

Abstract
The DNA nucleotide thymidylate is synthesized by the enzyme thymidylate synthase, which catalyzes the reductive methylation of deoxyuridylate using the cofactor methylene-tetrahydrofolate (CH2H4folate). Most organisms, including humans, rely on the thyA- or TYMS-encoded classic thymidylate synthase, whereas, certain microorganisms, including all Rickettsia and other pathogens, use an alternative thyX-encoded flavin-dependent thymidylate synthase (FDTS). Although several crystal structures of FDTSs have been reported, the absence of a structure with folates limits understanding of the molecular mechanism and the scope of drug design for these enzymes. Here we present X-ray crystal structures of FDTS with several folate derivatives, which together with mutagenesis, kinetic analysis, and computer modeling shed light on the cofactor binding and function. The unique structural data will likely facilitate further elucidation of FDTSs’ mechanism and the design of structure-based inhibitors as potential leads to new antimicrobial drugs.
Keywords: methylene transfer, thymine biosynthesis, crystallography
Unlike other deoxynucleotides, thymidylate (2′-deoxythymidine-5′-monophosphate; dTMP) cannot be directly synthesized by a ribonucleotide reductase, and its de novo biosynthesis requires the enzyme thymidylate synthase (1–3). Thymidylate synthases use N5,N10-methylene-5,6,7,8-tetrahydrofolate (CH2H4folate) to reductively methylate dUMP, producing dTMP. Two general classes of thymidylate synthases are known (4). The enzymes encoded by the thyA gene (TYMS in humans and other mammals) are well studied and are known as classic thymidylate synthases (TSase) (1, 3). Certain bacteriological and archeological organisms have been found to lack any gene coding for such TSase, as well as dihydrofolate reductase and thymidine kinase (5), yet can survive in thymidine-deprived environments. This observation led to the identification of alternative flavin-dependent thymidylate synthases (FDTSs), which are encoded by the thyX gene and have no sequence or structure homology to classic TSase enzymes (2, 6, 7). Furthermore, multiple studies have identified key differences in the molecular mechanism of catalysis between FDTSs and classic TSases (2, 4, 8–21). These differences, along with the fact that the thyX gene is present in many human pathogens (e.g., bacteria causing Anthrax, Tuberculosis, Typhus, and other diseases), renders these flavo-enzymes as potential targets for rational inhibitor design, possibly affording compounds that might be effective antimicrobial drugs (11, 22–24).
The catalytic mechanism of classic TSase is well established and has facilitated in the identification of several inhibitors, some of which are in clinical use as anticancer drugs [e.g., 5-fluoro-uracil, tomudex (Raltitrexed)] (1, 25, 26); this is owed greatly to the large amount of chemical, kinetic, and structural information. An abundant amount of X-ray crystal structures have been solved for classic TSase, including ternary complexes with various combinations of substrate and folate cofactor, along with their analogs (1, 3). Studies of the FDTS reaction mechanism on the other hand, are still in their infancy, and critical structures of FDTS with any folate moiety have not been reported. Additionally, FDTS requires a nicotinamide cofactor to reduce the bound flavin, but a reactive binding site for nicotinamides has not been identified or proposed.
FDTS enzymes use a flavin adenine dinucleotide (FAD) prosthetic group to catalyze the redox chemistry, which can be divided into reductive and oxidative-half reactions. The FAD can be reduced to FADH2 by nicotinamides, ferrodoxin, and other small molecule reductants (e.g., dithionite). Although the reductive half-reaction is activated by dUMP the conversion of dUMP to dTMP occurs during the oxidative half-reaction (FADH2 →FAD) (8, 10, 14, 18, 27). In contrast to classic TSase, where the cofactor CH2H4folate provides both the H− and methylene, in the FDTS reaction the H− is provided by the FADH2 and CH2H4folate is used only as a source for the methylene moiety (18, 20, 28).
Several different mechanisms for methylene-transfer have been proposed, suggesting either a direct methylene transfer (Fig. 1A) from CH2H4folate to dUMP (4, 8, 10, 13–15, 18–20), or transfer of the methylene via an arginine residue (Arg174 in Thermatoga maritima FDTS; TmFDTS) before its delivery to dUMP (Fig. 1B) (16). These studies may have been complicated by the competing oxidase activity (FADH2 reaction with O2 to form H2O2) (10, 14, 27) and by other kinetic complexities, such as possible positive cooperativity (15). Published structures of FDTSs (7, 9, 15, 29, 30) have not been reported with folates, making further differentiation between these methylene transfer mechanisms difficult. Structural information regarding folate binding would not only provide a better understanding of the methylene transfer step, but will also present a new FDTS binding mode that might serve as a potential target for inhibitor design.
Fig. 1.

Mechanisms for FDTS catalyzed methylene transfer. (A) A recently proposed mechanism for FDTS enzymes involving direct transfer of the methylene between CH2H4folate and dUMP (4, 8). (B) A mechanism proposed for methylene transfer that involves an enzymatic arginine residue (16). Please note that the methylene transfer is the focus of this figure, and the methylene could in principle be transfered to the reduced dUMP (as drawn), to the oxidized dUMP as proposed in ref. 8, or to dUMP activated by Michael addition of Ser to C6, as originally suggested by ref. 16. R, 2′-deoxyribose-5′-phosphate; R′, (p-aminobenzoyl)-glutamate.
In this work we present X-ray crystal structures of TmFDTS in complex with FAD, dUMP, and either CH2H4folate, folinic acid, or Tomudex (Raltitrexed; an antifolate). The observed location for folate binding does not allow direct methylene transfer from CH2H4folate to dUMP and could be an ideal binding site for NADPH during the reductive-half reaction. The previously proposed mechanism where the methylene is transferred through an arginine residue was tested and eliminated by site-directed mutagenesis and kinetic analysis, warranting further examination of direct methylene transfer mechanisms. In the past we hypothesized that the isoalloxazine ring of the FAD may rotate away from dUMP, allowing room for CH2H4folate to bind in the active site (20), but it was not clear how this may happen. In the present work, we identified an additional FAD conformation when solving the structures of the E144R and R174K mutants of TmFDTS, indicating that the flavin can occupy more than one conformational state during catalysis. Using the active-site configuration of the quaternary complex, and FAD conformation observed in the E144R and R174K mutants, we modeled alternative binding options and identified a potential configuration where the CH2H4folate is bound adjacent to dUMP suitable for direct methylene transfer.
Results
Folate Binding Pocket.
In the FDTS-catalyzed reaction CH2H4folate provides the methylene moiety for the conversion of dUMP to dTMP (Fig. 1). Although several studies have published structures of FDTS from various organisms in complex with dUMP and dUMP analogs along with FAD (7, 9, 15, 21, 30), and even a nonreactive complex with NADP+ (29), a structure with CH2H4folate or any other folates has been an unrealized goal. Here we report the structures of the TmFDTS with the cofactor CH2H4folate and other folate derivatives in complex with FAD and dUMP (Fig. 2). We solved crystal structures of TmFDTS-FAD-dUMP complex with CH2H4folate at 1.39 Å resolution, an intermediate analog (leucovorin or folinic acid) at 1.50 Å resolution, and an antifolate (the anticancer drug Raltitrexed or tomudex) at 1.70 Å resolution (Fig. 3 and Tables S1–S3; see SI Materials and Methods for experimental details).
Fig. 2.
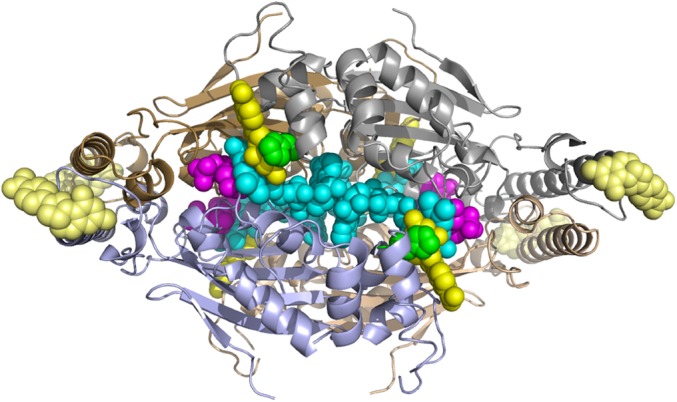
A view of the TmFDTS in complex with FAD, dUMP, and CH2H4folate. The CH2H4folate is in yellow, dUMP in magenta, FAD in cyan, and His53 in green spheres to emphasize the packing of these residues in the active site. Four other CH2H4folate per subunit (in lighter yellow, illustrating low-electron density) are found remote from the active site; these may play allosteric role, as discussed in the main text. PDB code 4GT9.
Fig. 3.

An active site view of crystal structures of TmFDTS in complex with FAD, dUMP, and folate derivatives. A view of the omit maps contoured at 3σ for CH2H4folate (A), folinic acid (B), and Raltitrexed (C). Ribbon drawings for the three protein chains constructing the active site (light gray, light blue, and wheat) and stick representation for FAD (cyan), dUMP (magenta), folate (yellow), and His53 (green). Note that the glutamate moiety of the tail is not defined in any of the crystal structures. The tail portion of the folate points to the solvent accessible region, and we also modeled the missing glutamic portion into the current structures and it fits well without any clashes with the protein residues. The PDB codes are 4GT9, 4GTA, and 4GTB for A, B, and C, respectively.
In these structures, the pterin moiety is stacked between the isoalloxazine of FAD and the imidizole of His53, and on the opposite side of the flavin relative to the substrate dUMP. It is important to note that without the folate bound, His53 shows weaker electron density and crystallizes in various conformations (7, 21, 30). We mutated this residue to alanine and solved the crystal structure of this mutant with FAD, dUMP, and folate at 1.77Ǻ (Tables S1–S3). The H53A mutant structure showed similar folate binding for the pterin ring (Fig. S1), but the activity of both H53A and H53D mutants was dramatically reduced (Table S4).
The residues near the folate binding site are highly conserved amino acids highlighting the importance of this newly found binding pocket (i.e., Ala27, Arg28, Ser30, Phe31, Leu44, Leu48, His53, Thr55, Pro56, Asn85, and Tyr91, where Ser30 could be Thr). (See Figs. S2 and S3 for an Alignment Chart and close view of that site, respectively). The CH2H4folate makes eight H-bonding interactions involving seven water-mediated H-bonds and one direct H-bond to the protein (OG1 of Thr55). The structure with folinic acid (Fig. 3B) shows a similar hydrogen bonding pattern with additional hydrogen bonds involving the formyl group. It is important to note that folinic acid is good mimic of the reactive form of CH2H4folate in any proposed mechanism (e.g., the N5-iminium, which follows the first step in Fig. 1 mechanisms). Raltitrexed (tomudex) also binds FDTS in the same binding pocket with similar interactions (Fig. 3C). Structural comparison with our previously solved structures of TmFDTS-FAD-dUMP complex (e.g., PDB code 1O26) showed that the positions of FAD and dUMP are not affected by the binding of the different folate compounds.
In addition to the folate bound in the active site, the electron-density map also showed evidence for an additional CH2H4folate molecule bound in the 108–111 loop region (light yellow in Fig. 2). Although the electron density at this region was weak, the pterin ring of this folate appears to make a hydrogen bonding interaction with the main chain atoms of Leu106, Tyr109, and Thr111 and a water molecule (Fig. S4). The 108–111 loop is at the N terminus of the long 25-residue helix of the protein and this loop region also showed weak density in the previously reported dUMP structure of TmFDTS (21). A comparison with the other folate structures in the I4122 space group reported in this article, suggests that the ordering of the 108–111 loop is because of packing effects. The remote folate binding site found here might represent crystallographic trapping (of no functional implications). Alternatively, it could indicate an allosteric binding site with impact on the reaction kinetics, which may explain some of the kinetic complexity (10, 22, 24, 28) and cooperativity reported for this enzyme (15).
Examination of the Protein Methylation Mechanism (Fig. 1B).
The observed folate binding at the active site (Fig. 3) places the reactive methylene ∼8 Å from the C5 of dUMP and does not support a direct methylene transfer mechanism between CH2H4folate and dUMP (Fig. 1A). This finding could imply a methylene transfer involving an arginine of the protein as previously suggested (Fig. 1B) (16). In this mechanism the iminium form of CH2H4folate condenses with the guanidine group of an enzymatic arginine (Fig. 1B, step 2), elimination of H4folate results in an enzymatic Shiff-base (Fig. 1B, step 3), which following conformational changes in the protein can, in principle, be attacked by the activated dUMP (Fig. 1B, step 4) regenerating the catalytic arginine and completing methylene transfer. In T. maritima the residue proposed by ref. 16 is Arg174, which is proximal to dUMP in the crystal structures and shares hydrogen bonds with the C4 and C2 carbonyls of dUMP. We tested the activity of both R174K and R174A mutant of FDTS (Table S4). Notably, although the activity of R174A mutant was much slower than wild-type FDTS, dTMP was produced (as confirmed by formation of 14C-dTMP), demonstrating this mutant’s capability of catalyzing methylene transfer without Arg174. The reduced activity of R174A makes sense in light of the important role of this residue in binding and orientation of dUMP (21, 30). The only other amino acid, beside arginine, that could function as the methylene carrier is lysine, but R174K was not active. The lack of activity of R174K might be attributed to the hydrogen bonds between this Lys174 and His79 leading to active site deformation (Fig. 4B). These and other findings (8–10, 14, 15, 18–20) seem to eliminate a mechanism where the methylene is transferred through a protein residue, and further examination of direct methylene transfer mechanisms was necessary.
Fig. 4.

An alternative conformation where the flavin is in the opposite side of the active site, and away from the substrate dUMP binding-site. (A) The flavin binding mode observed in one of the active sites of E144R mutant (PDB code: 4GTD), with the 2Fo-Fc electron density contoured at 1.5σ. (B) The flavin binding mode observed in two of the actives sites of R174K mutant (PDB code: 4GTL), with the 2Fo-Fc electron density contoured at 1.5σ shown for FAD, H53, K174, and H79. Superposition with the wild-type enzyme in complex with dUMP (PDB code: 1O26) is shown in pink.
Identification of an Alternative Flavin Conformation.
For the CH2H4folate to bind closer to the dUMP, the isoalloxazine ring would have to move to the other side of the active site as we hypothesized in an earlier mechanistic study (20). While we were determining the structures of various mutants of TmFDTS, two of the mutants (E144R and R174K) in complex with FAD fortuitously showed an additional conformation where the isoalloxazine moiety is stacked with the imidizole of His53, which is rotated relative to its common orientation (Fig. 4). Only one of the active sites of the E144R and two of the active sites of the R174K tetramer showed this alternative flavin conformation. Additionally these mutants had greatly reduced activity (Table S4), perhaps because of a loss of the stable wild-type FAD conformation. Nevertheless, the flavin occupying this unique conformation showed better electron density than the other isoalloxazine moieties of FAD in the tetramer. As it has been pointed out earlier, the isoalloxazine of FAD is either disordered or shows very weak electron density in structures without dUMP (7, 9, 15, 21, 30). Interestingly, the newly observed binding site of the isoalloxazine moiety is almost identical to the binding site of the pterin moiety of the folate derivatives presented in Fig. 3. This finding is consistent with the chemical and structural similarities between these two moieties. From these observations, we suggest that conformational switching of the flavin and the folate could be catalytically significant. The stacking of the flavin with the rotated imidizole of His53, as depicted in E144R and R174K mutants, may stabilize the alternative conformation of the isoalloxazine ring, which enables CH2H4folate access to the dUMP.
Attempts to get a structure of the folate bound between the flavin, in its newly identified “flipped” conformation, and dUMP were not successful even when attempting to soak folates into the E144R-FAD crystals. The electron density maps for the resulting structure (Fig. S5) showed very weak electron density that could be part of folate bound between the flavin and dUMP, as expected for direct methylene transfer to dUMP. Although this observed electron density was too low to be considered significant or conclusive, it did indicate that the appropriate space is available for folates to bind close to dUMP, enabling direct methylene transfer.
Modeling of an Alternative Folate Binding Mode.
To test whether the “flipped” isoalloxazine ring of FAD observed in Fig. 4 could facilitate binding of the CH2H4folate cofactor proximal to the substrate dUMP, we modeled the iminium form of CH2H4folate (the reactive intermediate ready for methylene transfer as illustrated in Fig. 1) into the solved FDTS-FAD-dUMP-CH2H4folate structure. Then, by rotation of His53, as well as the C2′-C3′ and C1′-N10 bonds of FAD, the isoalloxazine ring was moved into a new binding site with the same conformation and functional group interactions as observed in the E144R and R174K structures discussed above. The iminium CH2H4folate was modeled into the site previously occupied by the flavin (as described in detail in SI Materials and Methods). After minimization, the iminium CH2H4folate intermediate fits well between FAD and dUMP. Fig. 5 presents the preferred binding orientation, and additional conformations with a short encounter distance between the iminium methylene and the C5 of dUMP were found and are presented in Fig. S6. Notably, the position of dUMP was not significantly modified in any of the minimized structures. The small energy differences between these binding modes suggest a broad and flat binding potential that would enable several orientations where the methylene could be transferred directly to the dUMP. The details of the modeling procedures and methods are described in detail in SI Materials and Methods.
Fig. 5.
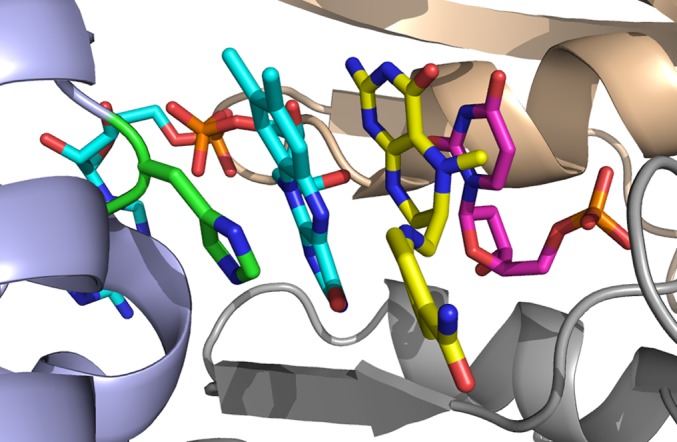
Modeled reactive structure for the FDTS-dUMP-FAD-CH2H4folate intermediate complex. FAD’s isoalloxazine ring and His53 are rotated as in Fig. 4 and Fig. S5. The iminium-CH2H4folate intermediate is docked into the site as described in the main text. Chain A is colored in gray, chain C in purple, and chain D in wheat color.
The outcome of these modeling efforts suggests that such ligand binding conformation is feasible. For all of the ligand binding conformations modeled (Fig. 5 and Fig. S6) the activated methylene of CH2H4folate is at a reasonable distance (3.3–3.5 Å) and orientation (N5-CH2-C5 of 100° ± 20°) to enable a direct methylene transfer to the dUMP substrate. The main structural modifications involved are the rotation of the FAD’s isoalloxazine ring and His53 (chain C), as well as small rearrangements of residues Asn85 (chain A) and Tyr91 (chain A). The rotated isoalloxazine ring interacts with Asn85 (chain A), Arg78 (chain D), and Thr55 (chain C). These structures are substantially stabilized by the stacking of the FAD’s isoalloxazine ring with the imidizole ring of His53, which is rotated as in the crystal structures of the E144R and R174K mutants.
Folate Inhibition of TmFDTS-FAD Reduction by NADPH.
The folate binding site observed here (the re-face of the isoalloxazine of FAD, opposite to the dUMP), could be an ideal site for NADPH binding during the reductive-half reaction. Such binding would accord with other structures of nicotinamide cofactors bound to flavo-enzymes (31, 32). To test this hypothesis, we examined the effect of folate moieties on the single reductive-half reaction of FAD with NADPH. Steady-state competitive CH2H4folate inhibition has been reported for the oxidase activity of FDTS (10, 14), indicating that NADPH and folate cannot both be bound to the active site at the same time, thus supporting the current suggestion. Nevertheless, such competitive inhibition under steady-state conditions could be complicated by the reported positive cooperativity (15), and the possible allosteric binding site (light yellow in Fig. 2 and Fig. S4). Consequently, more direct evidence was needed to suggest that folate moieties could inhibit the reductive-half reaction of FAD via presteady-state measurements. This experiment was carried out in the presence of dUMP, because it is a substantial activator of the reductive-half reaction of FAD (10, 14, 18, 27). Because we could not use the CH2H4folate cofactor, which would react with the dUMP, we used folinic acid to inhibit the reduction of the FDTS-FAD-dUMP by NADPH. These measurements indicated that the rate of flavin reduction decreased over 100-fold in the presence of folinic acid (Fig. S7), supporting the hypothesis that folates in Fig. 3 are occupying the site where NADPH binds during the reductive half reaction. To further examine this possibility, we modeled NADPH into the space occupied by folate derivatives in the crystal structures presented in Fig. 3. Several minimized NADPH orientations (e.g., Fig. S8) indicated that this pocket can bind NADPH with no steric clashes in more than one way, which is in accordance with the low stereospecificity, binding promiscuity, and high KM for nicotinamide cofactors (20). Our attempts to grow crystals with NADP+, NADPH, and few other nicotinamide derivatives were not successful, and soaking crystals with NADP+ has been reported to result in the replacement of FAD by NADP+ (29).
Discussion
The structural characterization of folate binding for the classic TSase has enabled understanding of the reaction mechanism and has been extensively exploited for the design of highly specific inhibitors and drugs (1, 3, 25, 26, 33). Similarly, the identification of the folate binding site for FDTS is crucial to understand the reaction mechanism and to develop folate-based inhibitors.
Here we report crystal structures of FDTS with CH2H4folate and other folate derivatives, together with the commonly observed ligands FAD and dUMP (Figs. 2 and 3). These structures show an extended stacking configuration between the pyrimidine, isoalloxazine, pterin, and imidazole rings of dUMP, FAD, CH2H4folate, and His53, respectively. His53 is highly conserved in FDTSs and has also been suggested to stabilize the binding of substrates like folate (30). Although the mutant H53A enzyme resulted in the same folate binding configuration (Fig. S1) and still produced dTMP, its overall catalytic activity was greatly reduced (Table S4). Similar stacking of folate pterin ring between the isoalloxazine of FAD and the imidazole of an enzymatic histidine was also observed in the folate/FAD-dependent tRNA T54 methyltransferase (TrmFO) (34). It is notable that TrmFO and FDTS catalyze the same net reductive methylation reaction of an uridyl moiety and, therefore, this unique stacking could be a general feature of such flavoenzymes. The main difference between the folate binding in the TrmFO enzyme vs. FDTS is that the si-face instead of the re-face of the flavin is stacked with the folate in the current structure.
Crystal structures of classic TSase have been solved with both CH2H4folate, and tomudex (1, 3, 33, 35). A comparison of these structures with the FDTS-folate structures presented here shows major differences in the folate binding site. The most striking difference is in the relative positions of the methylene donor (pterin ring) and the substrate (pyridmidine ring) (Fig. S9). The stacking of the pterin ring of the CH2H4folate and the pyrimidine ring of dUMP is essential for direct methylene transfer as proposed in Fig. 1A. We tested the proposed protein mediated methylene transfer mechanism (Fig. 1B) (16) using site-directed mutagenesis studies, which in accordance with previous studies (12), identified no strictly conserved arginine residue that is crucial for methylene transfer. Even Arg174 proposed specifically as methylene acceptor (16) when mutated to alanine was still capable of producing dTMP. Additionally, no other strictly conserved residues seem to be capable of catalyzing such chemistry (12). Furthermore, in contrast to the protein methylation suggested in ref. 16, we were unable to identify any methylene transfer to the protein using radiolabeled [14C]-CH2H4folate and following intermediate formation during the FDTS reaction (8). These findings, along with multiple kinetic data (10, 13–15, 18–20, 27), encouraged the examination of an alternative CH2H4folate binding mode that would enable direct methylene transfer to dUMP.
To explore a binding option for direct methylene transfer between the CH2H4folate and dUMP, we used the alternative “flipped” FAD conformation observed in our E144R and R174K structures (Fig. 4) and modeled new reactive structures with the CH2H4folate iminium intermediate now in direct contact with dUMP. The modeled structures (Fig. 5 and Fig. S6) place the reactive methylene within reasonable distance and angle from the C5 of dUMP for direct methylene transfer. The stacking interaction between dUMP, CH2H4folate, FAD, and the imidizole of His53, further emphasize the possible role of His53 in altering the correct configuration of ligands in the active site at different steps of the catalytic cascade. Similar stacking of the uracil of dUMP with the pterin of CH2H4folate is also observed for classic TSase (Fig. S9B), and might be a general structural requirement for direct transfer of the methylene.
Comparing the folate binding modes observed for the crystallographic and reactive-modeled structures of FDTS presented here with that of classic TSase indicate additional structural differences. The pterin of the folate in all FDTS structures (Figs. 2, 3, and 5) is situated in an antiparallel position relative to dUMP, opposite to the classic TS binding configuration (Fig. S9). Additionally, FDTS structures show folates bound in an extended conformation, whereas classic TSase binds folates with the para-aminobenzoyl moiety perpendicular to the pterin ring system. These unique binding differences for folate derivatives might present an avenue for rationally designed selectivity of folate-based inhibitors (i.e., antifolates) between human TSases and pathogenic FDTSs. Needless to say, such selectivity might be key for new antibiotic drugs with low toxicity.
The folate binding site observed in the current crystal structures (Figs. 2 and 3) could be ideal for nicotinamide binding during the reductive-half reaction, where NADPH reduces the dUMP-FAD-FDTS complex before CH2H4folate binding (8, 14, 18). The present measurements showing that folinic acid inhibits the reduction of the FDTS-FAD-dUMP complex by NADPH (Fig. S7) indicate that folate moieties can compete for the nicotinamide binding site. This suggestion is also consistent with the successful docking of NADPH into the newly identified folate binding site (Fig. S8) and with the CH2H4folate substrate-inhibition reported in the past (15, 19). The present structure also shows a secondary and weaker (as evident form lower electron density) binding site for CH2H4folate at the surface of the protein (light yellow in Fig. 2 and Fig. S4). This secondary binding site might be related to the positive cooperativity that has been reported for certain FDTSs (15), although more studies will be needed to completely understand how folate binding affects both oxidative and reductive-half reactions.
Conclusions
The potential impact of the findings presented here on our understanding of FDTS catalysis, its differences from classic TSase, and the resulting implications for antibiotic drug design are substantial. The crystal structures of TmFDTS with CH2H4folate, folinic acid, and tomudex presented here are, to our knowledge, unique in having folates bound to the FDTS. Although these structures do not indicate how the methylene can be transferred to the substrate dUMP, a protein-mediated transfer was excluded by site-directed mutagenesis of relevant residues. Additionally, most of the previously reported kinetic and mutagenic studies are consistent with mechanisms of direct methylene transfer from CH2H4folate to dUMP (i.e., a mechanism such as in Fig. 1A). Consequently, an alternative binding configuration was explored via computer modeling based on the “flipped” flavin conformation found for the E144R and R174K mutants, which enabled binding of the folate in direct contact with the substrate dUMP (Fig. 5). The folate binding site identified in the crystal structures is likely to represent the NADPH binding site during the reductive-half reaction, as supported by current and past kinetic data and modeling (Figs. S7 and S8).
In summary, many folate-based inhibitors that target classic TSase enzymes are in clinical use; however, these compounds have low affinity for FDTS enzymes. The development of these compounds for classic TSases has been greatly facilitated by structural studies with folate and its analogs (antifolates). Similarly, it is expected that new compounds taking advantage of the unique folate binding modes presented here for FDTS may provide new avenues for specific inhibitor design.
Materials and Methods
Detailed experimental and computational procedures are presented in SI Materials and Methods. In short: (i) Protein expression and purification has been previously described (6). (ii) Crystallization was conducted at 22 °C in 3–6% PEG 4K (wt/vol), 200 mM NaCl, 100 mM phosphate buffer (pH 6.58), and structure determination followed refs. 7 and 4. (iii) Modeling, in silico docking, and minimizations followed standard procedures using the Sander module of the Amber9 package, and solvation used the Generalized Born approach. (iv) The enzymatic activities of all mutants relative to the wild-type were determined using the 14C-dUMP (4, 20). (v) Single turnover reductive-half reaction rates were measured as previously described (14), with and without folinic acid.
Supplementary Material
Acknowledgments
We thank R. Moser (Eprova, Switzerland) for providing us with samples of the reduced folate derivatives. This work was supported by National Institutes of Health Grant R01 GM065368 and National Science Foundation Grant CHE 0715448 (to A.K.); the National Science Foundation Graduate Research Fellowship Program (E.M.K.); and Joint Center for Structural Genomics Grant U54GM074898 (to S.A.L.). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the US Department of Energy Office of Science by Stanford University. The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209), and the National Institute of General Medical Sciences.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4GT9, 4GTA, 4GTB, 4GTC, 4GTD, 4GTE, 4GTF, and 4GTL).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1206077109/-/DCSupplemental.
References
- 1.Finer-Moore JS, Santi DV, Stroud RM. Lessons and conclusions from dissecting the mechanism of a bisubstrate enzyme: Thymidylate synthase mutagenesis, function, and structure. Biochemistry. 2003;42:248–256. doi: 10.1021/bi020599a. [DOI] [PubMed] [Google Scholar]
- 2.Myllykallio H, et al. An alternative flavin-dependent mechanism for thymidylate synthesis. Science. 2002;297:105–107. doi: 10.1126/science.1072113. [DOI] [PubMed] [Google Scholar]
- 3.Carreras CW, Santi DV. The catalytic mechanism and structure of thymidylate synthase. Annu Rev Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 4.Koehn EM, et al. An unusual mechanism of thymidylate biosynthesis in organisms containing the thyX gene. Nature. 2009;458:919–923. doi: 10.1038/nature07973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myllykallio H, Leduc D, Filee J, Liebl U. Life without dihydrofolate reductase FolA. Trends Microbiol. 2003;11:220–223. doi: 10.1016/s0966-842x(03)00101-x. [DOI] [PubMed] [Google Scholar]
- 6.Lesley SA, et al. Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc Natl Acad Sci USA. 2002;99:11664–11669. doi: 10.1073/pnas.142413399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuhn P, et al. Crystal structure of thy1, a thymidylate synthase complementing protein from Thermotoga maritima at 2.25 A resolution. Proteins. 2002;49:142–145. doi: 10.1002/prot.10202. [DOI] [PubMed] [Google Scholar]
- 8.Mishanina TV, et al. Trapping of an intermediate in the reaction catalyzed by flavin-dependent thymidylate synthase. J Am Chem Soc. 2012;134:4442–4448. doi: 10.1021/ja2120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang K, et al. Crystal structure and enzymatic characterization of thymidylate synthase X from Helicobacter pylori strain SS1. Protein Sci. 2011;20:1398–1410. doi: 10.1002/pro.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, et al. Oxidase activity of a flavin-dependent thymidylate synthase. FEBS J. 2009;276:2801–2810. doi: 10.1111/j.1742-4658.2009.07003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costi MP, Ferrari S. Biochemistry: Anchors away. Nature. 2009;458:840–841. doi: 10.1038/458840a. [DOI] [PubMed] [Google Scholar]
- 12.Ulmer JE, Boum Y, Thouvenel CD, Myllykallio H, Sibley CH. Functional analysis of the Mycobacterium tuberculosis FAD-dependent thymidylate synthase, ThyX, reveals new amino acid residues contributing to an extended ThyX motif. J Bacteriol. 2008;190:2056–2064. doi: 10.1128/JB.01094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunter JH, Gujjar R, Pang CK, Rathod PK. Kinetics and ligand-binding preferences of Mycobacterium tuberculosis thymidylate synthases, ThyA and ThyX. PLoS ONE. 2008;3:e2237. doi: 10.1371/journal.pone.0002237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chernyshev A, Fleischmann T, Koehn EM, Lesley SA, Kohen A. The relationships between oxidase and synthase activities of flavin dependent thymidylate synthase (FDTS) Chem Commun (Camb) 2007;(27):2861–2863. doi: 10.1039/b700977a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graziani S, et al. Catalytic mechanism and structure of viral flavin-dependent thymidylate synthase ThyX. J Biol Chem. 2006;281:24048–24057. doi: 10.1074/jbc.M600745200. [DOI] [PubMed] [Google Scholar]
- 16.Griffin J, Roshick C, Iliffe-Lee E, McClarty G. Catalytic mechanism of Chlamydia trachomatis flavin-dependent thymidylate synthase. J Biol Chem. 2005;280:5456–5467. doi: 10.1074/jbc.M412415200. [DOI] [PubMed] [Google Scholar]
- 17.Leduc D, et al. Two distinct pathways for thymidylate (dTMP) synthesis in (hyper)thermophilic Bacteria and Archaea. Biochem Soc Trans. 2004;32:231–235. doi: 10.1042/bst0320231. [DOI] [PubMed] [Google Scholar]
- 18.Leduc D, et al. Functional evidence for active site location of tetrameric thymidylate synthase X at the interphase of three monomers. Proc Natl Acad Sci USA. 2004;101:7252–7257. doi: 10.1073/pnas.0401365101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graziani S, et al. Functional analysis of FAD-dependent thymidylate synthase ThyX from Paramecium bursaria Chlorella virus-1. J Biol Chem. 2004;279:54340–54347. doi: 10.1074/jbc.M409121200. [DOI] [PubMed] [Google Scholar]
- 20.Agrawal N, Lesley SA, Kuhn P, Kohen A. Mechanistic studies of a flavin-dependent thymidylate synthase. Biochemistry. 2004;43:10295–10301. doi: 10.1021/bi0490439. [DOI] [PubMed] [Google Scholar]
- 21.Mathews II, et al. Functional analysis of substrate and cofactor complex structures of a thymidylate synthase-complementing protein. Structure. 2003;11:677–690. doi: 10.1016/s0969-2126(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 22.Chernyshev A, Fleischmann T, Kohen A. Thymidyl biosynthesis enzymes as antibiotic targets. Appl Microbiol Biotechnol. 2007;74:282–289. doi: 10.1007/s00253-006-0763-1. [DOI] [PubMed] [Google Scholar]
- 23.Fivian-Hughes AS, Houghton J, Davis EO. Mycobacterium tuberculosis thymidylate synthase gene thyX is essential and potentially bifunctional, while thyA deletion confers resistance to p-aminosalicylic acid. Microbiology. 2012;158:308–318. doi: 10.1099/mic.0.053983-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mishanina TV, Koehn EM, Kohen A. Mechanisms and inhibition of uracil methylating enzymes. Bioorg Chem. 2012;43:37–43. doi: 10.1016/j.bioorg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cunningham D, et al. ‘Tomudex’ (ZD1694): A novel thymidylate synthase inhibitor with clinical antitumour activity in a range of solid tumours. ‘Tomudex’ International Study Group. Ann Oncol. 1996;7:179–182. doi: 10.1093/oxfordjournals.annonc.a010546. [DOI] [PubMed] [Google Scholar]
- 26.Takemura Y, Jackman AL. Folate-based thymidylate synthase inhibitors in cancer chemotherapy. Anticancer Drugs. 1997;8:3–16. doi: 10.1097/00001813-199701000-00001. [DOI] [PubMed] [Google Scholar]
- 27.Mason A, Agrawal N, Washington MT, Lesley SA, Kohen A. A lag-phase in the reduction of flavin dependent thymidylate synthase (FDTS) revealed a mechanistic missing link. Chem Commun (Camb) 2006;(16):1781–1783. doi: 10.1039/b517881a. [DOI] [PubMed] [Google Scholar]
- 28.Koehn EM, Kohen A. Flavin-dependent thymidylate synthase: A novel pathway towards thymine. Arch Biochem Biophys. 2010;493:96–102. doi: 10.1016/j.abb.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sampathkumar P, Turley S, Sibley CH, Hol WGJ. NADP+ expels both the co-factor and a substrate analog from the Mycobacterium tuberculosis ThyX active site: Opportunities for anti-bacterial drug design. J Mol Biol. 2006;360:1–6. doi: 10.1016/j.jmb.2006.04.061. [DOI] [PubMed] [Google Scholar]
- 30.Sampathkumar P, et al. Structure of the Mycobacterium tuberculosis flavin dependent thymidylate synthase (MtbThyX) at 2.0A resolution. J Mol Biol. 2005;352:1091–1104. doi: 10.1016/j.jmb.2005.07.071. [DOI] [PubMed] [Google Scholar]
- 31.Berkholz DS, Faber HR, Savvides SN, Karplus PA. Catalytic cycle of human glutathione reductase near 1 A resolution. J Mol Biol. 2008;382:371–384. doi: 10.1016/j.jmb.2008.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kyte J. Mechanisms in Protein Chemistry. Chicago, IL: Garland; 1995. [Google Scholar]
- 33.Sotelo-Mundo RR, et al. Crystal structures of rat thymidylate synthase inhibited by Tomudex, a potent anticancer drug. Biochemistry. 1999;38:1087–1094. doi: 10.1021/bi981881d. [DOI] [PubMed] [Google Scholar]
- 34.Nishimasu H, et al. Atomic structure of a folate/FAD-dependent tRNA T54 methyltransferase. Proc Natl Acad Sci USA. 2009;106:8180–8185. doi: 10.1073/pnas.0901330106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phan J, et al. Human thymidylate synthase is in the closed conformation when complexed with dUMP and raltitrexed, an antifolate drug. Biochemistry. 2001;40:1897–1902. doi: 10.1021/bi002413i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.