

Abstract
A dicobaloxime in which monomeric Co(III) units are linked by an octamethylene bis(glyoxime) catalyzes the reduction of protons from p-toluenesulfonic acid as evidenced by electrocatalytic waves at -0.4 V vs. the saturated calomel electrode (SCE) in acetonitrile solutions. Rates of hydrogen evolution were determined from catalytic current peak heights (kapp = 1100 ± 70 M-1 s-1). Electrochemical experiments reveal no significant enhancement in the rate of H2 evolution from that of a monomeric analogue: The experimental rate law is first order in catalyst and acid consistent with previous findings for similar mononuclear cobaloximes. Our work suggests that H2 evolution likely occurs by protonation of reductively generated CoIIH rather than homolysis of two CoIIIH units.
Keywords: hydrogen evolving catalysts, solar fuel
Efficient catalytic reduction of protons to dihydrogen is a requirement for one half of a functional solar water splitting system (1, 2). Progress has been made recently in identifying catalysts capable of producing hydrogen from acidic media using either small molecule mimics of hydrogenase active sites or other synthetic systems (3–11).
Our work has focused on difluoroboryl-bridged CoII-diglyoxime complexes that catalyze hydrogen evolution at low overpotentials (12–15). With these complexes, hydrogen evolution is initiated upon reduction to CoI, which then reacts with a proton donor to form a CoIII-hydride (CoIIIH). The CoIIIH intermediate can either undergo subsequent protonation to release H2 and a CoIII species that is reduced to regenerate the catalyst (heterolytic route) or react with another CoIIIH to eliminate H2 by a homolytic route. Alternatively, CoIIIH can be reduced further to form CoIIH, which can react via similar heterolytic or homolytic routes (16). Digital simulations of electrocatalytic waves performed by Hu et al. (17) indicated that the bimolecular homolytic CoIIIH route is the dominant pathway for hydrogen evolution. Our analysis of electron transfer rates in the catalytic cycle confirmed that the barriers to H2 evolution associated with this pathway are more favorable than that for protonation of CoIIIH (17, 18). Recent work in our group examining the mechanism of hydrogen evolution from a photogenerated hydridocobaloxime suggests that hydrogen evolution via protonation of CoIIH is favored under certain conditions (CoIIIH in low concentrations with reducing equivalents in excess) (16). Computational studies reported by Hammes-Schiffer (19, 20) and Muckerman (21) suggest that CoIIH protonation is a very favorable reaction pathway and that relative fluxes through the homolytic CoIIIH and heterolytic CoIIH channels depend on experimental conditions (relative concentrations of acid and catalyst).
In the low-barrier homolytic mechanism, two CoIIIH species must diffuse together in solution in order to react and release H2. It follows that immobilization onto an electrode surface would disfavor this pathway (7, 22, 23). It is of interest in this context that binuclear analogues of many catalysts that rely on bimetallic interactions show enhanced activity when compared to monomeric species, examples include certain Ru and Os diporphyrin H2 evolving catalysts with cofacial orientation (24, 25). In one noteworthy case, Jones and coworkers (26, 27) showed that a “bisalen” analogue of Jacobsen’s Co-(salen) catalyst for hydrolytic kinetic resolution of racemic epoxides (a reaction that also shows a second-order dependence on catalyst concentration) maintained activity when linked to a solid support, whereas the immobilized monomer was essentially inactive (26–28). Because one of our goals is construction of a catalytic cobaloxime-modified electrode for water reduction, we have investigated the reactivity of a covalently-linked dicobalt complex.
Results and Discussion
An octamethylene-linked bis(glyoxime) ligand, tetradecane-2,3,12,13-tetraone tetraoxime (LH4), was synthesized by a method similar to the procedure of Busch and coworkers (Scheme 1) (29). Reaction of the ligand with 2 eq. diphenylglyoxime (dpgH2) and 2 eq. cobalt(II) dichloride in ethanol, followed by addition of 4 eq. pyridine under aerobic conditions, yielded a mixture of products including the desired binuclear CoIIICl complex 1, mononuclear ClCoIII(dpgH)2py, and the singly metalated ligand, ClCoIII(dpgH)(LH3)py. Product purification difficulties prevented us from isolating the analogous binuclear species with a dimethylglyoxime (dmgH2) ligand.
Scheme 1.

The reaction mixture was separated via column chromatography and 1 was isolated in 10% yield. The oxime-linking hydrogen atoms of 1 were replaced by bridging BF2 groups, which enhance stability and shift reduction potentials to more positive values (17), by reflux with excess boron trifluoride in acetonitrile. Under these conditions, the axial pyridine is displaced by acetonitrile yielding the desired binuclear cobaloxime 2.
As a mononuclear analogue of 2, ClCoIII(dmgH)(dpgH)py was synthesized according to Gupta (30–32), and reaction with boron trifluoride diethyl etherate afforded ClCoIII(dmgBF2)(dpgBF2)(CH3CN) (3).
The cyclic voltammogram of 2 (see SI Text, Fig. S1) measured in 0.1 M 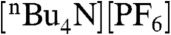 acetonitrile solution reveals an irreversible reduction at approximately 0.2 V vs. the saturated calomel electrode (SCE) attributed to one-electron CoIII reduction accompanied by the loss of an axial chloride and a reversible redox process at -0.37 V vs. SCE assigned to the CoII/I couple. Electrochemical measurements of 3 under similar conditions also show an irreversible CoIII/II couple at approximately 0.2 V vs. SCE and the CoII/I couple at -0.37 V vs. SCE (see SI Text, Fig. S2). The virtually identical CoII/I reduction potentials for 2 and 3 are due to the similarity in electronic properties of the respective ligands. The methyl and alkyl substituents of dimethylglyoxime and tetradecane-2,3,12,13-tetraone tetraoxime have similar electron-donating properties, and the redox potentials of the mixed dioxime complexes 2 and 3 fall, unsurprisingly, between the corresponding CoII/I potentials of Co(dpgBF2)2(CH3CN)2 and Co(dmgBF2)2(CH3CN)2, which appear at -0.28 and -0.55 V vs. SCE, respectively (17, 33). The CoII/I reductions of 2 and 3 are well-defined with peak separations similar to that of the reference Fc+/Fc0 couple (approximately 70 mV) even at low scan rates, and the peak current is linear with the square root of scan rate (see SI Text, Figs. S3 and S4) consistent with a reversible one-electron transfer process at each metal site with no electronic communication between the two cobalt centers (34, 35). The diffusion coefficients for 2 and 3 were estimated from the scan rate dependence of the CoII/I peak current in the absence of acid (see SI Materials and Methods). As expected, bulkier 2 has a substantially smaller diffusion coefficient (2.0 × 10-6 cm2 s-1) (Fig. S3) than the mononuclear complex 3 (8.0 × 10-6 cm2 s-1) (Fig. S4).
acetonitrile solution reveals an irreversible reduction at approximately 0.2 V vs. the saturated calomel electrode (SCE) attributed to one-electron CoIII reduction accompanied by the loss of an axial chloride and a reversible redox process at -0.37 V vs. SCE assigned to the CoII/I couple. Electrochemical measurements of 3 under similar conditions also show an irreversible CoIII/II couple at approximately 0.2 V vs. SCE and the CoII/I couple at -0.37 V vs. SCE (see SI Text, Fig. S2). The virtually identical CoII/I reduction potentials for 2 and 3 are due to the similarity in electronic properties of the respective ligands. The methyl and alkyl substituents of dimethylglyoxime and tetradecane-2,3,12,13-tetraone tetraoxime have similar electron-donating properties, and the redox potentials of the mixed dioxime complexes 2 and 3 fall, unsurprisingly, between the corresponding CoII/I potentials of Co(dpgBF2)2(CH3CN)2 and Co(dmgBF2)2(CH3CN)2, which appear at -0.28 and -0.55 V vs. SCE, respectively (17, 33). The CoII/I reductions of 2 and 3 are well-defined with peak separations similar to that of the reference Fc+/Fc0 couple (approximately 70 mV) even at low scan rates, and the peak current is linear with the square root of scan rate (see SI Text, Figs. S3 and S4) consistent with a reversible one-electron transfer process at each metal site with no electronic communication between the two cobalt centers (34, 35). The diffusion coefficients for 2 and 3 were estimated from the scan rate dependence of the CoII/I peak current in the absence of acid (see SI Materials and Methods). As expected, bulkier 2 has a substantially smaller diffusion coefficient (2.0 × 10-6 cm2 s-1) (Fig. S3) than the mononuclear complex 3 (8.0 × 10-6 cm2 s-1) (Fig. S4).
Upon addition of p-toluenesulfonic acid monohydrate (TsOH•H2O) to solutions of 2 or 3 in an inert atmosphere, catalytic waves were observed at a potential slightly negative of the formal reduction potential, E°′(CoII/I) of the species indicating catalytic hydrogen evolution (Fig. 1). Based on the standard potential for hydrogen evolution for TsOH•H2O in CH3CN (E°′(HA/H2) = -0.27 V vs. SCE) (36), 2 and 3 operate at an overpotential of approximately 100 mV. Similar to the electrocatalytic behavior of Co(dpgBF2)2(CH3CN)2 and Co(dmgBF2)2(CH3CN)2, catalytic waves at low acid concentrations exhibit peak-like shapes indicative of a process wherein catalysis occurs rapidly enough that current is controlled by diffusion of acid to the electrode surface.
Fig. 1.

Cyclic voltammograms of 0.372 mM 2 (Top) and 0.524 mM 3 (Bottom) in the absence (red curve) and presence (all other colors) of p-toluenesulfonic acid monohydrate (TsOH•H2O). Samples were measured at a scan rate of 100 mV s-1 in 0.1 M 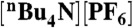 CH3CN solution.
CH3CN solution.
At higher acid-to-catalyst ratios, the catalytic waves approach a plateau. Under these conditions, acid concentration is not depleted in the reaction region and catalytic current plateau indicates that the rate of reduction of the catalytic species at the electrode is equivalent to the rate of its reoxidation by the acid. The plateau currents of 2 and 3 follow a first-order dependence on the concentration of acid (Fig. 2), but tend toward saturation at high catalyst concentrations, possibly indicating a change in rate-limiting step (Fig. 3). An analysis similar to that of Hu et al. (17) was used to calculate the overall rate constant for H2 evolution (vide infra).
Fig. 2.

Dependence of catalytic plateau current on acid concentration for 0.372 mM 2 (Top) and 0.524 mM 3 (Bottom). Data were fit to the function ic = b[TsOH]1/2 as suggested by Eq. 1. For 2: b = 4.8 × 10-4 A M-1/2, for 3: b = 6.3 × 10-4 A M-1/2. Samples were measured at a scan rate of 100 mV s-1 in 0.1 M 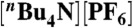 CH3CN solution.
CH3CN solution.
Fig. 3.

Dependence of catalytic plateau current on catalyst concentration for 2 with 12 mM p-toluenesulfonic acid monoydrate (Top) and 3 with with 20 mM p-toluenesulfonic acid monoydrate (Bottom). Samples were measured at a scan rate of 100 mV s-1 in 0.1 M 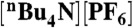 CH3CN solution.
CH3CN solution.
The overall rate constant for H2 evolution was estimated from the height of the catalytic plateau current as a function of acid concentration for 2 and 3. For a reaction that is first order in catalyst and acid, the current in the plateau region (ic, in A) is related to the concentrations of catalyst per metal atom ([Co], in mol cm-3) and acid ([TsOH•H2O] in M) by
| [1] |
where n is the number of electrons (n = 2), F is Faraday’s constant (96,485 C mol-1), A is the area of the electrode surface (cm2), D is the diffusion coefficient (cm2 s-1), and k is the reaction rate constant (M-1 s-1) (37, 38). There is no obvious increase in the apparent rate constant for 2, 1,100 M-1 s-1, compared to that for 3, 900 M-1 s-1, and both lie between the measured values for Co(dpgBF2)2(CH3CN)2 and Co(dmgBF2)2(CH3CN)2 (Table 1). Overall, the electrocatalysis of hydrogen evolution by 2 and 3 is closely analogous to that observed for Co(dmgBF2)2(CH3CN)2 (17).
Table 1.
Reduction potentials, apparent rate constants, and diffusion constants for cobaloxime complexes
If the homolytic reactivity of two CoIIIH species were the rate-limiting step for hydrogen evolution, the rate law would have a second-order dependence on the concentrations of CoI and acid as well as an inverse second-order dependence on the concentration of the conjugate base. Hence, an enhancement in rate would be expected for 2, which is not what we observed. We conclude that [CoIIIH] homolysis is less favorable than protonation of reductively generated CoIIH under the conditions employed for hydrogen production catalyzed by dicobaloxime 2.
Materials and Methods
General Procedures.
All synthetic procedures and electrochemical measurements were performed under anaerobic and anhydrous conditions under an atmosphere of argon or nitrogen with the exception of the preparation of the ligand. Dry acetonitrile was prepared by passing commercially available dry solvent over an activated alumina column and was stored on 4 Å sieves under nitrogen. Tetrabutylammonium hexafluorophosphate was recrystallized from ethanol before use. All other reagents were purchased from chemical suppliers and used as received. ClCo(dmgH)(dpgH)py (dmgH2 = dimethylglyoxime, dpgH2 = diphenylglyoxime, and py = pyridine) was prepared by the method of Gupta et al. (32).
1H and 19F NMR measurements were recorded on a Varian Mercury 300 MHz spectrometer and analyzed using MestReNova (version 6.1.1). 1H NMR spectra were referenced to internal solvents and 19F spectra were referenced using an external C6F6 standard.
Electrospray high resolution mass spectrometry measurements were recorded on a Finnigan LCQ ion trap mass spectrometer. High resolution mass spectrometry (FAB) measurements were recorded using a JEOL JMS-600H high resolution mass spectrometer.
Electrochemical measurements were made with a Pine Instruments WaveNow potentiostat and the data were processed using Igor Pro 5.01 (Wavemetrics). Electrochemical analysis was carried out in a three-electrode cell consisting of a glassy carbon working electrode (surface area = 0.07 cm2), a platinum wire counter electrode, and a freshly prepared Ag/AgNO3 (0.01 M) reference electrode filled with 0.1 M 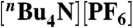 acetonitrile solution. All potentials were referenced to Fc+/Fc0 as an internal standard and converted to SCE (E°(Fc+/Fc0) = 0.380 V vs. SCE).
acetonitrile solution. All potentials were referenced to Fc+/Fc0 as an internal standard and converted to SCE (E°(Fc+/Fc0) = 0.380 V vs. SCE).
Tetradecane-2,3,12,13-Tetraone 3,12-Dioxime Synthesis.
Under an argon atmosphere, sodium metal (1.5 g, 65 mmol) was dissolved in 100 mL of absolute ethanol. Ethyl acetoacetate [8.28 mL (8.45 g), 65 mmol] was syringed into the stirring solution and allowed to react. After 10 min, the yellow solution was heated to reflux followed by the addition of 1,8-dibromooctane (8.02 g, 29.5 mmol) and exposure to atmosphere. The solution was allowed to react for 12 h until neutral followed by the removal of solvent via rotary evaporation to yield a viscous oil containing a white precipitate (NaBr). Enough water was added to the product to dissolve the precipitate, and the product was extracted with ether. The ether layers were combined and dried over anhydrous magnesium sulfate, and the ether was removed via rotary evaporation. To the resulting clear oil, an aqueous solution of potassium hydroxide (4.51 g, 80 mmol) was added, and hydrolysis was effected for 3 h at room temperature producing a cloudy, bright yellow solution. The mixture was cooled to 0 °C followed by the addition of sodium nitrite (4.26 g, 62 mmol) and then 10% sulfuric acid was added dropwise until an aliquot of the yellow solution turned the color of a potassium iodide:starch test dark blue for 15 min. Aqueous potassium hydroxide was added until the mixture became homogeneous, followed by extraction with ether to remove residual starting materials. The yellow mixture was cooled and 4 M sulfuric acid was added until the solution was acidified (pH ≈ 5). The resulting solid was extracted with ether, washed with water and a saturated sodium carbonate solution, and dried over MgSO4. Ether was removed via rotary evaporation to yield a cream solid (2.4 g, 28%). 1H NMR (300 MHz, (CD3)2CO): δ 10.4 (s, 2 H, oxime), δ 2.48 (t, 4 H, J = 7.3 Hz, α-CH2), δ 2.26 (s, 6 H, CH3), and δ 1.5–1.25 (m, 12 H, CH2) ppm.
Tetradecane-2,3,12,13-Tetraone Tetraoxime (LH4) Synthesis.
Tetradecane-2,3,12,13-tetraone 3,12-dioxime (2.4 g, 8.4 mmol), hydroxylamine hydrochloride (2.3 g, 33.5 mmol), and sodium carbonate (2.45 g, 17.7 mmol) were suspended in ethanol (100 mL). Water was added slowly (20 mL) until all solids dissolved and the solution was refluxed for 14 h. The white precipitate that formed was collected, washed with water until chloride was no longer detected in the filtrate, and dried (1.65 g, 63%).1H NMR (300 MHz, (CD3)2CO): δ 10.46 (s, 2 H, oximea), δ 10.41 (s, 2 H, oximeb), δ 2.60 (t, 4 H, J = 7.5 Hz, α-CH2), δ 1.96 (s, 6 H, CH3), and δ 1.41–1.30 (m, 12 H, CH2) ppm. Electrospray MS (50∶50 MeOH∶H2O), m/z calculated for C14H26N4O4∶314.2. Found: 315.0 (M), 296.9 (M–OH).
ClCo(dpgH)py-LH2-ClCo(dpgH)py (LH4 = Tetradecane-2,3,12,13-Tetraone Tetraoxime, py = Pyridine and dpgH2 = Diphenylglyoxime) (1) Synthesis.
Under an argon atmosphere, anhydrous pyridine (0.79 g, 10 mmol) was added by syringe to a refluxing solution of cobalt(II) chloride (0.45 g, 1.9 mmol), diphenylglyoxime (0.49 g, 2.0 mmol), and tetradecane-2,3,12,13-tetraone tetraoxime (0.3 g, 0.9 mmol) in degassed ethanol (50 mL). The heterogeneous mixture was cooled to room temperature, and air was bubbled through the solution for at least 6 h. The solvent was removed via rotary evaporation to yield a brown solid. This brown mixture was dissolved in a minimal amount of chloroform and loaded onto a silica gel column preeluted with chloroform. Three bands were observed with 1∶9 ethyl acetate:chloroform. The first band, corresponding to the monomeric species ClCo(dpgH)2py (as determined by 1H NMR), eluted with 1∶9 ethyl acetate:chloroform, and the second and third bands eluted together with 1∶1 ethyl acetate:chloroform. The second band corresponds to the half-metalated ligand, ClCo(dpgH)(LH3)py (as determined by 1H NMR), and the third to the desired product. Solvent was evaporated from this mixture and the resulting solid was dissolved in minimal chloroform. The two products were separated on a silica gel column with 15∶85 ethyl acetate:chloroform to yield the desired product as a brown crystalline solid (0.11 g, 10%). 1H NMR (300 MHz, CD3CN): δ 18.5 (s, 4 H, -OHO-), δ 8.29 [m, 4 H, py CH(2,6)], δ 7.86 [m, 4 H, py CH(3,5)], δ 7.39 [m, 2 H, py CH(4)], δ 7.4–7.1 (m, 20 H, Ph), δ 2.85 (m, 4 H, α-CH2), δ 2.39 (s, 6 H, CH3), and δ 1.55–1.05 (m, 12 H, CH2) ppm. HRMS (FAB+), m/z calculated for C52H56Cl2Co2N10O8∶1,136.23. Found: 1137.24 (M + H), 1022.3 (M - Cl - pyridine), 979.3 (M - C6H5 - pyridine - 2H) 907.3 (M - 2Cl - 2pyridine + H, M - 3 C6H5 - H), 783.2 (M - 4 C6H5 - Cl - CH3), 704.2 (M - 4 C6H5 - CH3 - Cl - pyridine), and 668.2 (M - 4 C6H5 - CH3 - 2Cl - pyridine - H).
ClCo(dpgBF2)(CH3CN)-L(BF2)2-ClCo(dpgBF2)(CH3CN) (LH4 = Tetradecane-2,3,12,13-Tetraone Tetraoxime and dpgH2 = Diphenylglyoxime) (2) Synthesis.
Under an atmosphere of argon, boron trifluoride diethyl etherate (0.153 g, 1.1 mmol) was syringed into a brown, refluxing solution of 1 (0.15 g, 0.13 mmol) in dry acetonitrile (25 mL). The solution turned red-brown and was allowed to reflux for 1 h, then the solvent was evaporated to yield a red-brown solid that was recrystallized from acetonitrile and diethyl ether (0.15 g, 91%). 1H NMR (300 MHz, CD3Cl): δ 7.4–7.2 (m, 20 H, Ph), δ 3.08 (m, 4 H, α-CH2), δ 2.67 (s, 6H, CH3), and δ 1.7–1.3 (m, 12 H, CH2) ppm. 19F NMR (300 MHz, CD3CN): δ - 151 (s) ppm.
ClCo(dmgBF2)(dpgBF2)(CH3CN) (dmg = Dimethylglyoxime and dpg = Diphenylglyoxime) (3) Synthesis.
Under an atmosphere of argon, ClCo(dmgH)(dpgH)py (0.24 g, 0.455 mmol) was dissolved in dry acetonitrile (15 mL). Boron trifluoride diethyl etherate (0.646 g, 4.55 mmol) was syringed into the brown solution. The resulting red solution was heated to reflux for 1 h then allowed to stir at room temperature for 3 h. The solvent was evaporated, and the resulting red oily solid was redissolved in acetonitrile (4 mL). Diethyl ether (15 mL) was added until a white solid crashed out. The solution was filtered, and the solvent was removed from the red-brown solution. The oily solid was dissolved in ethyl acetate (8 mL) and washed with water (2 mL) then dried over MgSO4. The solvent was evaporated to yield a red-brown solid (0.079 g, 30%).1H NMR (300 MHz, CD3CN): δ 7.5–7.3 (m, 10 H, Ph), δ 2.68 (s, 6 H, CH3) ppm. 19F NMR (300 MHz, CD3CN): δ - 151 (s) ppm. HRMS (FAB+), m/z calculated for C20H19BClCoF4N5O4∶585.06 Found: 585.06 (M), 566.05 (M - F), 509.03 (M - C6H5), 490.03 (M - F - C6H5), 461.04 (M - C6H5 - BF2 + H), and 442.01 (M - C6H5 - BF2 - F + H).
Supplementary Material
ACKNOWLEDGMENTS.
We thank Ian Stewart, Alex Miller, Bryan Stubbert, Charles McCrory, Xile Hu, and Jonas Peters for insightful discussions. This work was supported by the NSF Center for Chemical Innovation (Powering the Planet, CHE-0802907), the Arnold and Mabel Beckman Foundation, CCSER (Gordon and Betty Moore Foundation), and the BP MC2 program. CNV is grateful for support from the Caltech Summer Undergraduate Research Program and an Amgen Scholars Fellowship. JLD was supported by an NSF Graduate Research Fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1118329109/-/DCSupplemental.
References
- 1.Esswein AJ, Nocera DG. Hydrogen production by molecular photocatalysis. Chem Rev. 2007;107:4022–4047. doi: 10.1021/cr050193e. [DOI] [PubMed] [Google Scholar]
- 2.Lewis NS, Nocera DG. Powering the planet: chemical challenges in solar energy utilization. Proc Natl Acad Sci USA. 2006;103:15729–15735. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Appel AM, DuBois DL, Rakowski DuBois M. Molybdenum-sulfur dimers as electrocatalysts for the production of hydrogen at low overpotentials. J Am Chem Soc. 2005;127:12717–12726. doi: 10.1021/ja054034o. [DOI] [PubMed] [Google Scholar]
- 4.Artero V, Fontecave M. Some general principles for designing electrocatalysts with hydrogenase activity. Hydrogenases. 2005;249:1518–1535. [Google Scholar]
- 5.Gloaguen F, Lawrence JD, Rauchfuss TB. Biomimetic hydrogen evolution catalyzed by an iron carbonyl thiolate. J Am Chem Soc. 2001;123:9476–9477. doi: 10.1021/ja016516f. [DOI] [PubMed] [Google Scholar]
- 6.Justice AK, Linck RC, Rauchfuss TB, Wilson SR. Dihydrogen activation by a diruthenium analogue of the Fe-only hydrogenase active site. J Am Chem Soc. 2004;126:13214–13215. doi: 10.1021/ja0455594. [DOI] [PubMed] [Google Scholar]
- 7.Le Goff A, et al. From hydrogenases to noble metal-free catalytic nanomaterials for H2 production and uptake. Science. 2009;326:1384–1387. doi: 10.1126/science.1179773. [DOI] [PubMed] [Google Scholar]
- 8.Lyon EJ, Georgakaki IP, Reibenspies JH, Darensbourg MY. Coordination sphere flexibility of active-site models for Fe-only hydrogenase: Studies in intra-and intermolecular diatomic ligand exchange. J Am Chem Soc. 2001;123:3268–3278. doi: 10.1021/ja003147z. [DOI] [PubMed] [Google Scholar]
- 9.Mejia-Rodriguez R, Chong D, Reibenspies JH, Soriaga MP, Darensbourg MY. The hydrophilic phosphatriazaadamantane ligand in the development of H2 production electrocatalysts: Iron hydrogenase model complexes. J Am Chem Soc. 2004;126:12004–12014. doi: 10.1021/ja039394v. [DOI] [PubMed] [Google Scholar]
- 10.Sun Y, et al. Molecular cobalt pentapyridine catalysts for generating hydrogen from water. J Am Chem Soc. 2011;133:9212–9215. doi: 10.1021/ja202743r. [DOI] [PubMed] [Google Scholar]
- 11.Wilson AD, et al. Hydrogen oxidation and production using nickel-based molecular catalysts with positioned proton relays. J Am Chem Soc. 2006;128:358–366. doi: 10.1021/ja056442y. [DOI] [PubMed] [Google Scholar]
- 12.Baffert C, Artero V, Fontecave M. Cobaloximes as functional models for hydrogenases. 2. Proton electroreduction catalyzed by difluoroborylbis(dimethylglyoximato)cobalt(II) complexes in organic media. Inorg Chem. 2007;46:1817–1824. doi: 10.1021/ic061625m. [DOI] [PubMed] [Google Scholar]
- 13.Dempsey JL, Brunschwig BS, Winkler JR, Gray HB. Hydrogen evolution catalyzed by cobaloximes. Acc Chem Res. 2009;42:1995–2004. doi: 10.1021/ar900253e. [DOI] [PubMed] [Google Scholar]
- 14.Razavet M, Artero V, Fontecave M. Proton electroreduction catalyzed by cobaloximes: Functional models for hydrogenases. Inorg Chem. 2005;44:4786–4795. doi: 10.1021/ic050167z. [DOI] [PubMed] [Google Scholar]
- 15.Du P, Schneider J, Luo G, Brennessel WW, Eisenberg R. Visible light-driven hydrogen production from aqueous protons catalyzed by molecular cobaloxime catalysts. Inorg Chem. 2009;48:4952–4962. doi: 10.1021/ic900389z. [DOI] [PubMed] [Google Scholar]
- 16.Dempsey JL, Winkler JR, Gray HB. Mechanism of H2 evolution from a photogenerated hydridocobaloxime. J Am Chem Soc. 2010;132:16774–16776. doi: 10.1021/ja109351h. [DOI] [PubMed] [Google Scholar]
- 17.Hu X, Brunschwig BS, Peters JC. Electrocatalytic hydrogen evolution at low overpotentials by cobalt macrocyclic glyoxime and tetraimine complexes. J Am Chem Soc. 2007;129:8988–8998. doi: 10.1021/ja067876b. [DOI] [PubMed] [Google Scholar]
- 18.Dempsey JL, Winkler JR, Gray HB. Kinetics of electron transfer reactions of H2-evolving cobalt diglyoxime catalysts. J Am Chem Soc. 2010;132:1060–1065. doi: 10.1021/ja9080259. [DOI] [PubMed] [Google Scholar]
- 19.Solis BH, Hammes-Schiffer S. Theoretical analysis of mechanistic pathways for hydrogen evolution catalyzed by cobaloximes. Inorg Chem. 2011;50:11252–11262. doi: 10.1021/ic201842v. [DOI] [PubMed] [Google Scholar]
- 20.Solis BH, Hammes-Schiffer S. Substituent effects on cobalt diglyoxime catalysts for hydrogen evolution. J Am Chem Soc. 2011;133:19036–19039. doi: 10.1021/ja208091e. [DOI] [PubMed] [Google Scholar]
- 21.Muckerman JT, Fujita E. Theoretical studies of the mechanism of catalytic hydrogen production by a cobaloxime. Chem Commun. 2011;47:12456–12458. doi: 10.1039/c1cc15330g. [DOI] [PubMed] [Google Scholar]
- 22.Gray HB. Powering the planet with solar fuel. Nat Chem. 2009;1:7. doi: 10.1038/nchem.141. [DOI] [PubMed] [Google Scholar]
- 23.Berben LA, Peters JC. Hydrogen evolution by cobalt tetraimine catalysts adsorbed on electrode surfaces. Chem Commun. 2010;46:398–400. doi: 10.1039/b921559j. [DOI] [PubMed] [Google Scholar]
- 24.Collman JP, Ha Y, Wagenknecht PS, Lopez MA, Guilard R. Cofacial bisorganometallic diporphyrins: Synthetic control in proton reduction catalysis. J Am Chem Soc. 1993;115:9080–9088. [Google Scholar]
- 25.Collman JP, Wagenknecht PS, Lewis NS. Hydride transfer and dihydrogen elimination from osmium and ruthenium metalloporphyrin hydrides: Model processes for hydrogenase enzymes and the hydrogen electrode reaction. J Am Chem Soc. 1992;114:5665–5673. [Google Scholar]
- 26.Gill CS, Venkatasubbaiah K, Phan NTS, Weck M, Jones CW. Enhanced cooperativity through design: Pendant Co-III-salen polymer brush catalysts for the hydrolytic kinetic resolution of epichlorohydrin (salen = N,N ’-bis(salicylidene)ethylenediamine dianion) Chem-Eur J. 2008;14:7306–7313. doi: 10.1002/chem.200800532. [DOI] [PubMed] [Google Scholar]
- 27.Venkatasubbaiah K, Gill CS, Takatani T, Sherrill CD, Jones CW. A versatile co(bisalen) unit for homogeneous and heterogeneous cooperative catalysis in the hydrolytic kinetic resolution of epoxides. Chem-Eur J. 2009;15:3951–3955. doi: 10.1002/chem.200900030. [DOI] [PubMed] [Google Scholar]
- 28.Jacobsen EN. Asymmetric catalysis of epoxide ring-opening reactions. Acc Chem Res. 2000;33:421–431. doi: 10.1021/ar960061v. [DOI] [PubMed] [Google Scholar]
- 29.Lance KA, Goldsby KA, Busch DH. Effective new cobalt(II) dioxygen carriers derived from dimethylglyoxime by the replacement of the linking protons with difluoroboron(1+) Inorg Chem. 1990;29:4537–4544. [Google Scholar]
- 30.Gupta BD, Singh V, Yamuna R, Barclay T, Cordes W. Organocobaloximes with mixed dioxime equatorial ligands: A convenient one-pot synthesis. X-ray structures and cis-trans influence studies. Organometallics. 2003;22:2670–2678. [Google Scholar]
- 31.Gupta BD, Yamuna R, Singh V, Tiwari U. Synthesis and characterization of and cis-trans influence in cobaloximes with glyoxime as the equatorial ligand. Organometallics. 2003;22:226–232. [Google Scholar]
- 32.Gupta BD, et al. Organocobaloximes with mixed dioxime equatorial ligands: A convenient one-pot synthesis. X-ray crystal structures of BnCoIII(dmgH)(dpgH)Py and BnCoIII(chgH)(dpgH)py. J Organomet Chem. 2001;627:80–92. [Google Scholar]
- 33.Hu XL, Cossairt BM, Brunschwig BS, Lewis NS, Peters JC. Electrocatalytic hydrogen evolution by cobalt difluoroboryl-diglyoximate complexes. Chem Commun. 2005:4723–4725. doi: 10.1039/b509188h. [DOI] [PubMed] [Google Scholar]
- 34.Fourmond V, Jacques P-A, Fontecave M, Artero V. H2 Evolution and molecular electrocatalysts: Determination of overpotentials and effect of homoconjugation. Inorg Chem. 2010;49:10338–10347. doi: 10.1021/ic101187v. [DOI] [PubMed] [Google Scholar]
- 35.Shimakoshi H, Koga M, Hisaeda Y. Synthesis, characterization, and redox behavior of new dicobalt complexes having monoanionic imine/oxime-type ligands. B Chem Soc Jpn. 2002;75:1553–1558. [Google Scholar]
- 36.Kütt A, et al. A comprehensive self-consistent spectrophotometric acidity scale of neutral Brønsted acids in acetonitrile. J Org Chem. 2006;71:2829–2838. doi: 10.1021/jo060031y. [DOI] [PubMed] [Google Scholar]
- 37.Andrieux CP, Blocman C, Dumas-Bouchiat JM, M’Halla F, Savéant JM. Homogeneous redox catalysis of electrochemical reactions: Part V. cyclic voltammetry. J Electroanal Chem. 1980;113:19–40. [Google Scholar]
- 38.Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. New York: Wiley; 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.