

Abstract
The effective design of an artificial photosynthetic system entails the optimization of several important interactions. Herein we report stopped-flow UV-visible (UV-vis) spectroscopy, X-ray crystallographic, density functional theory (DFT), and electrochemical kinetic studies of the Re(bipy-tBu)(CO)3(L) catalyst for the reduction of CO2 to CO. A remarkable selectivity for CO2 over H+ was observed by stopped-flow UV-vis spectroscopy of [Re(bipy-tBu)(CO)3]-1. The reaction with CO2 is about 25 times faster than the reaction with water or methanol at the same concentrations. X-ray crystallography and DFT studies of the doubly reduced anionic species suggest that the highest occupied molecular orbital (HOMO) has mixed metal-ligand character rather than being purely doubly occupied 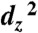 , which is believed to determine selectivity by favoring CO2 (σ + π) over H+ (σ only) binding. Electrocatalytic studies performed with the addition of Brönsted acids reveal a primary H/D kinetic isotope effect, indicating that transfer of protons to Re -CO2 is involved in the rate limiting step. Lastly, the effects of electrode surface modification on interfacial electron transfer between a semiconductor and catalyst were investigated and found to affect the observed current densities for catalysis more than threefold, indicating that the properties of the electrode surface need to be addressed when developing a homogeneous artificial photosynthetic system.
, which is believed to determine selectivity by favoring CO2 (σ + π) over H+ (σ only) binding. Electrocatalytic studies performed with the addition of Brönsted acids reveal a primary H/D kinetic isotope effect, indicating that transfer of protons to Re -CO2 is involved in the rate limiting step. Lastly, the effects of electrode surface modification on interfacial electron transfer between a semiconductor and catalyst were investigated and found to affect the observed current densities for catalysis more than threefold, indicating that the properties of the electrode surface need to be addressed when developing a homogeneous artificial photosynthetic system.
Keywords: carbon dioxide reduction, electrochemistry, kinetics, electrocatalyst
The development of artificial photosynthetic systems is of immediate concern in view of the world’s dependence on fossil fuels and the increasing emissions of CO2. Rapid industrial growth in developing nations will significantly increase the global energy demand in coming years, and although known reserves of fuels such as natural gas and coal are sufficient for the near future, they are becoming increasingly costly to obtain. As the use of fossil fuels is fundamentally unsustainable and generates greenhouse gases and other pollutants, the development of environmentally benign energy sources is important. Solar energy is an abundant alternative but suffers from being a diffuse energy source, and its availability varies by location and time of day. If we can capture solar energy and use CO2 as a C1 feedstock for liquid fuels, we can envision converting our global energy economy into a nearly carbon-neutral system (1).
Photosynthesis is one of the great triumphs of nature and is the cornerstone for advanced life on the planet. Mankind has yet to master nature’s ability to store sunlight as chemical energy by splitting CO2 and H2O to form C─C, C─H, and O─O bonds. The energy-dense liquid fuels formed by this process would have the advantage of conforming to the existing infrastructure. Photosynthesis can be divided into two parts: water splitting and reduction of carbon dioxide. Water oxidation has been reviewed extensively by others (2–4).
Our laboratory is currently exploring the development of CO2 reduction catalysts with the goal of implementing a directly photo-driven system. A few systems for the direct conversion of solar energy into chemical energy using a CO2 reduction catalyst at a semiconductor photoelectrode have been reported (5–9), and further development of this concept will require optimization of both the catalyst and the semiconductor components.
The reduction of CO2 to useful products is inherently proton-dependent. This dependence is true not only for the production of carbohydrates by natural photosynthesis, but also for the synthetic production of other value-added products. The protons required are susceptible to competitive, direct reduction to H2, a process that occurs at a more positive (i.e., more favorable) thermodynamic potential. A functional artificial photosynthetic system therefore requires a catalyst that is kinetically selective for CO2 reduction over H+ reduction.
Recently, we have explored complexes of the type Re(bipy-R)(CO)3(L) (where bipy-R is 4,4′-disubstituted-2,2′-bipyridine and L is a halide or a neutral ligand with -OTf) and reported that Re(bipy-tBu)(CO)3Cl 1 is a precatalyst for the electrochemical reduction of CO2 to CO at high turnover frequency (> 200 s-1) (10) compared with the previous reports on Re(bipy)(CO)3Cl (11, 12). Complex 1 has one of the highest turnover frequencies reported for the reduction of CO2 to CO, and exhibits nearly 100% faradic efficiency with very high turnover numbers. We have also successfully coupled the catalyst to a semiconductor electrode, p-type silicon (p-Si), which enables us to provide part of the thermodynamic energy for catalysis using solar illumination (8).
A fully operational photoelectrochemical device will require the coordination of photovoltaics, semiconductor electrodes, catalysts, and substrates. We believe that understanding the individual components of the system will accelerate research toward a functional device. Herein, we report detailed studies of the key components in an artificial photosynthetic system based on the Re(bipy-tBu)(CO)3(L) catalyst (Fig. 1) that are widely applicable to homogeneous photoelectrochemical catalysis. The first is the interaction between the catalyst and the two potential substrates, CO2 and H+. We show by stopped-flow UV-visible (UV-vis) spectroscopy that the active catalyst displays remarkable selectivity for CO2 over H+. The origins of this desirable activity were further investigated through crystallographic and density functional theory (DFT) characterization of the reduced catalytic intermediate. We also show that CO2 reduction is enhanced by the addition of Brönsted acids, and exhibits a primary H/D kinetic isotope effect (KIE). Whereas most electrocatalytic studies focus primarily on the catalyst/substrate interaction, it is important not to overlook the heterogeneous electron transfer from the electrode to the catalyst. To probe this interaction, we studied catalytic behavior at various electrode surfaces, including semiconductors.
Fig. 1.

Schematic for a device using a p-Si/ Re(bipy-tBu)(CO)3Cl semiconductor/molecular catalyst junction to photoelectrochemically reduce CO2.
Results and Discussion
Synthesis of [Re(bipy-tBu)(CO)3]-1.
Previously published results (10) suggested that the doubly reduced [Re(bipy-tBu)(CO)3]-1 anion is the species that actively binds CO2, rendering it the most immediately relevant intermediate in studies of CO2/H+ selectivity. We observed that solutions of this species could be generated by reduction of 1 with KC8. With careful handling, they could be maintained and characterized by a variety of techniques.
UV-Vis Stopped-Flow Spectroscopy: Probing the Selectivity of the Re Catalyst.
Recently published results demonstrated the remarkable selectivity of the [Re(bipy-tBu)(CO)3]-1 anion toward CO2 over H+ reduction (13). We investigated the CO2/H+ selectivity in detail using stopped-flow UV-vis experiments to quantify their relative rates of reaction with the Re catalyst. Although the exact products are not well-defined in these stoichiometric reactions, the relative rates of the reactions are still informative. The UV-vis spectrum of the reduced Re anion contains a strong absorption at 570 nm (14,000 M-1 cm-1) that presumably arises from a coordinated bipyridine-based radical. The persistence of the anion was thus monitored at this absorption. The parent complex, 1, however, has a λmax at 384 nm (3,900 M-1 cm-1) (14). Solutions of the reduced complex were mixed in the stopped-flow apparatus with either CO2-saturated tetrahydrofuran (THF) (0.20 M) (15) or with 0.20 M solutions of H2O, CH3OH, or CF3CH2OH (TFE) in THF.
The spectral changes for the CO2 reaction show rapid and complete decay of Re anion (Fig. 2). The absorbance at 570 nm follows first-order kinetics over three half-lives, with t1/2 = 20 ms and a pseudo first-order rate constant of 35 s-1 (Table 1). The reactions with water, CH3OH, and TFE were much slower and were not complete at the end of monitoring. Control reactions mixing the Re anion with pure THF (no CO2 or proton source) showed background degradation on this longer timescale. Remarkably, TFE did not appear to react with the Re anion on the timescale of the experiments (up to 150 s). These results mirror the electrochemical results that suggest that CO2 is more readily reduced than H+ by the Re anion, and suggest that this selectivity may have its origin in the initial reaction of the substrate with the doubly reduced Re species.
Fig. 2.

Reaction of [Re(bipy-tBu)(CO)3]-1 in THF with a solution of 200 mM CO2 in THF over 0.75 s in a stopped-flow UV-vis experiment. Scans were taken every 1.5 ms over the course of the experiment and the degradation of the peak at 570 nm was followed for the half-life and pseudo first-order rate constant determination.
Table 1.
Results from UV-vis stopped-flow experiments of [Re(bipy-tBu)(CO)3]-1
| Reactant |
Half-life (t1/2) |
Pseudo first-order rate constant |
| CO2 | 0.02 s | 35 s-1 |
| H2O | [0.5 s] | [∼1 s-1] |
| CH3OH | [1 s] | [∼1 s-1] |
The first three half-lives for the CO2 reaction fit well to pseudo first-order kinetics. The H2O and CH3OH reactions, however, as described in the text, are more complex. The values given above in square brackets are rough estimates of the time for the first half-life of reaction, estimated from the spectral changes in Figs. S1 and S2.
The H2O and CH3OH reactions were more complicated than simple first-order decay of the Re anion. The absorbance at 570 nm did not fully bleach, and new absorbances are observed at approximately 520 and 370 nm (Fig. S1). Until more detailed kinetic analyses can be completed, we have estimated half-lives for these reactions to provide a comparison with the well-behaved CO2 reaction: approximately 0.5 s for H2O, approximately 1 s CH3OH, and greater than or equal to 100 s for TFE. These values imply that the reaction with CO2 is about 25 times faster than that with H2O, and 50 times faster than that with CH3OH. The slower reaction with TFE than with H2O or CH3OH is surprising given the higher acidity of TFE and suggests that these reactions may be more complicated than simple protonation of the Re anion. Still, the conclusion is very clear that the Re anion reacts much more quickly with CO2 than with sources of H+.
Crystallography and DFT of the Re Anion: Origins of Selectivity.
With stable solutions of the [Re(bipy-tBu)(CO)3]-1 species in hand, the source of CO2 selectivity could be investigated through structural studies. Single crystals suitable for X-ray diffraction were grown from the vapor diffusion of pentane into a THF solution of the anion containing 18-crown-6 as a stabilizing agent. Upon reduction, complex 1 loses chloride and adopts a distorted trigonal bipyramidal structure (τ5 = 0.46) (16) in the solid state with the Re coordinated by three facial carbonyl ligands and a chelating bipyridine (Fig. 3, numbering scheme in Fig. S2, selected bond lengths and angles in Table S1, and crystallographic data and refinement information in Table S2). The potassium cation is encapsulated by the crown ether and has one bound THF molecule. It is also associated with the axial carbonyl [O3–K1, 2.945(7) Å]. The Re–N distance contracts and the bite angle of the bipyridine increases slightly compared with the unreduced starting material (Fig. S3). This behavior is indicative of an improved orbital overlap between the ligand and metal center in the anion.
Fig. 3.

Full structure of [Re(bipy-tBu)(CO)3] [K(18-crown-6)•THF]. Hydrogen atoms are omitted for clarity and ellipsoids are shown at 50% probability.
To obtain a better understanding of the electronics of this reduced state, we employed DFT calculations using the Amsterdam Data Functional (ADF) program 2007.1. The calculated highest occupied molecular orbital (HOMO) is a hybrid involving both the ligand and the metal center, containing substantial π∗ character (Fig. S4). This calculated orbital is consistent with the bond length alternation observed in the bipyridine rings of the Re anion crystal structure, which is similar to what is seen in crystal structures of reduced 2,2′-bipyridine (17), suggesting significant electron density on the ligand. The geometry of the HOMO diffuses the directional basicity of the Re anion, and thus other factors such as π interactions become important. We expect that the mixed character of the HOMO (bipy- + Re0) relative to a doubly occupied 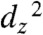 lone-pair is sufficient to cause CO2 binding to be more favorable than H+ binding. Similarly, a HOMO that is delocalized disfavors direct protonation of the Re
lone-pair is sufficient to cause CO2 binding to be more favorable than H+ binding. Similarly, a HOMO that is delocalized disfavors direct protonation of the Re 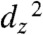 orbital to produce a hydride.
orbital to produce a hydride.
The nature of the binding of CO2 to the anion can also be better understood through DFT. The geometry-optimized electronic structure of a Re(bipy-tBu)(CO)3(CO2)K complex was calculated. The structure did not converge without the addition of a cation (H, Li, Na, K) to support the highly electronegative oxygen atoms. We observed that the 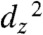 orbital can form a σ bond to the carbon atom of CO2 (HOMO), and the interaction with CO2 can be further stabilized by a π interaction of the metal dxz and dyz orbitals with p orbitals on the CO2 oxygen atoms (HOMO-4) (Fig. 4). This stabilizing interaction is clearly not available for the interaction with H+ and is similar to that calculated by Fujita and coworkers for [Co(macrocycle)(CO2)(CH3CN)]+, but the Re complex forms an extended bonding interaction with the CO2 oxygen atoms rather than with the carbon center (18).
orbital can form a σ bond to the carbon atom of CO2 (HOMO), and the interaction with CO2 can be further stabilized by a π interaction of the metal dxz and dyz orbitals with p orbitals on the CO2 oxygen atoms (HOMO-4) (Fig. 4). This stabilizing interaction is clearly not available for the interaction with H+ and is similar to that calculated by Fujita and coworkers for [Co(macrocycle)(CO2)(CH3CN)]+, but the Re complex forms an extended bonding interaction with the CO2 oxygen atoms rather than with the carbon center (18).
Fig. 4.

Calculated orbitals of Re(bipy-tBu)(CO)3(CO2)K using ADF 2007.1 showing (A) the HOMO orbital where Re (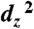 ) forms a σ bond to the carbon atom of CO2 (s and pz), and (B) the HOMO-4 orbital that forms π interactions between the dyz and dxz orbitals of Re and the p orbitals of the oxygen atoms on CO2.
) forms a σ bond to the carbon atom of CO2 (s and pz), and (B) the HOMO-4 orbital that forms π interactions between the dyz and dxz orbitals of Re and the p orbitals of the oxygen atoms on CO2.
Brönsted Acid Rate Enhancement and Kinetic Isotope Effects.
Having established a marked selectivity for CO2 over H+, we investigated the catalytic system in the presence of added proton sources. The parent catalyst system, 1, has a very negative catalytic potential of -2.23 V (vs. Fc/Fc+) relative to the thermodynamic potential of the reduction of CO2 to CO (approximately -1.2 V vs. Fc/Fc+ in water at pH 7). The high overpotential creates a situation where catalysis can occur without an added proton source. Previous reports have suggested that the oxygen from CO2 is transferred to an unknown oxide acceptor (19). The oxide acceptor can be a proton that can be extracted from acetonitrile (20) or even from the supporting electrolyte via Hoffman degradation (21). More recently, evidence pointing to protons as the oxide acceptor was reported by Wong et al. They showed that the catalytic current increased for the reduction of Re(bipy)(CO)3(py)(OTf) under CO2 in the presence of increasing concentrations of weak Brönsted acids (22). In their study, the rate of electrocatalytic CO2 reduction increased for the series: H2O < CH3OH < phenol < TFE. In all cases, the faradic efficiency for the production of CO remained greater than 95%.
The CO2 reduction behavior of the improved catalysts Re(bipy-tBu)(CO)3(MeCN)(OTf) 2 and Re(bipy-tBu)(CO)3(py)(OTf) 3 were studied in the presence of H2O, CH3OH, and TFE under an atmosphere of CO2. Under argon before the addition of protons, complex 2 exhibits one reversible reduction at -1.71 V followed by a quasireversible reduction at -1.94 V (vs. Fc/Fc+). Complex 3 exhibits one reversible reduction at -1.64 V and one quasireversible reduction at -1.95 V (vs. Fc/Fc+) (Fig. 5).
Fig. 5.

Re(bipy-tBu)(CO)3(py)(OTf) 2 under argon (black) and CO2 (red). The solution is 1 mM in catalyst with 0.1 M tetrabutylammonium hexafluorophosphate (TBAH) as the supporting electrolyte. A 1 mm diameter glassy carbon electrode, a Pt wire counter electrode, and an Ag wire pseudo reference were used with ferrocene added as an internal reference.
Given that 1 showed a substantial increase in the rate of catalysis over the original Re(bipy)(CO)3Cl catalyst (10), we expected to see even higher current densities upon the addition of protons to 2 and 3 in the presence of CO2. Upon addition of CH3OH or TFE, an increased icat/ip was observed compared with the results of Wong et al., whereas for water a similar icat/ip was observed (Table 2).
Table 2.
Summary of results for addition of Brönsted acids
| Precatalyst |
Brönsted acid |
Highest icat/ip* |
CO2 solubility at highest icat/ip |
| Re(bipy-tBu)(CO)3(MeCN)(OTf) 2 | TFE | 54.0 (1.6 M) | 0.19 M |
| Re(bipy-tBu)(CO)3(MeCN)(OTf) 2 | methanol | 41.9 (9.9 M) | 0.22 M |
| Re(bipy-tBu)(CO)3(MeCN)(OTf) 2 | water | 9.0 (10.0 M) | 0.18 M |
| Re(bipy-tBu)(CO)3(py)(OTf) 3 | TFE | 62.0 (1.5 M) | 0.27 M |
| Re(bipy-tBu)(CO)3(py)(OTf) 3 | methanol | 28.9 (8.0 M) | 0.23 M |
| Re(bipy-tBu)(CO)3(py)(OTf) 3 | water | 8.2 (7.3 M) | 0.19 M |
| Re(bipy)(CO)3(py)(OTf) † | TFE | 47.1 (0.77 M) | 0.27 M |
| Re(bipy)(CO)3(py)(OTf) † | phenol | 39.5 (0.57 M) | 0.30 M |
| Re(bipy)(CO)3(py)(OTf) † | methanol | 31.1 (7.20 M) | 0.24 M |
| Re(bipy)(CO)3(py)(OTf) † | water | 11.4 (10.4 M) | 0.17 M |
*Acid concentration in parenthesis.
†From Wong et al. (22).
A normal primary H/D kinetic isotope effect was observed in these experiments (data for CH3OH vs. CD3OD in Fig. 6). For complex 2, the KIE for H2O/D2O is 1.8( ± 0.1), the KIE for CH3OH/CD3OD is 1.8( ± 0.1), and the KIE for TFE/TFE-d3 is 1.2( ± 0.1). The decreased KIE for TFE is attributed to an increased mismatch between the pKa of the metal-bound CO2 and the pKa of the acid (23). The observation of a KIE suggests that proton transfer to the Re -CO2 adduct is likely involved in the rate limiting step in this reaction.
Fig. 6.

KIE study for CH3OH and CD3OD addition to the catalytic reaction of [Re(bipy-tBu)(CO)3]-1 with CO2. In each case we see a significant increase in the catalytic current upon addition of the Brönsted acid. The x axis is offset for clarity. These data are representative of the results for H2O/D2O and TFE/TFE-d3.
The Brönsted acid experiments also suggest that the catalytic reaction is second-order in acid. A linear region can be observed in a plot of the ratio of catalytic current (icat) to noncatalytic current (ip) vs. substrate concentration (Fig. S5). This behavior is expected for an EC’ catalytic mechanism, in which the relationship between substrate concentration and current ratio can be described by Eq. 1 (24, 25), as follows:
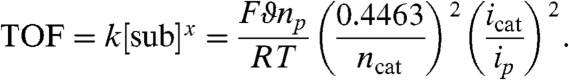 |
[1] |
All of the acids exhibit this behavior before tapering and establishing a steady icat/ip. Catalytic currents do not plateau in the cyclic voltammograms under CO2 (even at high H+ concentrations), but Eq. 1 applies because the catalytic current is scan rate independent. Additional experiments have established that the reaction is first-order in [CO2] (22), leading to the rate equation
| [2] |
For all Brönsted acids, the production of CO was nearly quantitative (greater than 95% current efficiency) even at acid concentrations where icat/ip was at its maximum, consistent with other data indicating that the catalyst remains very selective for CO2 even in the presence of a large excess of H+. Together, the KIE and second-order [H+] dependence of this catalytic reaction provide strong evidence for H2O formation from the second oxygen atom of the CO2 substrate.
Interfacial Charge Transfer Studies.
We then turned our attention to the heterogeneous charge transfer at the electrode/catalyst interface. In the system shown in Fig. 1, the electrons are generated via photoexcitation from the valence band to the conduction band of the p-Si electrode and move toward the interface between the p-Si and catalyst under the influence of a potential bias. They are then transferred to the catalyst, which goes on to reduce CO2 to CO.
No reduction peaks are observed for 1 under dark conditions. Under polychromatic light illumination, two one-electron reduction peaks can be observed that are about 600 mV more positive than those obtained at glassy carbon or platinum electrodes (Fig. S6A) (8). The limiting catalytic current density for the hydrogen-terminated p-Si/1 junction was 2.9 mA/cm2 (at 0.6 mM catalyst), which is lower than the 5.3 mA/cm2 measured at glassy carbon. Given that (i) these TOFs are measured in identical conditions in the same electrochemical cell, (ii) cyclic voltammograms (CVs) of the Re catalyst under Ar have been shown to be stable at illuminated p-Si, (iii) the faradic efficiency for the reduction of CO2 to CO is nearly 100% for both surfaces, and (iv) single crystalline silicon was used for this work, losses associated with the homogeneous catalysis and bulk semiconductor should be minimal and cannot account for the decrease in limiting catalytic current density. The lower current density for the hydrogen-terminated p-Si/1 junction can only be explained if the limiting factor involves heterogeneous charge transfer and/or the interaction between the p-Si and complex 1. This possibility was probed by modifying the surface of the photocathode to improve its charge transfer properties. Styrene and n-hexene were used separately to generate phenylethyl p-Si and hexyl p-Si surfaces using the method described by Buriak and Allen (26).
The current density for the photoelectrochemical reduction of CO2 at a styrene-modified p-Si electrode in the presence of complex 1 scales with the illumination intensity (Fig. S6B and Table S3). A direct comparison of the photocatalytic current densities shows a more than threefold increase for the styrene-modified p-Si surface over both the hydrogen-terminated surface (Fig. S6C) and the hexene-modified surface with no appreciable change in the onset potential for the catalysis. The hydrogen-terminated and hexene-modified surfaces had very similar current densities under the same catalyst and substrate concentrations. Based on these data, the styrene-modified p-Si outperforms the other semiconductor surfaces, as well as the glassy carbon/1 junction. The superior performance of the styrene-modified p-Si/1 is attributed to improved charge transport via π interactions between the aromatic rings on the styrene-modified p-Si surface and the bipyridine ligand of 1. These results illustrate the importance of heterogeneous charge transfer between the electrode and catalyst, and the optimization of this interaction should not be overlooked in a homogeneous electrocatalytic system.
Concluding Remarks
This work reinforces the importance of considering not only the interactions of an electrocatalyst with the targeted catalytic substrate but also the heterogeneous electron transfer from the electrode surface to the catalyst. We have shown that the Re(bipy-tBu)(CO)3(L) electrocatalytic system is very selective for the reduction of CO2, and that the rate of catalysis can be enhanced by the addition of a proton source. Stopped-flow UV-vis experimental data indicate that CO2 reacts about 25 times faster than water and about 50 times faster than methanol with [Re(bipy-tBu)(CO)3]-1. The reduced form presumed to be the active catalyst was studied by X-ray crystallography. The structural data, combined with DFT studies of the reduced complex, suggest that the HOMO contains mixed metal-ligand character. KIE and kinetic studies indicate that protons are involved in the rate-determining step for the reduction of CO2 to CO by Re(bipy-tBu)(CO)3(L). In addition, we have shown that the electrode surfaces make a large difference in the catalytic current density by controlling the electron transfer from the electrode to the catalyst. The catalytic current densities can be increased threefold by modifying the p-Si surface with phenylethyl groups as opposed to hexyl groups or hydrogen atoms. These results may help guide future catalyst design as well as the optimization of other artificial photosynthesis systems.
Materials and Methods
Complex 1 was synthesized by previously reported methods (10). Syntheses of complexes 2 and 3 are reported in the SI Text and the crystal structure of 3 is shown in Fig. S7. Reductions of complex 1 to the [Re(bipy-tBu)(CO)3]-1 were carried out using 2.1 equivalents of KC8 in THF. DFT calculations were performed with the ADF program suite (27, 28), version 2007.1 using the triple-ζ Slater-type orbital basis set. Geometry optimized xyz coordinates for [[Re(bipy-tBu)(CO)3]-1] and [Re(bipy-tBu)(CO)3(CO2)(K)] can be found in Tables S4 and S5, respectively. Further experimental procedures, including electrochemical details and p-Si surface modification techniques, as well as additional references are included in the SI Text.
Stopped-flow measurements were performed using a Hi-Tech/TgK Scientific SF-61DX2 CryoStopped-Flow system. A xenon-lamp illuminated the mixing chamber (1 cm path length, fused UV silica) in conjunction with a diode array detector. Samples were prepared under a nitrogen atmosphere in a dry box and were transported to the stopped-flow apparatus in gas-tight syringes. The concentration of [Re(bipy-tBu)(CO)3]-1 before mixing was 0.12 mM, CO2 was at saturation (approximately 200 mM) in THF. H2O, methanol, and TFE were all prepared at 200 mM in THF.
Supplementary Material
ACKNOWLEDGMENTS.
The authors would like to thank Prof. Arnie Rheingold and Dr. Curtis Moore for assistance with the X-ray crystallographic studies and Prof. Joshua Figueroa for his assistance with computations. This research was supported at University of California at San Diego by the Defense Advanced Research Projects Agency Surface Catalysis for Energy Program and by the Air Force Office of Scientific Research through the Multidisciplinary University Research Initiative program Award FA9550-10-1-0572. This research was supported at the University of Washington by a fellowship from the Camille and Henry Dreyfus Postdoctoral Program in Environmental Chemistry (A.J.M.M.), the National Institutes of Health stopped-flow apparatus, Grant GM-50422 (to J.M.M.), and the Center for Molecular Electrocatalysis, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Crystallography, atomic coordinates, and structure factors in this paper have been deposited in the Cambridge Structural Database, Crystallographic Data Center, Cambridge CB2 1EZ, United Kingdom http://www.ccdc.cam.ac.uk, (CSD reference nos. 871193, 871194, and 871195).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1119863109/-/DCSupplemental.
References
- 1.Olah GA, Prakash GKS, Goeppert A. Anthropogenic chemical carbon cycle for a sustainable future. J Am Chem Soc. 2011;133:12881–12898. doi: 10.1021/ja202642y. [DOI] [PubMed] [Google Scholar]
- 2.Dau H, et al. The mechanism of water oxidation: From electrolysis via homogeneous to biological catalysis. ChemCatChem. 2010;2:724–761. [Google Scholar]
- 3.Yagi M, Kaneko M. Molecular catalysts for water oxidation. Chem Rev. 2001;101:21–36. doi: 10.1021/cr980108l. [DOI] [PubMed] [Google Scholar]
- 4.Rüttinger W, Dismukes GC. Synthetic water-oxidation catalysts for artificial photosynthetic water oxidation†. Chem Rev. 1997;97:1–24. doi: 10.1021/cr950201z. [DOI] [PubMed] [Google Scholar]
- 5.Barton EE, Rampulla DM, Bocarsly AB. Selective solar-driven reduction of CO2 to methanol using a catalyzed p-GaP based photoelectrochemical cell. J Am Chem Soc. 2008;130:6342–6344. doi: 10.1021/ja0776327. [DOI] [PubMed] [Google Scholar]
- 6.Beley M, Collin J-P, Sauvage J-P, Petit J-P, Chartier P. Photoassisted electro-reduction of CO2 on p-GaAs in the presence of Ni cyclam2+ J Electroanal Chem. 1986;206:333–339. [Google Scholar]
- 7.Bradley MG, Tysak T, Graves DJ, Viachiopoulos NA. Electrocatalytic reduction of carbon dioxide at illuminated p-type silicon semiconducting electrodes. J Chem Soc Chem Commun. 1983;7:349–350. [Google Scholar]
- 8.Kumar B, Smieja JM, Kubiak CP. Photoreduction of CO2 on p-type silicon using Re(bipy-But)(CO)3Cl: Photovoltages exceeding 600 mV for the selective reduction of CO2 to CO. J Phys Chem C. 2010;114:14220–14223. [Google Scholar]
- 9.Petit PC J-P, Beley M, Sauvage J-P. Selective photoelectrochemical reduction of CO2 to CO in an aqueous medium on p-GaP, mediated by Ni cyclam2+ Nouv J Chim. 1987;11:751–752. [Google Scholar]
- 10.Smieja JM, Kubiak CP. Re(bipy-tBu)(CO)3Cl-improved catalytic activity for reduction of carbon dioxide: IR-spectroelectrochemical and mechanistic studies. Inorg Chem. 2010;49:9283–9289. doi: 10.1021/ic1008363. [DOI] [PubMed] [Google Scholar]
- 11.Hawecker J, Lehn JM, Ziessel R. Electrocatalytic reduction of carbon-dioxide mediated by Re(Bipy)(CO)3Cl (Bipy = 2,2′-Bipyridine) J Chem Soc Chem Commun. 1984:328–330. [Google Scholar]
- 12.Hawecker J, Lehn JM, Ziessel R. Photochemical and electrochemical reduction of carbon dioxide to carbon monoxide mediated by (2,2′-bipyridine)tricarbonylchlororhenium(I) and related complexes as homogeneous catalysts. Helv Chim Acta. 1986;69:1990–2012. [Google Scholar]
- 13.Kumar B, Smieja JM, Sasayama AF, Kubiak CP. Tunable, light-assisted cogeneration of CO and H2 from CO2 and H2O by Re(bipy-tbu)(CO)3Cl and p-Si in nonaqueous medium. Chem Commun. 2012;48:272–274. doi: 10.1039/c1cc16024a. [DOI] [PubMed] [Google Scholar]
-
14.Yam VWW, Lau VCY, Cheung KK. Synthesis and photophysics of luminescent rhenium(I) acetylides-precursors for organometallic rigid-rod materials—X-ray crystal structures of [
 ] and [
] and [ ] Organometallics. 1995;14:2749–2753. [Google Scholar]
] Organometallics. 1995;14:2749–2753. [Google Scholar] - 15.Gennaro A, Isse AA, Vianello E. Solubility and electrochemical determination of CO2 in some dipolar aprotic solvents. J Electroanal Chem. 1990;289:203–215. [Google Scholar]
- 16.Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulfur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J Chem Soc Dalton Trans. 1984;7:1349–1356. [Google Scholar]
- 17.Gore-Randall E, Irwin M, Denning MS, Goicoechea JM. Synthesis and characterization of alkali-metal salts of 2,2′- and 2,4′-bipyridyl radicals and dianions. Inorg Chem. 2009;48:8304–8316. doi: 10.1021/ic9009459. [DOI] [PubMed] [Google Scholar]
- 18.Schneider J, Jia H, Muckerman JT, Fujita E. Thermodynamics and kinetics of CO2, CO, and H+ binding to the metal center of CO2 reduction catalysts. Chem Soc Rev. 2012;41:2036–2051. doi: 10.1039/c1cs15278e. [DOI] [PubMed] [Google Scholar]
- 19.Sullivan BP, Bolinger CM, Conrad D, Vining WJ, Meyer TJ. One-electron and two-electron pathways in the electrocatalytic reduction of CO2 by fac- Re(2,2′-Bipyridine)(CO)3Cl. J Chem Soc Chem Commun. 1985;20:1414–1415. [Google Scholar]
- 20.Slater S, Wagenknecht JH. Electrochemical reduction of carbon dioxide catalyzed by Rh(diphos)2Cl. J Am Chem Soc. 1984;106:5367–5368. [Google Scholar]
- 21.Bolinger CM, et al. Electrocatalytic reduction of CO2 based on polypyridyl complexes of rhodium and ruthenium. J Chem Soc Chem Commun. 1985;12:796–797. [Google Scholar]
- 22.Wong K-Y, Chung W-H, Lau C-P. The effect of weak Brönsted acids on the electrocatalytic reduction of carbon dioxide by a rhenium tricarbonyl bipyridyl complex. J Electroanal Chem. 1998;453:161–169. [Google Scholar]
- 23.Dixon JE, Bruice TC. Dependence of the primary isotope effect (kH/kD) on base strength for the primary amine catalyzed ionization of nitroethane. J Am Chem Soc. 1970;92:905–909. [Google Scholar]
- 24.Savéant JM, Vianello E. Potential-sweep chronoamperometry theory of kinetic currents in the case of a first order chemical reaction preceding the electron-transfer process. Electrochim Acta. 1962;8:905–923. [Google Scholar]
- 25.DuBois DL, Miedaner A, Haltiwanger RC. Electrochemical reduction of carbon dioxide catalyzed by [Pd(triphosphine)(solvent)](BF4)2 complexes: Synthetic and mechanistic studies. J Am Chem Soc. 1991;113:8753–8764. [Google Scholar]
- 26.Buriak JM, Allen MJ. Lewis acid mediated functionalization of porous silicon with substituted alkenes and alkynes. J Am Chem Soc. 1998;120:1339–1340. [Google Scholar]
- 27.Fonseca Guerra C, Snijders JG, te Velde G, Baerends EJ. Towards an order-N DFT method. Theor Chem Acc. 1998;99:391–403. [Google Scholar]
- 28.te Velde G, et al. Chemistry with ADF. J Comput Chem. 2001;22:931–967. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.