

Abstract
Photodriving the activity of water-oxidation catalysts is a critical step toward generating fuel from sunlight. The design of a system with optimal energetics and kinetics requires a mechanistic understanding of the single-electron transfer events in catalyst activation. To this end, we report here the synthesis and photophysical characterization of two covalently bound chromophore-catalyst electron transfer dyads, in which the dyes are derivatives of the strong photooxidant perylene-3,4:9,10-bis(dicarboximide) (PDI) and the molecular catalyst is the Cp∗Ir(ppy)Cl metal complex, where ppy = 2-phenylpyridine. Photoexcitation of the PDI in each dyad results in reduction of the chromophore to PDI•- in less than 10 ps, a process that outcompetes any generation of 3∗PDI by spin-orbit-induced intersystem crossing. Biexponential charge recombination largely to the PDI-Ir(III) ground state is suggestive of multiple populations of the PDI•--Ir(IV) ion-pair, whose relative abundance varies with solvent polarity. Electrochemical studies of the dyads show strong irreversible oxidation current similar to that seen for model catalysts, indicating that the catalytic integrity of the metal complex is maintained upon attachment to the high molecular weight photosensitizer.
Keywords: photoinduced electron transfer, solar fuels, ultrafast optical spectroscopy, water oxidation
Artificial photosynthetic systems for solar fuels generation must integrate the functions of light harvesting, charge separation, and catalysis, with water as the source of electrons for reductive fuel-forming chemistry (1–5). The design of a light-driven water-splitting system based on molecular catalysts requires a fundamental understanding of the individual electron transfer steps involved in multielectron catalyst activation. To investigate the energetic and kinetic demands of coupling photodriven charge separation and catalysis, many research groups have studied the photophysical properties of covalently linked redox-active organic dyes and transition metal complexes (6–16). In several cases, electron transfer to or from the metal has been observed and characterized, though energy transfer and intersystem crossing to the chromophore triplet state can be significant competing processes. In this work, we use derivatives of perylene-3,4∶9,10-bis(dicarboximide) (PDI) linked to an iridium complex of the type Cp∗Ir(N-C)X to demonstrate light-driven single-electron oxidation of a highly active molecular catalyst precursor for water oxidation.
Derivatives of PDI are useful organic chromophores for solar device applications (5, 17, 18). Their high molar absorptivity, stability, low cost, ease of synthetic manipulation, and often advantageous self-assembly properties have led to their use in molecular electronics and a variety of solar energy conversion systems (19). For solar fuels applications, the mild reduction potentials of PDI derivatives make their excited states powerful photooxidants, though there are only a few literature examples in which 1∗PDI is used to produce higher-valence states of a metal complex (8, 9, 16). Rybtchinski, Wasielewski, and coworkers have shown that upon selective photoexcitation of PDI in a PDI-(bpy)Ru(II)-PDI complex, in which the bpy ligand is bound to two PDI units through acetylide linkages at their aromatic cores, charge transfer generates PDI-Ru(III)-PDI•- in < 150 fs, and the vibrationally hot ion-pair exhibits fast relaxation (τ = 3.9 ps) and charge recombination (τ = 63 ps) (8). In a side-to-face ruthenium porphyrin/PDI assembly studied by Iengo, Scandola, Würthner, and coworkers, in which two Ru porphyrins are coordinated to pyridyl groups at the PDI imide positions, charge transfer (τ = 5.6 ps) and recombination (τ = 270 ps) are observed following PDI photoexcitation (16). Related work from the groups of Guldi, Torres, and Wasielewski has demonstrated electron transfer with a ruthenium phthalocyanine/PDI assembly in which the metal complexes are coordinated to the organic dye through pyridyloxy groups at its 1, 6, 7, and 12 positions (9).
PDI has a fluorescence quantum yield that is close to unity and thus a very low intrinsic triplet yield due to spin-orbit-induced intersystem crossing (SO-ISC) (20). Nevertheless, the competitiveness of heavy atom-induced SO-ISC is a concern for PDI/metal complex assemblies, and previous studies by the Castellano group of PDI linked to platinum(II) metal centers through acetylide groups at its 1-position have reported SO-ISC from 1∗PDI to 3∗PDI in 2–4 ps (7, 12). However, when Pd(II) is directly attached to the PDI aromatic core through metal-carbon σ-bonds, strong PDI fluorescence is observed and the yield of 3∗PDI is estimated to be only 6%, which is explained by the relatively weak coupling between the metal-based orbitals and the frontier π-orbitals of PDI (14). Also, Pd(II)-PDI and Pt(II)-PDI complexes in which the metal centers are bound through pyridyl groups at the PDI imide positions—through which the PDI frontier orbitals each have a nodal plane—have fluorescence quantum yields of 86% and 88%, respectively (15). Once again, the heavy-atom effect is diminished when the electronic coupling of the metal to the chromophore is weak.
The Cp∗Ir(N-C)X complex used in our studies, where (N-C) = 2-phenylpyridine and X = chloride, is a highly active catalyst precursor for water oxidation, with turnover frequencies on the order of 10 turnovers per minute and activity that continues over hours (21, 22). It catalyzes water oxidation both electrochemically and with cerium(IV) as a 1-electron oxidant, which suggests that catalyst activity could be driven with photoinduced electron transfer (23). In addition, the activity of the catalyst is homogeneous and not simply a precursor to catalytically active iridium oxides, as determined by piezoelectric gravimetry during water oxidation (24). The parent Cp∗Ir(ppy)Cl complex, which has an Ir(III) valence state, displays three irreversible waves in electrochemical studies at about 0.85, 1.4, and 1.8 V versus SCE. The 0.85 V oxidation of Cp∗Ir(ppy)Cl has been attributed to oxidation of the chloride-bound complex, which is followed by oxidation at higher potentials of a cationic solvent-bound species (23). In this work, we are using the 1∗PDI excited state to oxidize the chloride-bound complex, seeking to generate higher-valence states of the catalyst using light.
There has been one PDI/iridium complex dyad reported in the literature by Ortí and Gierschner, in which the metal is complexed by one PDI-linked 1,10-phenanthroline and two 2-phenylpyridine ligands (6, 25). Upon selective excitation of the PDI moiety, the dyad emits with a quantum yield of ΦF = 0.55, reduced from ΦF = 0.86 of the PDI model compound, a decrease which the authors attribute to SO-ISC induced by the iridium atom (6). The fluorescence lifetime of the 1∗PDI excited state is still long at 3.0 ns, which suggests that SO-ISC should not be competitive with fast electron transfer when an iridium complex is bound to PDI through an imide position.
We have prepared PDI/metal catalyst dyads 1 and 2 from ligands 3 and 4, in which PDI is covalently appended to the phenylpyridine ligand (Fig. 1). Two different PDI structures are considered: The first includes 3,5-di-t-butylphenoxy groups at the 1,7 “bay region” positions, while the second generation of this compound uses 3,5-bis(trifluoromethyl)phenyl groups to make the PDI a more powerful photooxidant, as well as a 3-pentyl tail at one imide position to enhance solubility in highly polar organic solvents relevant to solar fuels applications, like acetonitrile or propylene carbonate (26). While both PDI variants undergo electron transfer with the iridium complex, the more highly oxidizing 1∗PDI excited state in 2 will likely be necessary to access the putative higher-valent iridium(V) state implicated in the proposed catalytic cycle (21).
Fig. 1.

PDI/Ir complex dyads 1 and 2 and their respective ligands 3 and 4.
Results and Discussion
Synthesis and Characterization.
The syntheses of ligands 3 and 4 and dyads 1 and 2 are described in the SI Text. Briefly, each PDI-based ligand was prepared by condensing the corresponding imide anhydride with an amino-functionalized 2-phenylpyridine unit. Metalation was accomplished by stirring each ligand with the iridium pentamethylcyclopentadienyl dichloride dimer in CH2Cl2 in the presence of sodium acetate trihydrate (27). The steady-state absorption and emission spectra of each ligand (Fig. S1) resemble those of most PDI compounds (18). Upon metalation, the absorption spectrum of the chromophore is only slightly perturbed; λmax of 3 shifts from 551 to 553 nm, while λmax of 4 shifts from 541 to 544 nm (Fig. S1). Absorption to the blue of 450 nm is enhanced in both 1 and 2 due to absorption by the metal complex (Fig. S1). The spectrum of each dyad is a sum of their individual components, indicating that the Ir(III) complex and the PDI core are not strongly coupled. Importantly, the PDI unit can be selectively excited with visible wavelengths greater than 450 nm. The solubility of the ligands and their respective metal complexes are comparable: 1 and 3 are very soluble in toluene, CH2Cl2, and benzonitrile, but they are less soluble in butyronitrile and only 3 is soluble in acetonitrile, and in these two latter solvents the UV-Vis spectra show evidence of PDI aggregation (28, 29). Dyad 2 and ligand 4 are soluble in the entire range of solvents, as a result of the 3-pentyl tail at the imide that hinders π-π stacking of PDI aromatic cores (30) and the direct attachment of the phenyl side-groups, which may cause even greater distortion of the PDI core from planarity relative to the phenoxy-substituted dyes (31, 32).
Using cyclic voltammetry, the two reductions of the PDI chromophores are reversible, whereas the oxidation waves of the Ir complex are irreversible, as has been previously reported (22). The redox potentials of each dyad were thus determined with differential pulse voltammetry (Table 1), and the potentials of the PDI and the metal complex components remain unchanged relative to their potentials as individual molecules. The bis(CF3)phenyl-substituted PDI in 2 is 220 mV easier to reduce than the phenoxy-substituted PDI in 1, without a significant difference in the energy of 1∗PDI, enhancing the driving force for electron transfer from the iridium complex to the chromophore.
Table 1.
Redox potentials (V vs. SCE) of the PDI/Ir complex dyads
| Dyad | Eox (IrIII/IV) | Ered (PDI0/-) | Ered (PDI-/2-) |
| 1 | 0.80 | −0.57 | −0.79 |
| 2 | 0.80 | −0.35 | −0.61 |
Electron Transfer Energetics.
The ion-pair energies within 1 and 2, shown schematically in Fig. 2, were determined using the expression developed by Weller (33), as described in the SI Text. To determine the free energy for charge separation (ΔGCS) in each dyad, the lowest-lying singlet excited state energy of each PDI ligand (ES)—estimated by averaging the energies of the lowest energy transition in the absorption spectrum and highest energy transition in the emission spectrum—was subtracted from ΔGIP. Table 2 summarizes this data for dyads 1 and 2. In each solvent considered, electron transfer from the iridium complex to PDI should occur upon photoexcitation of the chromophore.
Fig. 2.

Energy level diagrams for dyads 1 (A) and 2 (B), featuring the calculated ion-pair energies in different solvents.
Table 2.
Singlet excited state energies, ion-pair energies and electron transfer driving forces for the PDI/Ir complex dyads
| Dyad | Ligand λabs * (nm) | Ligand λems * (nm) | ES * (eV) | Solvent | ΔGIP (eV) | ΔGCS (eV) |
| 1 | 551 | 576 | 2.20 | toluene | 1.75 | −0.45 |
| CH2Cl2 | 1.40 | −0.80 | ||||
| 2 | 541 | 579 | 2.22 | toluene | 1.53 | −0.69 |
| CH2Cl2 | 1.18 | −1.04 | ||||
| PhCN | 1.10 | −1.12 | ||||
| CH3CN | 1.09 | −1.13 |
*Measured in CH2Cl2.
Time-Resolved Spectroscopy.
Femtosecond transient absorption measurements were performed on 1–4 in a range of solvents of varying polarity using 150 fs, 550 nm pulses to selectively excite the PDI. Ligand 3 demonstrates 1∗PDI excited state relaxation to the ground state with τ = 3.3 ns and 3.8 ns in toluene and CH2Cl2, respectively, as determined by the decay kinetics of the 1∗PDI signal at 700 nm (Fig. 3 and Fig. S2). There is a shorter decay component in each solvent (6.7 ps in toluene and 4.3 ps in CH2Cl2) that accounts for 15% of the signal amplitude at 700 nm, and we attribute this to vibrational relaxation of hot 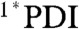 , which is supported by the red-shift of the stimulated emission feature at about 620 nm. For dyad 1 in both solvents, the initial
, which is supported by the red-shift of the stimulated emission feature at about 620 nm. For dyad 1 in both solvents, the initial 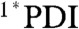 signal red-shifts into a peak at 725 nm (Fig. 3 and Fig. S2), which is the absorption of PDI•- as has been observed in numerous donor-acceptor systems containing 1,7-diphenoxy-PDI derivatives (34, 35). Charge separation, as also evidenced by the fast disappearance of stimulated emission at 620 nm, occurs with τ = 1.8 ps and 3.6 ps in toluene and CH2Cl2, respectively. These time scales of charge separation match those found by monitoring the fluorescence of 1 at 585 nm (Table 3 and Fig. S3).
signal red-shifts into a peak at 725 nm (Fig. 3 and Fig. S2), which is the absorption of PDI•- as has been observed in numerous donor-acceptor systems containing 1,7-diphenoxy-PDI derivatives (34, 35). Charge separation, as also evidenced by the fast disappearance of stimulated emission at 620 nm, occurs with τ = 1.8 ps and 3.6 ps in toluene and CH2Cl2, respectively. These time scales of charge separation match those found by monitoring the fluorescence of 1 at 585 nm (Table 3 and Fig. S3).
Fig. 3.

Femtosecond transient absorption spectra of 1,7-diphenoxy-PDI compounds in CH2Cl2 with 550 nm laser excitation. (A) Spectra of ligand 3 (inset: kinetic trace at 700 nm, with a nonlinear least-squares fit). (B) Spectra of dyad 1 (inset: kinetic trace at 725 nm, with a nonlinear least-squares fit).
Table 3.
Photophysical parameters for electron transfer in PDI/Ir complex dyads
| Dyad | Solvent | τCS* (ps) | τCS † (ps) | τCR * (ps) |
| 1 | toluene | 1.8 ± 0.2 | 1.6 ± 0.1 | 4.1 ± 0.9 (54%) 21 ± 1 (46%) |
| CH2Cl2 | 3.6 ± 0.2 | 3.9 ± 0.2 | 13 ± 1 (78%) | |
| 65 ± 5 (22%) | ||||
| 2 | toluene | 0.46 ± 0.01 | 0.20 ± 0.05 (75%) 1.3 ± 0.3 (25%) | 15 ± 1 (46%) 56 ± 2 (54%) |
| CH2Cl2 | 1.4 ± 0.1 | 1.5 ± 0.1 | 6.2 ± 0.1 (80%) | |
| 83 ± 2 (20%) | ||||
| PhCN | 1.5 ± 0.1 | 1.7 ± 0.2 | 11 ± 1 (88%) | |
| 125 ± 10 (12%) | ||||
| CH3CN | 1.5 ± 0.1 | 2.2 ± 0.2 | 5.3 ± 0.2 (83%) | |
| 72 ± 3 (17%) |
*Determined by transient absorption.
†Determined by time-resolved fluorescence.
The decay of PDI•- is biexponential in both solvents, as noted in Table 3. To help distinguish the spectral features of the species associated with each charge recombination time scale, the transient spectra were subjected to singular value decomposition (SVD) and global fitting using a sum of exponentials to obtain the principal kinetic components and their associated spectra. The two decay-associated spectra in CH2Cl2 (Fig. 4) have time scales of 14 ps (81%) and 75 ps (19%), which correlate with the two time scales of charge recombination from the transient absorption kinetics. Fig. 4 shows that the loss of stimulated emission and the rise of PDI•-, features that match previously reported spectra for PDI derivatives (29, 34, 35), are associated with τCS = 3.6 ps, while the recovery of the ground state bleach and decay of PDI•- are associated with both of the longer time scales (τCR = 14 ps and 75 ps), implying that two different ion-pair populations are generated upon charge separation and recombine with different rates. Similar results are obtained from SVD and global fitting of the data in toluene (Fig. S3). The possibility of two structural conformers of each of the starting complexes is unlikely, since the 1H and 13C NMR spectra of the dyads are each consistent with a singular structure, and the observed charge separation kinetics are monoexponential. Structural changes resulting in two ion-pair populations may follow the fast electron transfer and are more likely to occur at the Ir complex, since most donor-acceptor studies with 1,7-diphenoxy-PDI involve a single time constant for charge recombination (35–37). Yet, because the ground state of this Ir(III) complex and the Ir(IV) state generated by electron transfer are spectroscopically silent in the 430–820 nm visible probe range of the transient absorption experiment, it is difficult to draw definitive conclusions about geometry changes at the metal center. One possibility to consider is dissociation of the chloride ligand in some fraction of the molecules upon oxidation to Ir(IV), as changes in the electronic structure of the metal complex would likely affect ligand binding. The Jahn-Teller d5 configuration of the iridium(IV) complex in the ion-pair state would have unequal filling of degenerate orbitals, which could lead to enhanced ligand exchange rates. Sykes and coworkers have reported that [Ir(H2O)6]3+ is an inert ion, but oxidation to Ir(IV) accelerates ligand exchange and the formation of new complexes (38, 39). Furthermore, rates of water substitution are enhanced with Cp∗ as a ligand, as the ligand exchange rate for [Cp∗Ir(H2O)3]2+ is enhanced by up to 14 orders of magnitude relative to [Ir(H2O)6]3+ (40). There are no changes to the UV-Vis spectrum of the dyad following each laser experiment, strongly suggesting there is no photodecomposition. This observation is consistent with previous electrochemical studies with the Cp∗Ir(ppy)Cl model catalyst, which indicate that equilibration of chloride- and solvent-bound Ir(III) is relatively fast. Thus, following any ligand changes upon oxidation to the Ir(IV) transient, the initial chloride ligation can be restored (23).
Fig. 4.

Spectra associated with the kinetic components obtained by global analysis of the transient absorption spectra of 1 in CH2Cl2.
The transient spectra of 1 in toluene (Fig. S2) show a population of PDI triplet at long times, with a yield of approximately 8% (14, 20). The yield is smaller in CH2Cl2—a modest 2%—which is reasonable in the context of electron transfer theory given that the ion-pair energy lies higher than the PDI triplet energy in toluene than it does in CH2Cl2. Nanosecond transient absorption, performed by using 7 ns, 570 nm pulses to selectively excite the PDI, confirms the presence of a long-lived PDI triplet (> 45 μs) in toluene (Fig. S4).
Focusing on the bis(CF3)phenyl compounds, PDI ligand 4 demonstrates 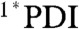 relaxation to the ground state in toluene, CH2Cl2, and benzonitrile with τ ∼ 4 ns (Fig. 5 and Fig. S5). The
relaxation to the ground state in toluene, CH2Cl2, and benzonitrile with τ ∼ 4 ns (Fig. 5 and Fig. S5). The 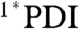 signal is red-shifted relative to the excited state of phenoxy PDI derivatives, appearing as a peak at 745 nm in CH2Cl2 and as a broad shoulder in that region in toluene and benzonitrile. As with the 1,7-diphenoxy-PDI ligand, there is a fast decay component that ranges from 4.5–13 ps (16–19%) in these solvents, attributed to vibrational relaxation. For dyad 2 in toluene, CH2Cl2, benzonitrile, or acetonitrile, the initial
signal is red-shifted relative to the excited state of phenoxy PDI derivatives, appearing as a peak at 745 nm in CH2Cl2 and as a broad shoulder in that region in toluene and benzonitrile. As with the 1,7-diphenoxy-PDI ligand, there is a fast decay component that ranges from 4.5–13 ps (16–19%) in these solvents, attributed to vibrational relaxation. For dyad 2 in toluene, CH2Cl2, benzonitrile, or acetonitrile, the initial 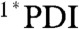 signal red-shifts into a peak at 720–730 nm, consistent with the results from dyad 1 and indicating electron transfer to generate PDI•- (Fig. 5 and Fig. S5). The UV-Vis absorption spectra of an electrochemically reduced bis(CF3)phenyl PDI model compound (Fig. S6) confirm the assignment of this signal. Charge separation, which again is evidenced by the fast disappearance of stimulated emission, occurs with τ = 0.46 ps in toluene, 1.4 ps in CH2Cl2, and 1.5 ps in the nitrile solvents, values determined by fitting the kinetic data at 725 nm. These time scales are faster than the corresponding values for dyad 1, which is expected given the additional 220 mV of driving force conferred by the 3,5-bis(trifluoromethyl)phenyl PDI side-groups. The charge separation time scales correlate well with those found by monitoring the fluorescence of 2 at 595 nm (Table 3 and Fig. S7), with the exception of a 200 fs fluorescence decay component in toluene, most likely due to S2 → S1 internal conversion in PDI given that the fluorescence data is obtained using 415 nm excitation.
signal red-shifts into a peak at 720–730 nm, consistent with the results from dyad 1 and indicating electron transfer to generate PDI•- (Fig. 5 and Fig. S5). The UV-Vis absorption spectra of an electrochemically reduced bis(CF3)phenyl PDI model compound (Fig. S6) confirm the assignment of this signal. Charge separation, which again is evidenced by the fast disappearance of stimulated emission, occurs with τ = 0.46 ps in toluene, 1.4 ps in CH2Cl2, and 1.5 ps in the nitrile solvents, values determined by fitting the kinetic data at 725 nm. These time scales are faster than the corresponding values for dyad 1, which is expected given the additional 220 mV of driving force conferred by the 3,5-bis(trifluoromethyl)phenyl PDI side-groups. The charge separation time scales correlate well with those found by monitoring the fluorescence of 2 at 595 nm (Table 3 and Fig. S7), with the exception of a 200 fs fluorescence decay component in toluene, most likely due to S2 → S1 internal conversion in PDI given that the fluorescence data is obtained using 415 nm excitation.
Fig. 5.

Femtosecond transient absorption spectra of bis(CF3)phenyl-PDI compounds in CH2Cl2 with 550 nm laser excitation. (A) Spectra of ligand 4 (inset: kinetic trace at 745 nm, with a nonlinear least-squares fit). (B) Spectra of dyad 2 (inset: kinetic trace at 725 nm, with a nonlinear least-squares fit).
The PDI•- decay is biexponential in all four solvents as noted in Table 3, mirroring the behavior of dyad 1. These transient spectra were also analyzed with SVD and global fitting, and the principal kinetic components correlate with the values in Table 3 (Fig. S8). The spectra associated with PDI•- decay, with features corresponding to radical anion decay and recovery of ground state bleach, again suggest the presence of two different ion-pair populations, each with its own recombination rate. The same process that results in two ion-pairs in 1 appears to be operative in 2, since the amplitudes of the two decay components in both toluene and CH2Cl2 are similar for 1 and 2. If chloride loss is creating the additional ion-pair population, then the stability and potential abundance of the effectively dicationic Ir(IV) metal center would be more significantly influenced by solvent polarity, relative to the Ir(IV) center with bound chloride. This difference is reflected in the relative contributions of the two recombination time scales, which vary strongly with solvent: the shorter time scale becomes more prominent as solvent polarity increases from toluene to CH2Cl2 to nitriles. This variation in time scale contributions also argues against multiple ground state conformers as the source of different ion-pair populations. Triplet PDI is again generated to a larger extent in toluene than in the more polar solvents, consistent with the greater driving force in toluene for recombination to 3∗PDI.
Catalytic Activity of PDI/Ir Complex Dyads.
Having thus demonstrated that 1∗PDI can oxidize the Ir(III) complex in a covalent system, we were next interested in confirming that the catalyst moiety of these dyads is still competent for water-oxidizing chemistry. Because oxidation of the chloride ligand to chlorine or hypochlorite is possible under oxidizing conditions, which can be an interfering signal in oxygen-evolution studies (21), dyad 1 was studied as a solvent-bound derivative, with acetonitrile displacing the chloride. Ligand substitution was accomplished by stirring dyad 1 in acetonitrile with AgPF6 (41), generating the now positively charged dyad as a PF6 salt (1-CH3CN). Cyclic voltammetry was used to investigate the oxidation chemistry of the dyad in both the absence and presence of water. As a comparison, we investigated the properties of the analogous control catalyst Cp∗Ir(ppy)OTf (22), which does not bear the light-absorbing PDI moiety.
As recently reported by the Meyer group, we used propylene carbonate as the oxidation-resistant solvent for the electrochemical investigation (26). The solvent-bound dyad and model catalyst are both soluble in propylene carbonate, and the solution can accommodate several mass percent of added water. The cyclic voltammograms of both compounds in propylene carbonate are unremarkable, showing low currents up to 2 V versus NHE in the absence of water (Fig. 6 and Fig. S9). In the case of Cp∗Ir(ppy)OTf, there is little above-background current in the absence of water. However, upon addition of water, a strong irreversible oxidation current is observed for both 1-CH3CN and Cp∗Ir(ppy)OTf, consistent with evolution of oxygen* (Fig. 6 and Fig. S9). Analogous experiments with the chloro-bound compound show a similar above-background oxidative current.
Fig. 6.

Cyclic voltammograms of solvent-bound dyad 1-CH3CN in (A) propylene carbonate and (B) propylene carbonate with 6% water by volume.
Recently, we reported a method for probing the ambiguity between homogeneous and heterogeneous water-oxidation catalysis by use of an electrochemical quartz crystal nanobalance (EQCN) (24). In this method, piezoelectric gravimetry is used to monitor the electrode mass in real time during catalyst oxidation. If a layer of catalytically active iridium oxide is deposited, the EQCN will register this as an increase in electrode mass. If the catalyst remains in solution, there will be no such mass deposition. In order to study the properties of Cp∗Ir(ppy)OTf by EQCN, we have now extended our work to the propylene carbonate/water mixture used for the electrochemistry as described above. Although there is significant above-background current ascribable to catalysis (Fig. S9), there is no corresponding increase in electrode mass. Thus, we feel that we can confidently ascribe the oxidation of Cp∗Ir(ppy)OTf—and by extension, the solvent-bound PDI/Ir complex dyad—to be a solution-based process.
Conclusion
In covalently bound chromophore-catalyst dyads 1 and 2, electron transfer to 1∗PDI occurs in less than 10 ps upon excitation of the dye, with unity quantum yields. The rates of electron transfer are increased when bis(CF3)phenyl groups are appended to the bay positions of PDI, which make the chromophore a stronger photooxidant and more soluble in polar, water-miscible organic solvents relevant to the field of solar fuels. Charge recombination is also relatively rapid, though biexponential decay of PDI•- suggests two different ion-pair populations, perhaps a result of structural changes at the iridium center upon oxidation. Electrochemical experiments with dyad 1 and its solvent-bound derivative show oxidative current consistent with catalytic water oxidation by the metal complex, suggesting that the attachment of the chromophore does not preclude catalytic activity. Current research directions include lengthening the charge-separated state lifetime to be able to characterize high-valent states of iridium using time-resolved electron paramagnetic resonance spectroscopy, as well as studying the electron transfer behavior of dye-catalyst dyads bound to semiconductors, a prerequisite to building a photoelectrochemical water-splitting cell.
Materials and Methods
Detailed synthetic procedures and characterization of all compounds, including dyads 1 and 2 and ligands 3 and 4, are described in the SI Text. All solvents were spectroscopic grade and dried using a Glass Contour solvent system or distilled prior to use.
Electrochemical measurements for basic characterization were performed using a CH Instruments Model 622 electrochemical workstation. All measurements were performed under an argon atmosphere in benzonitrile containing 0.1 M tetra-n-butylammonium hexafluorophosphate (TBAPF6). TBAPF6 was recrystallized twice from ethanol prior to use. A 1.0 mm diameter platinum disk, platinum wire, and silver wire were employed as working, auxiliary, and pseudoreference electrodes, respectively. The ferrocene/ferrocenium redox couple (Fc/Fc+, 0.47 V versus SCE) was used as an internal reference for all measurements. Spectroelectrochemistry was performed using a platinum mesh working electrode, a platinum wire counter electrode, and silver wire pseudoelectrode in a 2-mm cell.
Cyclic voltammetry (CV) measurements to assess catalytic activity were made on a Princeton Applied Research Versastat 4–400 or model 2273 potentiostat/galvanostat using a standard three-electrode configuration. A basal-plane graphite (surface area: 0.09 cm2) or gold (surface area: 0.017 cm2) electrode was used as the working electrode to minimize background oxidation current. The preparation and treatment of the basal-plane graphite electrode have been described previously (21). A platinum wire was used as the counter electrode, and a Ag/AgCl electrode (Bioanalytical Systems, Inc.) was used as the reference (Ag/AgCl versus NHE: +197 mV). Experiments were carried out in the mixed solvent solution containing 0.1 M TBAPF6 as the supporting electrolyte. The instrumentation and methodology for simultaneous CV and piezoelectric gravimetry measurements have been described previously (24) and are included in the SI Text.
Steady-state absorption spectroscopy was performed using a Shimadzu (UV-1801) spectrophotometer with a 1 cm quartz cuvette, and fluorescence measurements were performed using a Photon Technology International photon-counting spectrofluorimeter in a right angle configuration with a 1 cm quartz cuvette.
Femtosecond transient absorption measurements were made using the 550 nm, 110 fs output of an optical parametric amplifier using techniques described earlier (32). Samples were prepared with an optical density between 0.65 and 0.75 at 550 nm in a 2 mm cuvette, and they were irradiated with 1.0 μJ/pulse focused to a 200-μm spot. The total instrument response function for the pump-probe experiments was 150 fs. Transient absorption kinetics were fit to a sum of exponentials convolved with a Gaussian instrument function by using Levenberg-Marquardt least-squares fitting. The three-dimensional datasets of ΔA versus time and wavelength were subjected to singular value decomposition and global fitting to obtain the kinetic time constants and their associated spectra using Surface Xplorer software (Ultrafast Systems LLC, Sarasota, FL).
The sample for nanosecond transient absorption spectroscopy was prepared in a 10 mm path length quartz cuvette and degassed with five freeze-pump-thaw cycles. The sample was excited with 7 ns, 2.0 mJ, 570 nm laser pulses using the frequency-tripled output of a Continuum Precision II 8000 Nd:yttrium-aluminum-garnet laser pumping a Continuum Panther optical parametric oscillator, a setup described earlier (10). The excitation pulse was focused to an 8 mm diameter spot and matched to the diameter of the probe pulse. The total instrument response function is 7 ns and is determined primarily by the laser pulse duration.
Pump pulses at 415 nm for femtosecond time-resolved fluorescence experiments were generated using a home-built cavity-dumped Ti:sapphire laser system (center wavelength, 830 nm; spectral width, 55 nm; pulse duration, 25 fs; repetition rate, 820 KHz) followed by frequency doubling in a 200 μm thick lithium triborate crystal. The remaining fundamental served as the gate pulses for the fluorescence upconversion experiment, which delivered sub-50 fs time resolution by utilizing a noncollinear sum frequency generation scheme, which has been described in detail elsewhere (42, 43).
Supplementary Material
Acknowledgments.
This material is based upon work supported as part of the ANSER Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001059. Further funding from the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through grant DE-FG02-84ER13297 (R.H.C. and N.D.S.; synthesis and characterization of Cp*Ir(ppy)OTf) and the U.S. National Science Foundation through grant CHE-1110942 (F.D.; electrochemical quartz crystal nanobalance studies) is gratefully acknowledged. S.W.E. was supported by the IMI program of the National Science Foundation under award no. DMR-08-43962. M.T.V. acknowledges support from an NSF Graduate Research Fellowship and a National Defense Science and Engineering Graduate Fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1202075109/-/DCSupplemental.
*Although we were interested in observing the evolution of oxygen with a Clark-type electrode as we have done previously, the organic solvent used in the electrochemical investigation is not compatible with the membrane on the Clark electrode.
References
- 1.Alstrum-Acevedo JH, Brennaman MK, Meyer TJ. Chemical approaches to artificial photosynthesis 2. Inorg Chem. 2005;44:6802–6827. doi: 10.1021/ic050904r. [DOI] [PubMed] [Google Scholar]
- 2.Gust D, Moore TA, Moore AL. Solar fuels via artificial photosynthesis. Acc Chem Res. 2009;42:1890–1898. doi: 10.1021/ar900209b. [DOI] [PubMed] [Google Scholar]
- 3.Meyer TJ. Chemical approaches to artificial photosynthesis. Acc Chem Res. 1989;22:163–170. [Google Scholar]
- 4.Song WJ, et al. Making solar fuels by artificial photosynthesis. Pure Appl Chem. 2011;83:749–768. [Google Scholar]
- 5.Wasielewski MR. Energy, charge, and spin transport in molecules and self-assembled nanostructures inspired by photosynthesis. J Org Chem. 2006;71:5051–5066. doi: 10.1021/jo060225d. [DOI] [PubMed] [Google Scholar]
- 6.Costa RD, et al. A deep-red-emitting perylenediimide-iridium-complex dyad: Following the photophysical deactivation pathways. J Phys Chem C Nanomater Interfaces. 2009;113:19292–19297. [Google Scholar]
- 7.Danilov EO, Rachford AA, Goeb S, Castellano FN. Evolution of the triplet excited state in Pt(II) perylenediimides. J Phys Chem A. 2009;113:5763–5768. doi: 10.1021/jp9012762. [DOI] [PubMed] [Google Scholar]
- 8.Gunderson VL, et al. Photoinduced singlet charge transfer in a ruthenium(II) perylene-3,4∶9,10-bis(dicarboximide) complex. J Phys Chem B. 2011;115:7533–7540. doi: 10.1021/jp2016374. [DOI] [PubMed] [Google Scholar]
- 9.Jimenez AJ, et al. Synthesis, characterization, and photoinduced energy and electron transfer in a supramolecular tetrakis (ruthenium(II) phthalocyanine) perylenediimide pentad. Chem Eur J. 2011;17:5024–5032. doi: 10.1002/chem.201002963. [DOI] [PubMed] [Google Scholar]
- 10.Poddutoori P, et al. Photoinitiated multistep charge separation in ferrocene-zinc porphyrin-diiron hydrogenase model complex triads. Energy Environ Sci. 2011;4:2441–2450. [Google Scholar]
- 11.Qvortrup K, et al. Perylenediimide-metal ion dyads for photo-induced electron transfer. Chem Commun. 2008:1986–1988. doi: 10.1039/b719243f. [DOI] [PubMed] [Google Scholar]
- 12.Rachford AA, Goeb S, Castellano FN. Accessing the triplet excited state in perylenediimides. J Am Chem Soc. 2008;130:2766–2767. doi: 10.1021/ja800333y. [DOI] [PubMed] [Google Scholar]
- 13.Samuel APS, Co DT, Stern CL, Wasielewski MR. Ultrafast photodriven intramolecular electron transfer from a zinc porphyrin to a readily reduced diiron hydrogenase model complex. J Am Chem Soc. 2010;132:8813–8815. doi: 10.1021/ja100016v. [DOI] [PubMed] [Google Scholar]
- 14.Weissman H, et al. Palladium complexes of perylene diimides: Strong fluorescence despite direct attachment of late transition metals to organic dyes. Inorg Chem. 2007;46:4790–4792. doi: 10.1021/ic700539b. [DOI] [PubMed] [Google Scholar]
- 15.Wurthner F, You CC, Saha-Moller CR. Metallosupramolecular squares: From structure to function. Chem Soc Rev. 2004;33:133–146. doi: 10.1039/b300512g. [DOI] [PubMed] [Google Scholar]
- 16.Prodi A, et al. Wavelength-dependent electron and energy transfer pathways in a side-to-face ruthenium porphyrin/perylene bisimide assembly. J Am Chem Soc. 2005;127:1454–1462. doi: 10.1021/ja045379u. [DOI] [PubMed] [Google Scholar]
- 17.Jones BA, et al. High-mobility air-stable n-type semiconductors with processing versatility: Dicyanoperylene-3,4 : 9,10-bis(dicarboximides) Angew Chem Int Ed. 2004;43:6363–6366. doi: 10.1002/anie.200461324. [DOI] [PubMed] [Google Scholar]
- 18.Wurthner F. Perylene bisimide dyes as versatile building blocks for functional supramolecular architectures. Chem Commun. 2004:1564–1579. doi: 10.1039/b401630k. [DOI] [PubMed] [Google Scholar]
- 19.Huang C, Barlow S, Marder SR. Perylene-3,4,9,10-tetracarboxylic acid diimides: Synthesis, physical properties, and use in organic electronics. J Org Chem. 2011;76:2386–2407. doi: 10.1021/jo2001963. [DOI] [PubMed] [Google Scholar]
- 20.Ford WE, Kamat PV. Photochemistry of 3,4,9,10-perylenetetracarboxylic dianhydride dyes 3. Singlet and triplet excited-state properties of the bis(2,5-di-tert-butylphenyl)imide derivative. J Phys Chem. 1987;91:6373–6380. [Google Scholar]
- 21.Blakemore JD, et al. Half-sandwich iridium complexes for homogeneous water-oxidation catalysis. J Am Chem Soc. 2010;132:16017–16029. doi: 10.1021/ja104775j. [DOI] [PubMed] [Google Scholar]
- 22.Hull JF, et al. Highly active and robust Cp∗ iridium complexes for catalytic water oxidation. J Am Chem Soc. 2009;131:8730–8731. doi: 10.1021/ja901270f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brewster TP, et al. An iridium(IV) species, [Cp∗Ir(NHC)Cl]+, related to a water-oxidation catalyst. Organometallics. 2011;30:965–973. [Google Scholar]
- 24.Schley ND, et al. Distinguishing homogeneous from heterogeneous catalysis in electrode-driven water oxidation with molecular iridium complexes. J Am Chem Soc. 2011;133:10473–10481. doi: 10.1021/ja2004522. [DOI] [PubMed] [Google Scholar]
- 25.Costa RD, et al. Efficient deep-red light-emitting electrochemical cells based on a perylenediimide-iridium-complex dyad. Chem Commun. 2009:3886–3888. doi: 10.1039/b905367k. [DOI] [PubMed] [Google Scholar]
- 26.Chen ZF, et al. Nonaqueous catalytic water oxidation. J Am Chem Soc. 2010;132:17670–17673. doi: 10.1021/ja107347n. [DOI] [PubMed] [Google Scholar]
- 27.Boutadla Y, Al-Duaij O, Davies DL, Griffith GA, Singh K. Mechanistic study of acetate-assisted C-H activation of 2-substituted pyridines with [MCl2Cp∗]2 (M = Rh, Ir) and [RuCl2(p-cymene)]2. Organometallics. 2009;28:433–440. [Google Scholar]
- 28.Ahrens MJ, et al. Self-assembly of supramolecular light-harvesting arrays from covalent multi-chromophore perylene-3,4∶9,10-bis(dicarboximide) building blocks. J Am Chem Soc. 2004;126:8284–8294. doi: 10.1021/ja039820c. [DOI] [PubMed] [Google Scholar]
- 29.Giaimo JM, et al. Excited singlet states of covalently bound, cofacial dimers and trimers of perylene-3,4 : 9,10-bis(dicarboximide)s. J Phys Chem A. 2008;112:2322–2330. doi: 10.1021/jp710847q. [DOI] [PubMed] [Google Scholar]
- 30.Rajasingh P, Cohen R, Shirman E, Shimon LJW, Rybtchinski B. Selective bromination of perylene diimides under mild conditions. J Org Chem. 2007;72:5973–5979. doi: 10.1021/jo070367n. [DOI] [PubMed] [Google Scholar]
- 31.Sivalmurugan V, et al. Synthesis and Photophysical Properties of Glass-Forming Bay-Substituted Perylenediimide Derivatives. J Phys Chem B. 2010;114:1782–1789. doi: 10.1021/jp907697f. [DOI] [PubMed] [Google Scholar]
- 32.Ramanan C, Smeigh AL, Anthony JE, Marks TJ, Wasielewski MR. Competition between singlet fission and charge separation in solution-processed blend films of 6,13-bis(triisopropylsilylethynyl)-pentacene with sterically-encumbered perylene-3,4∶9,10-bis(dicarboximide)s. J Am Chem Soc. 2012;134:386–397. doi: 10.1021/ja2080482. [DOI] [PubMed] [Google Scholar]
- 33.Weller A. Photoinduced electron-transfer in solution—Exciplex and radical ion-pair formation free enthalpies and their solvent dependence. Z Phys Chem. 1982;133:93–98. [Google Scholar]
- 34.Colvin MT, et al. Competitive electron transfer and enhanced intersystem crossing in photoexcited covalent TEMPO-perylene-3,4∶9,10-bis(dicarboximide) dyads: Unusual spin polarization resulting from the radical-triplet interaction. J Phys Chem A. 2010;114:1741–1748. doi: 10.1021/jp909212c. [DOI] [PubMed] [Google Scholar]
- 35.Goldsmith RH, et al. Wire-like charge transport at near constant bridge energy through fluorene oligomers. Proc Natl Acad Sci USA. 2005;102:3540–3545. doi: 10.1073/pnas.0408940102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rybtchinski B, Sinks LE, Wasielewski MR. Photoinduced electron transfer in self-assembled dimers of 3-fold symmetric donor-acceptor molecules based on perylene-3,4 : 9,10-bis(dicarboximide) J Phys Chem A. 2004;108:7497–7505. [Google Scholar]
- 37.van der Boom T, et al. Charge transport in photofunctional nanoparticles self-assembled from zinc 5,10,15,20-tetrakis(perylenediimide)porphyrin building blocks. J Am Chem Soc. 2002;124:9582–9590. doi: 10.1021/ja026286k. [DOI] [PubMed] [Google Scholar]
- 38.Castillo-Blum SE, Richens DT, Sykes AG. Oxidation of hexaaquairidium(III) and related studies—preparation and properties of iridium(III), iridium(IV), and iridium(V) dimers as aqua ions. Inorg Chem. 1989;28:954–960. [Google Scholar]
- 39.Castillo-Blum SE, Sykes AG, Gamsjager H. Substitution inertness of [Ir(H2O)6]3+ Polyhedron. 1987;6:101–103. [Google Scholar]
-
40.Cayemittes S, et al. Mechanistic investigation on the water substitution in the η5-organometallic complexes
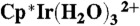 and
and 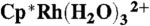 . Inorg Chem. 1999;38:4309–4316. [Google Scholar]
. Inorg Chem. 1999;38:4309–4316. [Google Scholar] - 41.Park-Gehrke LS, Freudenthal J, Kaminsky W, DiPasquale AG, Mayer JM. Synthesis and oxidation of Cp∗Ir(III) compounds: Functionalization of a Cp∗ methyl group. Dalton Trans. 2009:1972–1983. doi: 10.1039/b820839e. [DOI] [PubMed] [Google Scholar]
- 42.Kim CH, Joo T. Ultrafast time-resolved fluorescence by two photon absorption excitation. Opt Express. 2008;16:20742–20747. doi: 10.1364/oe.16.020742. [DOI] [PubMed] [Google Scholar]
- 43.Rhee H, Joo T. Noncollinear phase matching in fluorescence upconversion. Opt Lett. 2005;30:96–98. doi: 10.1364/ol.30.000096. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.