google Scholar]
- 17.Blakemore JD, et al. Half-sandwich iridium complexes for homogeneous water-oxidation catalysis. J Am Chem Soc. 2010;132:16017–16029. doi: 10.1021/ja104775j. [DOI] [PubMed] [Google Scholar]
- 18.Jiao F, Frei H. Nanostructured manganese oxide clusters supported on mesoporous silica as efficient oxygen-evolving catalysts. Chem Commun. 2010;46:2920–2922. doi: 10.1039/b921820c. [DOI] [PubMed] [Google Scholar]
- 19.Kanan MW, Nocera DG. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+ Science. 2008;321:1072–1075. doi: 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]
- 20.Concepcion JJ, Jurss JW, Templeton JL, Meyer TJ. Mediator-assisted water oxidation by the ruthenium “blue dimer” cis,cis-[(bpy)2(H2O)RuORu(OH2)(bpy)2]4+ Proc Natl Acad Sci USA. 2008;105:17632–17635. doi: 10.1073/pnas.0807153105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magnuson A, et al. Mimicking electron transfer reactions in photosystem II: Synthesis and photochemical characterization of a Ruthenium(II) Tris(bipyridyl) complex with a covalently linked tyrosine. J Am Chem Soc. 1997;119:10720–10725. [Google Scholar]
- 22.Sun L, et al. Hydrogen-bond promoted intramolecular electron transfer to photogenerated Ru(III): A functional mimic of tyrosinez and histidine 190 in Photosystem II. J Am Chem Soc. 1999;121:6834–6842. [Google Scholar]
- 23.Magnuson A, et al. A biomimetic model system for the water oxidizing triad in Photosystem II. J Am Chem Soc. 1999;121:89–96. [Google Scholar]
- 24.Sjödin M, Styring S, Åkermark B, Sun L, Hammarström L. Proton-coupled electron transfer from tyrosine in a tyrosine-Ruthenium-Tris-bipyridine complex: Comparison with tyrosinez oxidation in Photosystem II. J Am Chem Soc. 2000;122:3932–3936. [Google Scholar]
- 25.Moore GF, et al. A bioinspired construct that mimics the proton coupled electron transfer between P680•+ and the Tyr(z)-His190 pair of photosystem II. J Am Chem Soc. 2008;130:10466–10467. doi: 10.1021/ja803015m. [DOI] [PubMed] [Google Scholar]
- 26.Hara M, Waraksa CC, Lean JT, Lewis BA, Mallouk TE. Photocatalytic water oxidation in a buffered Tris(2,2′-bipyridyl)Ruthenium complex-colloidal IrO2 system. J Phys Chem A. 2000;104:5275–5280. [Google Scholar]
- 27.Hoertz PG, Kim YI, Youngblood WJ, Mallouk TE. Bidentate dicarboxylate capping groups and photosensitizers control the size of IrO2 nanoparticle catalysts for water oxidation. J Phys Chem B. 2007;111:6845–6856. doi: 10.1021/jp070735r. [DOI] [PubMed] [Google Scholar]
- 28.Harriman A, Thomas JM, Millward GR. Catalytic and structural-properties of Iridium-Iridium dioxide colloids. New J Chem. 1987;11:757–762. [Google Scholar]
- 29.Zhao YX, Hernandez-Pagan EA, Vargas-Barbosa NM, Dysart JL, Mallouk TE. A high yield synthesis of ligand-free Iridium oxide nanoparticles with high electrocatalytic activity. J Phys Chem Lett. 2011;2:402–406. [Google Scholar]
- 30.Zhao YX, Vargas-Barbosa NM, Hernandez-Pagan EA, Mallouk TE. Anodic deposition of colloidal iridium oxide thin films from hexahydroxyiridate(IV) solutions. Small. 2011;7:2087–2093. doi: 10.1002/smll.201100485. [DOI] [PubMed] [Google Scholar]
- 31.Castillo-Blum SE, Richens DT, Sykes AG. Oxidation of hexaaquairidium(III) and related studies: Preparation and properties of iridium(III), iridium(IV), and iridium(V) dimers as aqua ions. Inorg Chem. 1989;28:954–960. [Google Scholar]
- 32.Kuwabara T, et al. Characterization and analysis of self-assembly of a highly active colloidal catalyst for water oxidation onto transparent conducting oxide substrates. J Phys Chem C. 2008;112:3774–3779. [Google Scholar]
- 33.Nakagawa T, Bjorge NS, Murray RW. Electrogenerated IrOx nanoparticles as dissolved redox catalysts for water oxidation. J Am Chem Soc. 2009;131:15578–15579. doi: 10.1021/ja9063298. [DOI] [PubMed] [Google Scholar]
- 34.Yagi M, Tomita E, Sakita S, Kuwabara T, Nagai K. Self-assembly of active IrO2 colloid catalyst on an ITO electrode for efficient electrochemical water oxidation. J Phys Chem B. 2005;109:21489–21491. doi: 10.1021/jp0550208. [DOI] [PubMed] [Google Scholar]
- 35.Lee SHA, et al. Coupling of titania inverse opals to nanocrystalline titania layers in dye-sensitized solar cells. J Phys Chem B. 2008;112:14415–14421. doi: 10.1021/jp802692u. [DOI] [PubMed] [Google Scholar]
-
36.Ito S, et al. Control of dark current in photoelectrochemical (
 ) and dye-sensitized solar cells. Chem Commun. 2005;34:4351–4353. doi: 10.1039/b505718c. [DOI] [PubMed] [Google Scholar]
) and dye-sensitized solar cells. Chem Commun. 2005;34:4351–4353. doi: 10.1039/b505718c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.