

Abstract
DNA cytosine methylation is a central epigenetic marker that is usually mutagenic and may increase the level of sequence divergence. However, methylated genes have been reported to evolve more slowly than unmethylated genes. Hence, there is a controversy on whether DNA methylation is correlated with increased or decreased protein evolutionary rates. We hypothesize that this controversy has resulted from the differential correlations between DNA methylation and the evolutionary rates of coding exons in different genic positions. To test this hypothesis, we compare human–mouse and human–macaque exonic evolutionary rates against experimentally determined single-base resolution DNA methylation data derived from multiple human cell types. We show that DNA methylation is significantly related to within-gene variations in evolutionary rates. First, DNA methylation level is more strongly correlated with C-to-T mutations at CpG dinucleotides in the first coding exons than in the internal and last exons, although it is positively correlated with the synonymous substitution rate in all exon positions. Second, for the first exons, DNA methylation level is negatively correlated with exonic expression level, but positively correlated with both nonsynonymous substitution rate and the sample specificity of DNA methylation level. For the internal and last exons, however, we observe the opposite correlations. Our results imply that DNA methylation level is differentially correlated with the biological (and evolutionary) features of coding exons in different genic positions. The first exons appear more prone to the mutagenic effects, whereas the other exons are more influenced by the regulatory effects of DNA methylation.
Keywords: methylation-associated mutation, exon evolution, genomics, deep sequencing, bioinformatics
DNA methylation is a common form of epigenetic modification that is important for a variety of biological functions, including transcriptional silencing (1), genomic imprinting (2), X-chromosome inactivation (3), the silencing of transposons (4), tumorigenesis (5), and the differentiation of pluripotent cells (6). DNA methylation is unevenly distributed in the human genome. For example, the CpG islands near the promoter regions of active genes tend to be unmethylated (7–10), because methylation in these regions is strongly correlated with transcriptional suppression (8). It has also been reported that exons tend to be more methylated than introns, and that sharp transitions of methylation occur at exon–intron boundaries in human (6). Moreover, the level of DNA methylation also varies among exonic regions. The levels of DNA methylation in the first exons were reported to be lower than in downstream exons, and tightly linked to transcriptional silencing (11). In addition, the level of DNA methylation exhibits a slight but sharp step-down at the transcriptional terminal sites (6).
In terms of molecular evolution, DNA methylation is relevant in two aspects. First, DNA methylation can significantly increase the rate of spontaneous C-to-T mutations at CpG dinucleotides (12–14). Therefore, the level of DNA methylation should be positively correlated with mutation rate. Because DNA methylation is unevenly distributed within the gene body, the rate of mutation may vary significantly among different coding exons of the same genes. However, whether the uneven distribution of DNA methylation is correlated with intragene variations in evolutionary rate, and to what extent, remains unexplored. Second, the level of DNA methylation of gene bodies has been reported to be positively correlated with gene expression (6, 15) (although DNA methylation in promoter regions is known to be related to transcriptional repression) (8). Of note, highly expressed genes are subject to strong selective constraints, and tend to evolve slowly (16–18). Hence, highly methylated genes should have evolved slowly. In addition, methylated genes have been suggested to be more functionally important than unmethylated genes, and tend to evolve slowly (19, 20). However, these observations contradict the proposition that DNA methylation may increase evolutionary rates because of its mutagenic effects. Considering the multifaceted biological roles and the uneven intragene distributions of DNA methylation, we reason that the level of DNA methylation (as well as its spatiotemporal dynamics) may be significantly correlated with intragene variations in evolutionary rate. Such correlations, if any, may result either from the mutagenic effect of DNA methylation, or from its involvement in regulating selection-targeted biological features (e.g., the expression level) of coding exons. Therefore, it is of interest to investigate how this duality of DNA methylation is reconciled in exon evolution, and how DNA methylation is correlated with exonic evolutionary rate.
In this study, we systematically examine the correlations between regional (exonic) DNA methylation levels and the exonic evolutionary rates of mammalian coding exons. We divide coding exonic regions into three groups: first, internal, and last exons. We then use experimentally determined DNA methylation datasets derived from different human cells to address the following questions: (i) Does the correlation between the DNA methylation level and the extent of CpG depletion (a measurement of methylation-induced C-to-T mutation rate) vary among coding exons? (ii) Is the exonic DNA methylation level related to variations in exonic expression level within the same gene? (iii) How is the exonic DNA methylation level separately associated with synonymous substitution rates (dS), nonsynonymous substitution rates (dN), and the dN/dS ratio of individual exons? and (iv) Is the sample specificity of DNA methylation level correlated with the three preceding evolutionary measurements? We demonstrate that exonic evolutionary rates are significantly correlated with the regional variations in DNA methylation level. These correlations may be ascribed to the role of DNA methylation in increasing mutation rate, and possibly to its involvement in the regulations of exonic expression. Interestingly, the two factors appear to be differentially correlated with the evolutionary rates of exons in different genic positions: the first exons are more prone to the increase in mutation rate, whereas the internal and last exons seem to be more affected by the regulatory effects of DNA methylation. Thus, our results reveal that DNA methylation, a prevalent epigenetic modification, may be significantly correlated with exon evolution in an unexpectedly complex pattern.
Results
Distribution of DNA Methylation in Coding Sequences.
To investigate the level of DNA methylation in human coding sequences (CDSs), we retrieved single-base resolution DNA methylation data from six human cell lines (6, 7, 21) (Table 1). These cell lines span multiple spatial (i.e., blood cells, ES cells, and different fibroblasts) and temporal dimensions (i.e., undifferentiated human ES cells, ES cell-derived fibroblasts, and neonatal fibroblasts). The CpGs that are experimentally determined to be methylated are designated as “mCGs” (Materials and Methods). The CpGs examined in this study represent the majority of CpG dinucleotides in the analyzed human CDSs (Table 1). Throughout this study, the DNA methylation level is measured by the density of mCG (9).
Table 1.
Base resolution DNA methylation data used in this study
| Sample | Description (Ref.) | No. of exons | Average CpG coverage, %* |
| S1 | Peripheral blood mononuclear cells (21) | 8,471 | 63.38 |
| S2 | H1 human ES cells (7) | 19,188 | 87.85 |
| S3 | IMR90 fetal lung fibroblasts (7) | 21,665 | 92.32 |
| S4 | WA09 human ES cells (6) | 20,198 | 94.45 |
| S5 | Fibroblastic differentiated derivatives of WA09 human ES cells (6) | 18,798 | 91.81 |
| S6 | Neonatal human foreskin fibroblasts (6) | 19,167 | 92.36 |
*Coverage of the CpG dinucleotides for each exon is the no. of the sampled CpG dinucleotides/(no. of the sampled CpG dinucleotides + no. of the nonsampled CpG dinucleotides).
It has been reported that DNA methylation is unevenly distributed in different genic regions (which was not measured for individual exons) (7, 9, 10). To analyze the correlations between DNA methylation and evolutionary rates at the exon level, we should confirm the uneven distribution of DNA methylation for individual coding exons. To this end, we first examine the exonic mCG density of the six cell lines. We observe significant variations in exonic DNA methylation levels among cell types (Fig. S1A). This observation is consistent with the results of previous studies (7, 22, 23). Next, we divide coding exons into three groups: first, last, and internal exons (Table S1), and compare the average mCG densities of these three groups for each cell type. We show that in all of the six examined cell lines, the lowest mCG density occurs in the first exons, followed by the last exons, and finally by the internal exons (Fig. S1B). This result appears consistent with the previously reported decline in methylation density near the transcriptional start sites (7), and the slight but sharp step-down of DNA methylation level at the transcriptional terminal sites (6). Because the majority of DNA methylation was reported to occur in regions of low CpG density (defined as the number of CpG dinucleotides per 100 bp) (6, 7), we then examine whether the average CpG density differs among the three groups of exons. Indeed, we observe the highest average CpG density in the first exons, followed by the last exons, and then by the internal exons (Fig. S1C). Overall, we show that when individual exons are considered, the previously reported distribution patterns of DNA methylation hold well.
Because DNA methylation has been implicated in the regulation of mRNA splicing, an interesting question is whether different exon types in alternative splicing differ in the level of DNA methylation (24). We therefore examine the mCG densities separately for alternatively spliced exons (ASEs) and constitutively spliced exons (CSEs; Table S1). Here, CSEs are exons that always occur in the transcript isoforms of a gene, whereas ASEs do not (Materials and Methods). We find that ASEs tend to have a higher level of average mCG density than CSEs (Fig. S2A). Though it remains unclear how DNA methylation might affect alternative splicing, our observation appears consistent with a previous report that CpG hypermethylation occurs frequently in alternatively spliced sites (25). We further examine the mCG density separately for ASEs and CSEs in the first, last, or internal coding exons. Our result indicates that regardless of the CSE/ASE exon type, the internal exons have the highest level of mCG density, whereas the first exons have the lowest (Fig. S2B). This observation suggests that the CSE/ASE exon type and the relative positions of an exon are both significantly correlated with exonic DNA methylation level.
Mutagenic Effect of DNA Methylation Varies Among Coding Exons.
We next analyzed the correlation between the exonic mCG density and the CpGO/E ratio (i.e., the ratio of the observed-to-expected number of CpG dinucleotides; SI Text). Of note, a low CpGO/E indicates a high level of C-to-T mutations, which has been reported to result mostly from DNA methylation (18, 26, 27). Therefore, a negative correlation between DNA methylation level and CpGO/E is expected (10, 26, 27). We assume that the coefficient of correlation between the two measurements reflects the proportion of methylated CpGs having actually undergone mutation. A larger absolute value of the coefficient may indicate a greater number of methylated CpGs having been mutated. Accordingly, we first examine the Pearson’s coefficient of correlation (r) between CpGO/E and mCG density separately for the six cell lines. Fig. 1A shows that the coefficient falls between −0.18 and −0.35 (all P values < 10−15). We then evaluate the correlation separately for the first, last, and internal exons. Of interest, as illustrated in Fig. 1B, the highest correlation between CpGO/E and mCG density occurs in the first exons, followed by the last exons, and finally by the internal exons. Notably, this order is observed in all of the six examined cell lines, despite considerable variations in mCG density (Fig. S1A) and variations in the CpGO/E–mCG density correlation (Fig. 1A). Our results imply that the mutagenic effect of DNA methylation is the strongest in the first exons and the weakest in the internal exons. Hence, DNA methylation may more easily induce C-to-T mutations in the first coding exons, despite the low levels of methylation in this exon group (Fig. S1B). By contrast, the mutagenic effect appears to be strongly inhibited in the internal exons even though they exhibit the highest mCG density among the three exon groups (Fig. S1B). This observation is suggestive of strong selective constraints on the internal exons.
Fig. 1.

(A) Pearson’s r between CpGO/E and the mCG density for the six analyzed cell lines. (B) Pearson’s r between CpGO/E and the mCG density for the first, last, and internal exons in the six cell lines. (C) Comparison of the median evolutionary measurements (dN/dS, dN, and dS) in the first, last, and internal exons. Statistical significance was estimated using Pearson’s r (A and B) and a two-tailed Wilcoxon rank-sum test (C): *P < 0.05, **P < 0.01, and ***P < 0.001. NS, not significant. The dashed curves in C indicate the statistical significance in comparisons between the different exon groups.
To further investigate the selection pressures imposed on different exon groups, we calculated dN/dS, dN, and dS (SI Text) for the three groups of exons based on human–rhesus macaque and human–mouse orthologous genes. We find the highest median dN/dS and dN in the first exons, followed by the last exons, and then by the internal exons (Fig. 1C). This observation appears consistent with our inference that the internal exons are subject to strong negative selection. However, for dS, the differences between exon groups are less clear (Fig. 1C), which may be partly explained by alternative splicing, because synonymous sites of ASEs and CSEs have been suggested to be subject to different evolutionary forces (28) (Fig. S3 and SI Text).
Position-Dependent Correlations Between DNA Methylation and Coding Exon Evolution.
We have demonstrated that coding exons in different genic positions have different levels of mCG density, and are subject to different levels of selective constraint. We are then interested to know whether such differences are reflected in the correlations between mCG density and the evolutionary measurements (i.e., dN, dS, and the dN/dS ratio) in the first, last, and internal exons. As shown in Fig. 2 A and B, Left, the mCG density is positively correlated with dN/dS and dN in the first exons in both human–macaque and human–mouse comparisons. By contrast, for the last and internal exons, the correlations are negative. Meanwhile, the correlations between mCG density and dS are less clear (Fig. 2C, columns labeled “Before control”). However, these results should be treated cautiously because several confounding factors have not been controlled. For example, CpG density is known to be negatively correlated with DNA methylation level (6) (Fig. S1C). In addition, both G + C content and exon length have been shown to be positively correlated with evolutionary rates (27, 29). Furthermore, the factor of CSE/ASE exon type should also be controlled because mCG density is associated with the CSE/ASE exon type (Fig. S2 A and B), and because ASEs tend to evolve faster than CSEs (28, 30–32). Thus, we reevaluate the above correlations by using partial correlation analyses (33) to simultaneously control for these four stated factors: CpG density, G + C content, exon length, and CSE/ASE exon type. Of interest, the negative correlations between protein evolutionary rates (i.e., dN/dS and dN) and mCG density are maintained in the last and internal exons (Fig. 2 A and B, columns labeled “After control”). However, the positive correlations between dN/dS and mCG density become less significant in the first exons (Fig. 2A, columns labeled “After control”), probably because both dN and dS increase with mCG density in the first exons when the potential confounding factors are controlled. In other words, for the first exons, the increased dN in highly methylated regions may be partly explained by the increase in dS, thus weakening the correlation between dN/dS and the level of DNA methylation. By contrast, mCG density remains negatively correlated with both dN/dS and dN, and positively correlated with dS for the last and internal exons, even when the four potential confounding factors are controlled. This observation suggests that for the last and internal exons, highly methylated regions are subject to intensified selection pressures at the amino acid level even though the mutation rate may have been increased (leading to elevated dS) in these regions.
Fig. 2.

Spearman’s rank coefficient of correlation (ρ) between mCG density and (A) dN/dS; (B) dN; and (C) dS. The left and right columns under each heading illustrate the correlations in human–macaque and human–mouse comparisons before and after controlling for the four potential confounding factors (CpG density, G + C content, exon length, and the CSE/ASE exon type). *P < 0.05, **P < 0.01, and ***P < 0.001.
Sample Specificity of DNA Methylation Level Is Positively Correlated with dN/dS and dN.
In addition to mCG density, the sample specificity of DNA methylation level (τm) may also be related to evolutionary rates, because this measurement reflects the spatiotemporal variations in DNA methylation level, which in turn may indicate the level of biological importance and potential selective constraints. Of note, τm of an exonic region is defined as the heterogeneity of its DNA methylation level (Materials and Methods). A higher τm value indicates a greater variation in the mCG density across the examined samples (i.e., higher sample specificity of DNA methylation level). Table S2 shows that dN/dS and dN are both positively correlated with τm in all three exon groups. The correlations are maintained when the four stated confounding factors are controlled. One potential caveat here is that τm may be correlated with DNA methylation level. We thus examine the correlation between the two methylation measurements. Interestingly, the two measurements are positively correlated in the first exons but negatively correlated in the last and internal exons (Table 2). Because τm is positively correlated with both dN/dS and dN (Table S2), the results in Table 2 are consistent with our observation that the correlations between mCG density and dN are positive in the first exons but negative in the last and internal exons (Fig. 2).
Table 2.
Spearman’s ρ between mCG density and sample specificity of mCG density (τm)
| S1 | S2 | S3 | S4 | S5 | S6 | |
| First exon | 0.517*** | 0.576*** | 0.390*** | 0.534*** | 0.556*** | 0.374*** |
| Last exon | −0.301*** | −0.161*** | −0.457*** | −0.258*** | −0.311*** | −0.465*** |
| Internal exon | −0.370*** | −0.227*** | −0.546*** | −0.337*** | −0.406*** | −0.556*** |
***P < 0.001.
To clarify which of the two features (τm or mCG density) is more important for determining exonic evolutionary rates, we evaluate the partial correlations between each of the three evolutionary measurements (dN/dS, dN and dS) and τm (or the average mCG density) by simultaneously controlling for the average mCG density (or τm) and the four potential confounding factors. Our results show that for the first exons, dN/dS and dN are more strongly correlated with τm than with the average mCG density, whereas the reverse is true for dS (Fig. S4, Top). Meanwhile, for the internal and last exons, dN/dS and dN are more strongly correlated with the average mCG density, and dS has similar levels of correlation with both of the methylation measurements (Fig. S4, Middle and Bottom). These results indicate that the first exons and nonfirst exons may be affected by different evolutionary forces. Furthermore, our observations indicate that τm is more important than the average mCG density in affecting dN/dS and dN for the first exons. By contrast, the average mCG density is more important for the nonfirst exons in this regard.
Discussion
In this study, we use experimentally determined DNA methylation data to analyze the correlation between DNA methylation levels and evolutionary rates in coding exons. We first show that the first and nonfirst exons are subject to different evolutionary forces and different levels of DNA methylation. The order of average mCG density is first exons < last exons < internal exons (Fig. S1B), whereas the reverse order applies for the average CpG density (Fig. S1C), the mutagenic effect of DNA methylation (Fig. 1B), and dN/dS and dN values (Fig. 1C). In addition, dN and dS are both positively correlated with mCG density in the first exons (Fig. 2), suggesting that more densely methylated first exons evolve more rapidly. There are two possible explanations for this observation. First, the first exons, which usually contain N-terminal signal peptides, appear to be under more relaxed selection pressure than other exons (34). This relaxed selection pressure may cause the mutagenic effect of DNA methylation to be more evident, leading to higher dS and dN in this exon group. Second, the first exons tend to be part of CpG islands (35, 36), in which a strong negative correlation between CpGO/E and mCG density has been previously reported (13). Indeed, when we remove the first exons that overlap with CpG islands by ≥50% of the exon length, the absolute values of the coefficient of correlation decrease considerably (Fig. S5). However, in general, the correlations remain statistically significant (Fig. S5).
For the nonfirst exons (i.e., the internal and last exons), interestingly, the story is quite different. The mutagenic effect of DNA methylation is relatively weak in these exons (Fig. 1B). Although highly methylated nonfirst exons have a higher dS, the opposite is observed for dN (Fig. 2 B and C). Thus, the dN/dS ratios are negatively correlated with mCG density for these exons (Fig. 2A). In other words, dN/dS and dN decrease with increasing levels of CpG methylation in the nonfirst exons. The differences between the first and nonfirst exons in the mCG density–evolutionary rate correlations may be explained if mCG density influences the splicing/expression level differentially in different groups of exons. Because the RNA sequencing (RNA-seq) data produced by high-throughput transcriptome sequencing are available for samples S2 and S3 (Table 1), we may evaluate the correlation between mCG density and exonic expression level (SI Text) for these two samples. As expected, Spearman’s rank correlation tests show that the exonic expression level is negatively correlated with dN/dS and dN in all of the three exon groups in both of human–macaque and human–mouse comparisons (Table S3). Interestingly, we also find that mCG density and exonic expression levels are negatively correlated in the first exons but positively correlated in the nonfirst exons (Table S4). Thus, our results suggest that mCG density is differentially associated with the expression level in different exon groups. These results may partly explain why dN and CpG methylation are positively correlated in the first exons but negatively correlated in the nonfirst exons. Of note, the first exons are considered by some researchers as part of CpG islands (35, 36), or as part of the promoter region (37, 38). Therefore, the observed differences between the first exons and nonfirst exons may be viewed as the differential influences of DNA methylation on the potential promoter regions and the rest of the coding sequences.
Furthermore, a previous study by Laurent et al. (6) indicated that the level of gene expression (from microarray analysis) is negatively correlated with DNA methylation around the transcription start sites, but is positively correlated with mCG density in the gene body and in the regions around the transcription termination sites (6). Although our results appear to echo this previous report, our study presents the first work to demonstrate that the exonic DNA methylation level is differentially associated with the expression level and evolutionary rates of exons at different relative positions in the gene. We use RNA-seq data to achieve this exon-level resolution of mRNA expression. By contrast, the study by Laurent et al. (6) measured the mRNA expression level of the gene as a whole using the microarray approach; therefore, their results are applicable for entire genes, rather than individual exons, as demonstrated in this study.
Meanwhile, the correlation between DNA methylation level and RNA-seq–based gene expression level was also explored in a study by Lister et al. (7), the dataset of which was included in our study (samples S2 and S3 in Table 1). Lister et al. (7) explored the correlation between gene body methylation level and mRNA expression level for genes as a whole. They found the correlation to be positive in one cell line (sample S3), whereas the correlation was statistically insignificant in the other (sample S2) (7). In the current study, we took the research one step further by analyzing the same correlation at the exon level. Interestingly, we obtained consistent results for both cell lines: the correlation between exonic mCG density and exonic expression level is negative for the first exons but positive for the nonfirst exons (Table S4). This difference between the first and nonfirst exons may partly explain why Lister et al. (7) obtained inconsistent results from different cell lines: the positive and negative correlations of different exon groups may occasionally cancel each other out, leading to a lack of correlation when a gene is considered as a whole.
One interesting finding in our study is that the sample specificity of τm is positively correlated with dN/dS and dN in all three exon groups (Table S2). The observation that τm and mCG density are positively correlated in the first exons but are negative correlated in the nonfirst exons (Table 2) is interesting. We speculate that the lower mCG density of the first exons may indicate less epigenetic suppression of transcriptional initiation (8), and therefore a higher level of exonic expression (Table S4). Furthermore, highly expressed genes tend to have lower tissue specificity of expression (18, 39), which may be reflected in the low tissue specificity of τm in the first exons. These interconnections between biological features may have led to the positive correlation between mCG density and τm. By contrast, a low level of DNA methylation in the nonfirst exons is associated with a low exonic expression level (Table S4), but a high τm (Table 2). We speculate that the DNA methylation level may be associated with the splicing and/or exonic expression levels of the nonfirst exons, which in turn are correlated with exonic evolutionary rates. The correlations among mGC density, τm, exonic expression level, and exonic evolutionary rates are summarized in Fig. 3.
Fig. 3.

The correlations among mCG density, evolutionary rates, sample specificity of mCG Density (τm), and exonic expression level in (A) the first exons and (B) the last or internal exons. Green and red lines represent positive and negative correlations, respectively. NS, not significant.
Our study analyzes the relationship between experimentally determined CpG methylation level and exon-level sequence evolution in mammals. Our results indicate that (i) highly methylated coding regions tend to have higher dS values than lowly methylated regions, regardless of the relative positions of exons (i.e., first, internal, or last exons); and (ii) highly methylated first exons tend to have higher dN values, higher levels of exonic expression, and lower levels of sample specificity of mCG density than lowly methylated ones, whereas the reverse applies for the internal and last exons. The differences between the first exons and nonfirst exons may result from the differential biological effects of DNA methylation in different groups of exons. In the first exons, DNA methylation appears to induce spontaneous C-to-T mutations more easily (Fig. 1B), thus increasing both dS and dN (Fig. 2 B and C). By contrast, DNA methylation-induced mutations in the nonfirst exons seem to be strongly inhibited, leading to a weaker correlation between mCG density and CpGO/E (Fig. 1B). Furthermore, the levels of DNA methylation and exonic expression are negatively correlated in the first exons, but positively correlated in the nonfirst exons. Therefore, our study indicates that DNA methylation may be correlated with the evolution of mammalian coding exons in an unexpectedly complex, position-dependent pattern.
Materials and Methods
Data Retrieval.
The base-resolution DNA methylation data from six human cell lines (Table 1) was generated with bisulfite (samples S1, S4, S5, and S6) or methylC (samples S2 and S3) sequencing, and was downloaded from NGSmethDB (http://bioinfo2.ugr.es/meth/NGSmethDB.php) (40). To ensure accuracy, only the CpG dinucleotides that are covered by five or more bisulfite/methylC reads were retained (such CpG dinucleotides are designated as “sampled CpGs”). The methylation status of a CpG site was expressed as a 0–100% frequency (defined as the percentage of reads that support the methylation status at the CpG site). Only the CpGs with a methylation frequency of ≥80% were regarded as methylated (6, 41), and designated as “mCGs.” To ensure that the examined CDSs contain sufficient information for estimations of the methylation level, only the CDSs that contained ≥10 sampled CpGs were considered. Furthermore, because the accuracy of evolutionary rate measures may be compromised in the case of short exons (e.g., <50 bp) (29, 30, 42), we only considered the CDSs with ≥50 bp in length. In fact, more than 90% of the examined CDSs are ≥100 bp in length. Thus, the potential noise in evolutionary rate estimates should be limited. The RNA-seq data derived from samples S2 (H1 human ES cells) and S3 (IMR90) were downloaded from the study by Lister et al. (7). The CpG island data were downloaded from the University of California at Santa Cruz genome browser (http://genome.ucsc.edu/). The human gene annotations and the corresponding coding sequences were downloaded from the Ensembl genome browser (http://www.ensembl.org/), version 59. According to the relative positions of exons in the Ensembl-annotated genes, the retrieved coding exonic regions were divided into three groups: first, internal, and last exons. The CDSs that overlap with noncoding RNAs or pseudogenes were excluded. Single-exon genes were also excluded. In addition, the CSEs were defined as exonic regions that are annotated as CDSs in all alternatively spliced transcripts of a gene, whereas ASEs were defined as exonic regions that are annotated as CDSs in some alternatively spliced transcripts, but as introns in other transcripts of a gene. All of the retrieved alternatively spliced transcripts (transcripts encoded by the same Ensembl-annotated genes) are experimentally supported.
Measurement of mCG Density.
The methylation level of a particular exonic region was measured by calculating the density of mCG per 100 CpG dinucleotides, which was defined as
Measurement of Sample Specificity of mCG Density.
τm was defined as
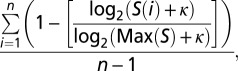 |
where n = 6 is the number of human samples examined in this study, S(i) indicates the mCG density of the exon of interest in sample i, and Max(S) is the highest mCG density of the exon across all examined samples. The measurement of τm value is similar to that usually applied for evaluating tissue specificity of gene expression (16). ĸ is a pseudocount arbitrarily set as 10 to avoid the occurrence of undefined values. τm ranges from 0 to 1, with higher τm values indicating greater variations (and higher sample specificities) in the level of mCG density across samples.
Supplementary Material
Acknowledgments
We thank Shuo-Huang Chen for programming assistance. This work was supported by the Genomics Research Center of Academia Sinica (T.-J.C.); National Science Council of Taiwan Grants NSC99-2628-B-001-008-MY3 (to T.-J.C.) and NSC101-2311-B-400-003 (to F.-C.C.); and intramural funding from the National Health Research Institutes (F.-C.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1208214109/-/DCSupplemental.
References
- 1.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 2.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 3.Heard E, Clerc P, Avner P. X-chromosome inactivation in mammals. Annu Rev Genet. 1997;31:571–610. doi: 10.1146/annurev.genet.31.1.571. [DOI] [PubMed] [Google Scholar]
- 4.Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- 5.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 6.Laurent L, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki MM, Bird A. DNA methylation landscapes: Provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 9.Feng S, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA. 2010;107:8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- 11.Brenet F, et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE. 2011;6:e14524. doi: 10.1371/journal.pone.0014524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ehrlich M, Wang RY. 5-Methylcytosine in eukaryotic DNA. Science. 1981;212:1350–1357. doi: 10.1126/science.6262918. [DOI] [PubMed] [Google Scholar]
- 13.Mugal CF, Ellegren H. Substitution rate variation at human CpG sites correlates with non-CpG divergence, methylation level and GC content. Genome Biol. 2011;12:R58. doi: 10.1186/gb-2011-12-6-r58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang DG, Green P. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc Natl Acad Sci USA. 2004;101:13994–14001. doi: 10.1073/pnas.0404142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ball MP, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao BY, Scott NM, Zhang J. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol. 2006;23:2072–2080. doi: 10.1093/molbev/msl076. [DOI] [PubMed] [Google Scholar]
- 17.Chen FC, Chen CJ, Li WH, Chuang TJ. Gene family size conservation is a good indicator of evolutionary rates. Mol Biol Evol. 2010;27:1750–1758. doi: 10.1093/molbev/msq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park J, Xu K, Park T, Yi SV. What are the determinants of gene expression levels and breadths in the human genome? Hum Mol Genet. 2012;21:46–56. doi: 10.1093/hmg/ddr436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takuno S, Gaut BS. Body-methylated genes in Arabidopsis thaliana are functionally important and evolve slowly. Mol Biol Evol. 2012;29:219–227. doi: 10.1093/molbev/msr188. [DOI] [PubMed] [Google Scholar]
- 20.Sarda S, Zeng J, Hunt BG, Yi SV. The evolution of invertebrate gene body methylation. Mol Biol Evol. 2012;29:1907–1916. doi: 10.1093/molbev/mss062. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010;8:e1000533. doi: 10.1371/journal.pbio.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cedar H, Bergman Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 24.Shukla S, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anastasiadou C, Malousi A, Maglaveras N, Kouidou S. Human epigenome data reveal increased CpG methylation in alternatively spliced sites and putative exonic splicing enhancers. DNA Cell Biol. 2011;30:267–275. doi: 10.1089/dna.2010.1094. [DOI] [PubMed] [Google Scholar]
- 26.Bird AP, Taggart MH. Variable patterns of total DNA and rDNA methylation in animals. Nucleic Acids Res. 1980;8:1485–1497. doi: 10.1093/nar/8.7.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park J, et al. Comparative analyses of DNA methylation and sequence evolution using Nasonia genomes. Mol Biol Evol. 2011;28:3345–3354. doi: 10.1093/molbev/msr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen FC, Wang SS, Chen CJ, Li WH, Chuang TJ. Alternatively and constitutively spliced exons are subject to different evolutionary forces. Mol Biol Evol. 2006;23:675–682. doi: 10.1093/molbev/msj081. [DOI] [PubMed] [Google Scholar]
- 29.Chen FC, Pan CL, Lin HY. Independent effects of alternative splicing and structural constraint on the evolution of mammalian coding exons. Mol Biol Evol. 2012;29:187–193. doi: 10.1093/molbev/msr182. [DOI] [PubMed] [Google Scholar]
- 30.Chen FC, Chuang TJ. The effects of multiple features of alternatively spliced exons on the K(A)/K(S) ratio test. BMC Bioinformatics. 2006;7:259. doi: 10.1186/1471-2105-7-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen FC, Chuang TJ. Different alternative splicing patterns are subject to opposite selection pressure for protein reading frame preservation. BMC Evol Biol. 2007;7:179. doi: 10.1186/1471-2148-7-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xing Y, Lee C. Evidence of functional selection pressure for alternative splicing events that accelerate evolution of protein subsequences. Proc Natl Acad Sci USA. 2005;102:13526–13531. doi: 10.1073/pnas.0501213102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SH, Yi SV. Understanding relationship between sequence and functional evolution in yeast proteins. Genetica. 2007;131:151–156. doi: 10.1007/s10709-006-9125-2. [DOI] [PubMed] [Google Scholar]
- 34.Li YD, et al. The rapid evolution of signal peptides is mainly caused by relaxed selection on non-synonymous and synonymous sites. Gene. 2009;436:8–11. doi: 10.1016/j.gene.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 35.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 36.Larsen F, Gundersen G, Lopez R, Prydz H. CpG islands as gene markers in the human genome. Genomics. 1992;13:1095–1107. doi: 10.1016/0888-7543(92)90024-m. [DOI] [PubMed] [Google Scholar]
- 37.Aerts S, Thijs G, Dabrowski M, Moreau Y, De Moor B. Comprehensive analysis of the base composition around the transcription start site in Metazoa. BMC Genomics. 2004;5:34. doi: 10.1186/1471-2164-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci USA. 2006;103:1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang L, Li WH. Mammalian housekeeping genes evolve more slowly than tissue-specific genes. Mol Biol Evol. 2004;21:236–239. doi: 10.1093/molbev/msh010. [DOI] [PubMed] [Google Scholar]
- 40.Hackenberg M, Barturen G, Oliver JL. NGSmethDB: A database for next-generation sequencing single-cytosine-resolution DNA methylation data. Nucleic Acids Res. 2011;39(Database issue):D75–D79. doi: 10.1093/nar/gkq942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meissner A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nekrutenko A, Makova KD, Li WH. The K(A)/K(S) ratio test for assessing the protein-coding potential of genomic regions: An empirical and simulation study. Genome Res. 2002;12:198–202. doi: 10.1101/gr.200901. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.