

Abstract
Background
Despite increasing evidence that neuroanatomical abnormalities underlie pathological anxiety, social anxiety disorder (SAD), although among the most common of anxiety disorders, has received little attention. Using Magnetic Resonance Imaging, we (1) examined grey matter (GM) differences between generalized SAD and healthy control groups; (2) retested the findings in an independent clinical sample; and (3) tested for specificity by contrasting the SAD group to a separate group of panic disorder (PD) subjects.
Methods
The primary SAD group (N=16) was required to meet DSM-IV criteria for SAD, with onset by age 30; controls (N=20) had no lifetime history of anxiety. The replication sample included 17 generalized SAD and 17 control subjects. The PD comparison group (N=16) was required to have no lifetime SAD. Images were acquired on a 1.5Tesla GE Signa MRI scanner using a 3D T1-weighted spoiled gradient recalled pulse sequence. Morphological differences were determined using voxel based morphometry, in SPM8.
Results
After adjusting for age, gender, and total intracranial volume, SAD (as compared to control) subjects had greater GM in the left parahippocampal and middle occipital, and bilateral supramarginal and angular cortices, and left cerebellum; and lower GM in bilateral temporal poles and left lateral orbitofrontal cortex. Cerebellar, parahippocampal, and temporal pole differences were observed in both samples, survived whole brain corrections, and were not observed in the PD group, pointing to relative specificity to SAD.
Conclusions
These findings parallel the functional literature on SAD, and suggest structural abnormalities underlying the functional disturbances.
Keywords: social anxiety disorder/social phobia, magnetic resonance imaging (MRI), voxel based morphometry (VBM), panic disorder, cerebellum, temporal pole, parahippocampal gyrus
Introduction
Anxiety disorders, as defined by the current Diagnostic and Statistical Manual (DSM-IV)(1), are among the most common psychiatric disorders. They share prominent anxiety as a clinical feature, as well as some abnormalities in brain circuitry associated with fear processing(2). Anxiety is also clinically heterogeneous(3–4), however, and identifying abnormalities in brain structure and function that pertain to the different diagnoses may help our understanding of the bases of this heterogeneity. Social anxiety disorder (SAD) although among the most common anxiety disorders(5), has however received relatively little attention in this context.
SAD is characterized by significant and persistent fear of social situations wherein the individual might be exposed to unfamiliar persons or situations, or to scrutiny by others(1). Lifetime prevalence is approximately 5–12%, with higher rates among females, and with mean onset in late childhood/early adolescence(6–7). Persons suffering from SAD typically either avoid the feared situations, or endure them with intense anxiety or distress, leading to significant functional impairment(8). Generalized SAD— the subtype involving experience of fear and avoidance in multiple situations – is associated with greater severity, comorbidity, and impairment, and may have greater genetic heritability(9–10).
Imaging studies have reported hyperactivity within limbic regions in SAD patients, particularly the amygdala, hippocampal region, and insula, when viewing emotionally charged faces(11–12). These paradigms have particular face-validity for SAD, where fear of scrutiny and negative evaluation, and avoidance of eye contact are core symptoms(13). Disturbances in frontal, and particularly anterior cingulate, cortex have been reported as well, although specificity and directionality of findings have been inconsistent(11). Other functional paradigms targeting anticipation of public speaking(14–15), gaze or eye contact(16), and judgment of self- versus non-self relevant information(17–18) have yielded generally similar patterns.
Data from functional paradigms, however, are dependent on the type of task performed, as well as the subject’s current state. This is of particular concern in studies of SAD, as the scanning environment may exacerbate performance anxiety— a common feature of the disorder— and impair task performance. Measures of brain structure, in contrast, are largely state- independent, and can complement functional studies by identifying morphological vulnerabilities that are robust to task parameters. Structural studies of SAD, however, have been extremely limited. A 2008 review of structural imaging studies of anxiety(19) identified only one report for SAD(20). That example failed to detect any differences between SAD cases and controls, but was restricted to examination of the thalamus, putamen, and an overall index of grey matter (GM)(20). A subsequent meta-analysis of anxiety disorders failed to find any studies of SAD that qualified for inclusion(21). Some studies have included SAD subjects within anxiety groups but without differentiating them from other fear-based disorders(22). Finally, a recent treatment study reported volume decreases in the cerebellum and superior temporal cortex in SAD patients following 12 weeks of treatment with the selective serotonin reuptake inhibitor (SSRI) escitalopram(23). SSRIs however, are broadly efficacious for multiple anxiety and mood disorders, so the extent to which the changes index social anxiety is unknown. These questions, coupled with the overall paucity of studies, invite additional investigation using complementary approaches and populations.
We used magnetic resonance imaging and voxel based morphometry(VBM) to identify brain abnormalities associated with SAD. Given the absence of well-established structural abnormalities for the disorder, we used a whole-brain approach involving three stages. First, we compared a primary group of persons with DSM-IV generalized SAD to a group of healthy control participants, to identify differences in GM differences between the two groups. We then re-examined the same measures in an independent clinical sample of generalized SAD patients and healthy controls, to replicate and evaluate the generalizability of our findings. And finally, we asked whether the abnormalities were specific to SAD, by contrasting the primary SAD group to subjects with a different anxiety disorder: panic disorder (PD). PD is a complex disorder characterized by recurrent episodes of unexpected and uncontrollable fear, accompanied by cardio-respiratory and other autonomic responses. Like SAD, it is more frequent among women and moderately heritable, although with later onset (24–25). Although the two disorders share some clinical symptoms as well as abnormal fear circuitry, (26–27), they also have distinguishing clinical and treatment profiles. Comparison to the PD group thus afforded us one mechanism to evaluate whether the aforementioned regional abnormalities specifically indexed social anxiety.
The goals of the study can thus be summarized as follows: (1) to identify brain abnormalities associated with SAD; (2) to retest the findings in an independent clinical population; and (3) to test specificity of these findings to social anxiety.
Methods
Primary Sample (“Sample 1”)
All subjects were 18–50 years of age. SAD cases were required to have a DSM-IV(1) diagnosis of generalized social anxiety disorder(28) with onset by age 30, and have a first-degree relative with an anxiety disorder. Controls were required to have no lifetime history of any psychiatric disorder, with exceptions for past minor depressive disorder, adjustment disorders, or brief periods of substance abuse (not dependence) in adolescence or college. Controls could also not have a history of an anxiety disorder in any first-degree relative. Neither group could have a personal or family history of schizophrenia or bipolar disorder.
Subjects were recruited through web advertisements (except 7 SAD subjects recruited from an ongoing genetic program project of anxiety(29)). Subjects responding to the advertisement were first screened by a RA using the anxiety screening modules of the SADS-LA-IV(30); subjects who screened positive for SAD then participated in a full DSM-IV interview (see below). Similar procedures were used for the PD group, except PD subjects could not have a lifetime diagnosis of SAD, and vice versa. All procedures were approved by the Columbia University/New York State Psychiatric Institute Institutional Review Boards, and all subjects provided written consent.
Diagnostic assessments were administered by clinically trained mental health professionals using the Schedule for Affective Disorders and Schizophrenia-Lifetime Version, modified for anxiety disorders and updated for DSM-IV (SADS-LA-IV)(30). Training and monitoring procedures have been previously described(29). Family history was obtained using the Family History Screen(31). Final diagnoses were made by an experienced clinician using the Best Estimate Procedure(32). Trait and state anxiety were assessed just prior to the scan using the Spielberger State-Trait Anxiety Inventory (STAI)(33).
Sample 2
Structural data were obtained for 17 SAD and 17 healthy control (age 20–52) subjects who were imaged as part of an unrelated study (Schneier, P.I)(16) using the same scanner. Subjects were recruited through media advertisements and clinical referrals, and interviewed using the Structured Clinical Interview for DSM-IV Axis 1disorders (SCIDIV)(34). Severity was also rated by a clinician using the Liebowitz Social Anxiety Scale (LSAS)(35). The SAD group was required to have current generalized SAD, but no other current Axis I disorder (except secondary diagnoses of generalized anxiety, dysthymia, or specific phobia). Controls were required to have no lifetime history of any Axis I disorder. Only images acquired at baseline (at which time all subjects had been medication-free for ≥4 weeks), were used.
Imaging and Data Analysis
Structural data were acquired on a 1.5Tesla GE Signa MRI scanner using a 3D T1-weighted spoiled gradient recalled (SPGR) pulse sequence with isomorphic voxels (1×1×1mm) in a 24cm field of view (256×256matrix; ~186 slices; TR:34ms; TE:3ms). Anatomical data were processed using whole-brain VBM(36–37), implemented in SPM8 software (http://www.fil.ion.ucl.ac.uk/spm) using Matlab v7.13. 3D T1-weighted images were segmented into the three main tissue classes (gray matter(GM), white matter(WM), and cerebrospinal fluid (CSF), using the SPM unified segmentation algorithm(38). GM and WM images were next spatially normalized to a group specific template and then to MNI space using a diffeomorphic image registration toolkit (DARTEL) in 1.5 mm cubic resolution(39). The images were modulated with the individual Jacobian determinants to preserve the local amount of GM and WM (40). Modulation was achieved by multiplying voxel values in the segmented images by the Jacobian determinants derived from the spatial normalization step. In effect, the analysis of modulated data tests for regional differences in the absolute amount of grey matter. Finally, images were smoothed with an 8mm full-width-at-half-maximum (FWHM) isotropic Gaussian kernel. This is the SPM default, is optimal for group inference(41), and is commonly used in studies of anxiety [e.g.(22, 42–43)], aiding future comparisons of our data with other studies.
Prior to statistical analysis, an inclusion mask was created by absolute thresholding which excluded all voxels with GM values < 0.2. Statistical analysis on processed GM images was carried out by means of whole brain multiple regression, using binary variables to code for SAD cases vs. controls. Sex, age, and total intracranial volume (TIV, which was the sum of GM, WM and CSF, for each subject normalized by 10,000) were always entered as covariates, as these are independently associated with GM differences in adults, and failure to adjust for these variables can result in false positives (44). For the combined dataset analysis, an additional variable coding for dataset was included in order to control for any possible systematic differences between samples. For whole-brain analyses, maps were thresholded at p=0.001 and cluster-size of 10(45). Additionally, significant clusters were identified by means non-stationary cluster extent correction using random fields(46) as implemented using the NS toolbox (http://fmri.wfubmc.edu/cms/software#NS) for SPM5. This correction method confers increased sensitivity to spatially extended signals while remaining valid when cluster-size distribution varies depending on local smoothness as is the case in VBM data(46).
Results
Sample 1
Demographic and Clinical Features
Sample characteristics are detailed in Table 1a. As compared to the controls, the SAD group had a higher proportion of female subjects, and reported higher state and trait anxiety. The groups did not differ by age or education. The most frequently co-morbid lifetime diagnoses were major depressive disorder and specific phobia. Three subjects reported taking medication for anxiety in the past, but no subject was on any psychoactive medication in the 10 weeks preceding the scan.
TABLE 1.
Sample Demographics and Clinical Features
| Diagnostic | Statistical | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| (a) SAMPLE 1 | SAD | CON | PD | SAD vs | PD vs CON | SAD vs |
|
|
|
|||||
| N = 16 | N = 20 | N=16 | ||||
| Gender [N(%), Female] | 13 (81) | 9 (45) | 13 | x2 = 4.9* | x2 = 3.8+ | x2 = 0.11 |
| Age [mean years, std] | 34.1 | 31.4 (7.8) | 31.8 (10) | t = 1.1 | t = .41 | t = .75 |
| Education [completed college, N(%)] | 11 (69) | 14 (70) | 9 | x2 = .007 | x2 = .72 | x2 = .53 |
| Age at onset [mean, std] | 11.0 | n/a | 18.4 (3.4) | t = | ||
| Trait Anxiety [mean, std] | 35.7 | 27 (6.1) | 39.4 (7.9) | t = 2.3* | t= 4.9**** | t = .99 |
| State Anxiety [mean, std] | 39 (11.9) | 26 (4.1) | 35.5 (10) | t = 4.0**** | t = 3.5*** | t = .88 |
| Lifetime Comorbid Diagnoses [N,%] | ||||||
| MDD | 5 (31) | 0 | 3 | x2 = .83 | ||
| GAD | 2 (12) | 0 | 2 | x2 = .004 | ||
| SP | 4 (25) | 0 | 5 | x2 = 0.08 | ||
| OCD | 1 (6) | 0 | 2 | x2 = 0.1 | ||
| DUD | 1 (6) | 0 | 4 | x2 = 1.9 | ||
| AUD | 0 | 0 | 4 | x2 = .3 | ||
| Lifetime Psych. Medication Use [N,%] | 3 (20) | 0 | 9 | x2 = 3.7+ | ||
|
|
|
|||||
| (b) SAMPLE 2 | SAD | CON | PD | SAD vs | ||
|
|
|
|||||
| N = 17 | N = 17 | n/a | ||||
| Gender [N(%), Female] | 11 (64) | 10 (59) | x2 = 0.1 | |||
| Age [mean years, std] | 29.1 | 31.3 | t = .66 | |||
| Education [completed college, N(%)] | 12 (71) | 10 (60) | x2 = .52 | |||
| State Anxiety [mean, std] | 44.7 | 24.4 (7.1) | t = 6.9**** | |||
| Liebowitz Social Anxiety Scale [mean, | 81.4 | 8.1 [5.4] | t = | |||
| Lifetime Comorbid Diagnoses [N,%] | ||||||
| MDD | 6 (35) | 0 | ||||
| GAD | 3 (18) | 0 | ||||
| AUD | 2 (12) | 0 | ||||
| Lifetime Psych. Medication Use [N,%] | 6 (35) | 2 (12) | ||||
|
|
|
|||||
| (C ): COMBINED SAMPLE | SAD | CON | PD | Sample | Group | Sample x |
|
|
|
|||||
| N = 33 | N = 37 | n/a | (1 vs 2) | (SAD vs | ||
| Gender [N(%), Female] | 24 (73) | 19 (51) | x2 = 0.03 | x2 = 3.4+ | x2 = 1.8 | |
| Age [mean years, std] | 31.5 | 31.4 (9.1) | t = 1.3 | t = .01 | F = 1.5 | |
| Education [completed college, N(%)] | 23 (70) | 24 (65) | x2 = 0.3 | x2 = 0.18 | x2 = 0.01 | |
| State Anxiety [mean, std] | 42.0 | 25.6 (5.7) | t = 1.5 | t = 64.9**** | F = 2.8 | |
p .1;
p ≤ .05;
p ≤ .01;
p ≤ .005;
p ≤.001
Abbreviations:
AUD: Alcohol Use Disorder (abuse or dependence); DUD: Drug Use Disorder (abuse or dependence); GAD: Generalized Anxiety Disorder; MDD = Major Depressive Disorder; OCD: Obsessive Compulsive Disorder; SP: Specific phobia.
Medication frequencies only include those prescribed for a psychiatric condition.
Grey Matter Differences associated with SAD
We first examined grey matter (GM) differences between the SAD and healthy control groups across the entire brain. Significant group differences (10 or more voxels, p ≤ .001) are identified in Table 2a. All analyses were adjusted for age, gender, and total intracranial volume (TIV). There were no overall differences in total grey or white matter between the SAD and control groups.
TABLE 2.
Gray Matter Abnormalities Associated with Social Anxiety Disorder
|
|
||||||
|---|---|---|---|---|---|---|
| (a) Sample 1 | BA | Size | x | y | z | t |
|
|
||||||
| SAD > Control | ||||||
| 1 L Cerebellum, Parahippocampal, Fusiform | 37, 36 | 451+ | - | - | −21 | 4.40 |
| 2 R Middle Frontal | 46, 10 | 224 | 45 | 51 | 9 | 4.95 |
| 3 R Lingual | 19 | 41 | 24 | - | −5 | 3.67 |
| 4 R Cerebellum | - | 35 | 15 | - | −44 | 3.95 |
| 5 L Middle Occipital | 19 | 11 | - | - | 8 | 3.98 |
| 6 L Lingual | 17 | 12 | −8 | - | −15 | 3.49 |
| Control > SAD | ||||||
| 1 R Precentral, Postcentral | 6,4 | 678* | 42 | - | 38 | −4.84 |
| 2 R Middle Cingulate | 24 | 199 | 14 | - | 48 | −4.64 |
| 3 L Middle Cingulate | 32 | 32 | - | 21 | 38 | −3.89 |
| 4 L Superior Temporal | 22 | 29 | - | 8 | 3 | −4.13 |
| 5 R Temporal Pole, Superior Temporal | 38 | 30 | 17 | −29 | −3.36 | |
| 6 R Medial Frontal, Middle Cingulate | 6,24 | 16 | 11 | −6 | 51 | −4.47 |
| 7 L Middle Cingulate | 24 | 11 | - | - | 44 | −3.59 |
| (b) Sample 2 | ||||||
| SAD > Control | ||||||
| 1 L, R Cerebellum | - | 701 | 2 | - | −12 | 4.92 |
| 2 L Inferior Parietal, Supramarginal | 40 | 214 | - | - | 53 | 3.92 |
| 3 R Paracentral Lobule, Supp. Motor Area | 6 | 5 | - | 48 | 4.24 | |
| 4 L Inferior Temporal | 20, 21 | 153 | - | - | −14 | 4.97 |
| 5 R Post Central Gyrus | 3,1,2 | 29 | 39 | - | 48 | 3.95 |
| Control > SAD | ||||||
| 1 R Temporal Pole, Superior Temporal | 38 | 603* | 38 | 17 | −29 | −4.91 |
| 2 R Middle Frontal, Orbitofrontal | 11,47 | 366+ | 33 | 47 | −9 | −5.66 |
| 3 L Temporal Pole, Superior Temporal | 38 | 31 | - | 20 | −27 | −3.71 |
| 4 L Inferior Frontal, Orbitofrontal | 11 | 22 | - | 33 | −9 | −4.14 |
| (c) Combined Sample | ||||||
| SAD > Control | ||||||
| 1 L Cerebellum, Parahippocampal, Fusiform | 37 | 1840* | 0 | - | −12 | 4.12 |
| 2 R Supramarginal, Angular | 40 | 192 | 53 | - | 36 | 3.81 |
| 3 L Supramarginal, Angular | 40 | 22 | - | - | 47 | 3.44 |
| 4 L Middle Occipital | 19 | 28 | - | - | 8 | 3.73 |
| Control > SAD | ||||||
| 1 R Temporal Pole, Superior Temporal | 38 | 851* | 32 | 17 | −30 | −5.22 |
| 2 L Temporal Pole, Superior Temporal | 38 | 97 | - | 15 | −30 | −3.56 |
| 3 L Inferior Frontal, Orbitofrontal | 47 | 18 | - | 35 | −9 | −3.69 |
| 4 R Superior Occipital | 10 | 24 | - | 29 | −3.55 | |
P < .001; k = 10
Sample 1: N = 36 (16 SAD, 20 Control);
Sample 2: N = 34 (17 SAD, 17 Control);
Combined Sample: N =70 (33 SAD, 37Control)
Clusters surviving whole brain correction are indicated as follows:
p < .05;
p < .1
Clusters are listed in order of descending size; coordinates refer to the voxel with the peak t value in the cluster.
The largest GM increases associated with SAD were in a left hemisphere cluster encompassing the cerebellum and fusiform/parahippocampal cortex [Brodmann’s Areas 37, 36]. Additional differences were detected in right and left lingual, middle occipital, and middle frontal gyri. The converse contrast (control>SAD) identified a cluster spanning right hemisphere primary motor and sensory cortices, multiple clusters in both hemispheres of the dorsal anterior cingulate, and a cluster in the temporopolar region of the left superior temporal cortex.
Sample 2
Demographic and Clinical Features
We next repeated the above analyses in an independently recruited and imaged clinical sample of SAD cases and healthy controls (sample 2). The SAD and control groups of sample 2 did not significantly differ on measures of age, gender, or education, either from each other (Table 1b), or from the corresponding groups in the first sample (Table 1c).
Grey Matter Differences associated with SAD
GM differences between the sample 2 SAD and control groups are listed in Table 2b, adjusted for age, gender, and TIV. Significantly greater GM among the SAD group was detected in the bilateral cerebellum, left supramarginal, right paracentral lobule and supplementary motor area, left inferior temporal and right post-central regions. The control>SAD contrast identified clusters in bilateral temporal pole, and regions of the middle and inferior frontal gyri encompassing orbitofrontal cortex. Similar clusters were identified when using an alternative continuous measure of social anxiety rather than a diagnosis (Supplement 1).
Combined Sample
We next combined the two samples into a single dataset and examined overall differences between SAD and control groups, adjusting for the previously noted variables, as well as for sample of origin. The final results, detailed in Table 2c, preserve a number of the regions observed in the individual samples. Specifically, the SAD>control contrast revealed large increases in the cerebellum, left parahippocampal and fusiform gyri, bilateral supramarginal and angular gyri, and left middle occipital gyrus. The control>SAD contrast identified lower temporal pole (both hemispheres, but predominantly right) and left inferior prefrontal/orbitofrontal GM in the SAD group. Cerebellum, parahippocampal and temporal pole differences were robust to multiple comparison correction at the whole-brain level (asterisked clusters). The main findings are illustrated in Figure 1, with clusters shown in red illustrating regions with greater GM volume among the SAD group than the controls, and clusters in blue, the converse. The numbering of regions in the figures corresponds to the clusters in Table 2. Importantly, group differences in the cerebellum/parahippocampal gyrus and temporal pole remained significant when we reran the above analysis using the same statistical thresholds and control subjects, but only including the sub-group of SAD subjects who had any history of any other psychiatric disorder over their lifetime. This further minimizes the possibility that neuroanatomical variation related to other lifetime anxiety disorders could have contributed to the observed findings. Additional corollary analyses further confirmed that regional findings from one sample were replicated in the other (Supplement 1). Finally, an exploratory analysis of ROIs that did not meet a priori criteria suggested lower right amygdala, bilateral insula, and left anterior cingulate GM in the SAD group (Supplement 1).
Figure 1. Grey Matter Differences Associated with Social Anxiety Disorder.
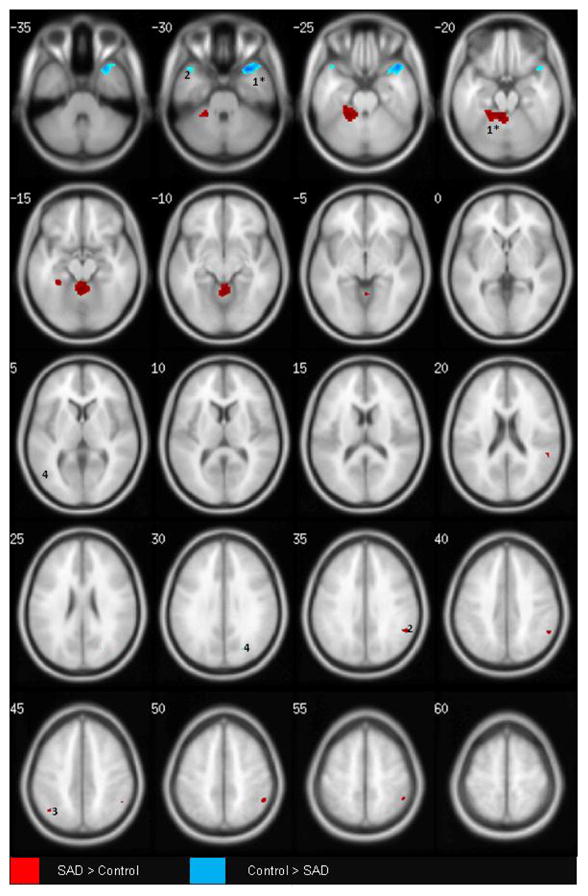
P ≤ .001; k =10; N =70 [33 SAD, 37 CONTROL]
T1 -weighted axial images; image left is brain left. Images Group differences are adjusted for differences in age, gender, intracranial volume, and sample source. Regions surviving multiple comparison correction at the whole brain level are asterisked.
Clusters are numbered corresponding to their listing in Table 2c.
SAD > Control (RED): 1: L Cerebellum, Parahippocampal, Fusiform; 2: R Supramarginal, Angular; 3: L Supramarginal, Angular; 4: L Middle Occipital
Control > SAD (BLUE): 1: R Superior Temporal, Anterior Temporal Pole; 2: L Superior Temporal, Anterior Temporal Pole; 3; L inferior Frontal (Orbitofrontal); 4: L Middle Occipital
Testing for Specificity
To further investigate whether the GM differences identified above were specific to SAD, we compared the SAD group from Sample 1 to a group of separate subjects from sample 1 with PD. The PD group and SAD groups did not differ on measures of age, gender, state or trait anxiety, or education (Table 1a), although age of onset for PD was later (18 vs. 11yrs), consistent with the epidemiology of the disorders.
We first examined differences between the PD and the control groups. As shown in Table 3a and Figure 2A, subjects with PD, as compared to controls, had large parieto-occipital GM increases— specifically, in bilateral cuneate and precuneate, lingual, and superior occipital cortices— as well as larger insular cortex. Conversely, a number of frontal cortical (right pre- and post-central gyri, left and right middle cingulate, supplementary motor area) as well as sub-cortical (thalamus, caudate) regions showed reduced GM among the PD cases.
TABLE 3.
Gray Matter Differences Between Panic Disorder and Social Anxiety Disorder
|
|
||||||
|---|---|---|---|---|---|---|
| (a) PD Vs. CONTROL | BA | Size | x | y | z | t |
|
|
||||||
| PD > Control | ||||||
| 1 L, R Cuneus, Lingual | 17,18 | 1620* | 0 | −98 | 6 | 5.03 |
| 2 L Insula | - | 379 | - | 9 | 5 | 4.44 |
| 3 L, R, Cuneus, Precuneus | 7 | 355 | 0 | −71 | 24 | 4.30 |
| 4 R Cuneus, Superior Occipital | 7 | 23 | 21 | −75 | 30 | 3.56 |
| Control > PD | ||||||
| 1 R Precentral, Postcentral | 6,1–4 | 884* | 44 | −14 | 42 | - |
| 2 R Middle Cingulate | 32 | 504* | 5 | 20 | 41 | - |
| 3 L Inferior Parietal | 40 | 104 | - | −45 | 42 | - |
| 4 R Middle Cingulate, R Supp. Motor Area | 24 | 29 | 11 | −6 | 50 | - |
| 5 L Caudate | - | 98 | - | 12 | 18 | - |
| 6 L Precentral | 6 | 40 | - | −5 | 36 | - |
| 7 R Middle Cingulate | 24 | 13 | 15 | −18 | 44 | - |
| 8 R Thalamus | - | 23 | 8 | −20 | 15 | - |
| (b) PD vs SAD | ||||||
| PD > SAD | ||||||
| 1 L Cuneus | 7 | 149 | −6 | −77 | 36 | 4.43 |
| 2 L Middle Frontal | 9 | 35 | − | 20 | 30 | 4.19 |
| 3 L, R Lingual | 18 | 25 | 2 | −87 | −8 | 3.56 |
| 4 R Superior Occipital | 18,31 | 12 | 23 | −74 | 29 | 3.74 |
| SAD > PD | ||||||
| 1 R Parahippocampal, Fusiform, Cerebellum | 37,36,19 | 534* | 30 | −48 | −11 | - |
| 2 L Parahippocampal, Fusiform | 37,36 | 402 | - | −41 | −17 | - |
| 3 R Middle Frontal, Inferior Frontal | 10 | 106 | 42 | 45 | 5 | - |
| 4 R Anterior Cingulate | 32 | 10 | 15 | 47 | 2 | - |
| 5 L Middle Frontal | 8 | 12 | - | 21 | 44 | - |
p < .001; k = 10
Table 3a: N = 16 PD, 20 Control; Table 3b: N = 16 SAD, 16 PD
Clusters surviving whole brain correction (p < .05) are asterisked.
Clusters are listed in order of descending size; coordinates refer to the voxel with the peak t value in the cluster.
Figure 2. Grey Matter Differences Between Social Anxiety Disorder (SAD) and Panic Disorder (PD).
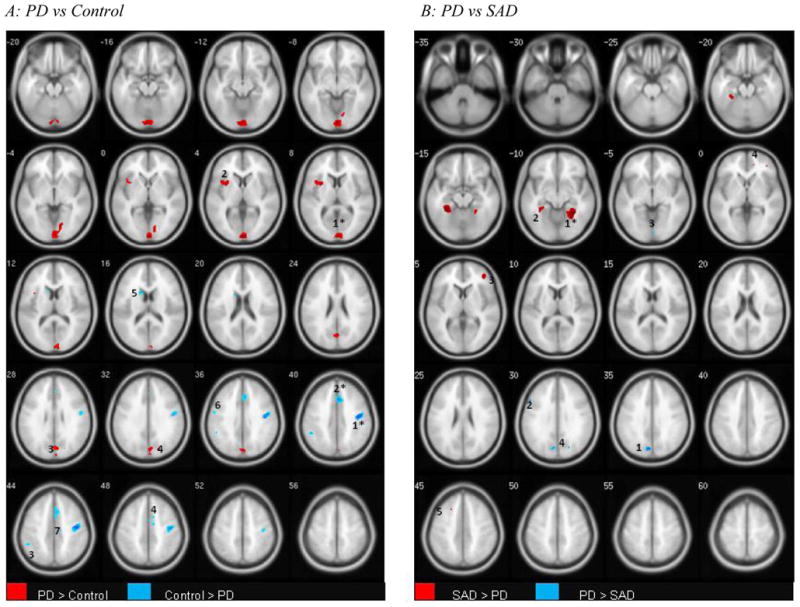
P ≤ .001; k =10
Figure 2A: N =16 PD, 20 Control; Figure 2B: N =16 SAD, 16 PD
T1 -weighted axial images; image left is brain left. Images Group differences are adjusted for differences in age, gender, and intracranial volume. Clusters surviving whole brain correction are indicated as follows: *(p < .05); + (p < 1).
Clusters are numbered corresponding to their listings in Tables 4a and b respectively.
Figure A:
PD > CONTROL (RED): 1: Bilateral Cuneate, Lingual; 2: L Insula; 3: Bilateral Cuneus, Precuneus; 4: R Cuneus, Superior Occipital
CONTROL > PD (BLUE) 1: R Precentral, Postcentral; 2: R Middle Cingulate; 3: L Inferior Parietal; 4: R Middle Cingulate, Supplementary Motor Area; 5: L Caudate; 6: Precentral; 7: R Middle Cingulate
Figure B:
SAD > PD (RED): 1: R Parahippocampal, Fusiform; 2: L Parahippocampal, Fusiform; 3: R Middle Frontal, Inferior Frontal; 4: R Anterior Cingulate; 5: L Middle Frontal
PD > SAD (BLUE) 1: L Cuneus; 2: L Middle Frontal; 3; L & R Lingual; 4: R Superior Occipital
We then formally contrasted the PD and SAD groups. As shown in Table 3b and Figure 2B, subjects with PD showed larger mean occipital GM volume, particularly in the cuneate cortex, calcarine sulcus, and lingual gyrus. Conversely, both hemispheres of the parahippocampal and fusiform gyri were significantly larger in the SAD group. Finally, right inferior frontal (orbitofrontal), and anterior cingulate were larger among the SAD groups, though both anxiety groups had reduced GM vis a vis healthy controls.
Discussion
Summary of Findings
We report here on morphological abnormalities associated with generalized SAD. We found that subjects with the disorder, as compared to healthy controls, had greater GM in the cerebellum and the left parahippocampal cortex, and lower GM in the temporal pole. Several observations strengthen our confidence in these findings. First, the differences were observed in both individual samples as well as in the combined dataset analysis. Second, the clusters remained significant after correction for multiple comparisons. Third, similar findings were obtained when using a severity index rather than a diagnosis of SAD (sample 2). And finally, subjects with panic disorder (PD) did not show these patterns, pointing to the relative specificity of these findings to SAD. We thus weight the ensuing discussion primarily toward the above regions. Other GM differences that were not observed in both samples, or did not survive corrections for multiple comparisons, may play a role, but such findings should be considered provisional.
Grey Matter Increases Associated with SAD
The largest differences were detected within the cerebellum. Resting state perfusion studies have reported both hyper- and hypo-perfusion in the cerebellum among subjects with SAD(47), and a Positron Emission Tomography study found anxiety induced in SAD patients to increase blood flow to the cerebellum(48). The aforementioned treatment study(23) reported decreased cerebellar volumes among SAD patients following three months of SSRI treatment, but because there was no control group, it is unclear whether the patients had abnormalities prior to being treated. Although the mechanisms are unclear, cerebellar abnormalities may increase vulnerability to anxiety states via modulation of arousal. Many cerebellar subdivisions, and the vermis in particular, project to the midbrain regions of the pons and medulla, which mediate the autonomic responses that are exaggerated in persons with anxiety(49).
Also having higher GM volume in the SAD group was the parahippocampal gyrus(PHG). The PHG consolidates memories and social communication cues, and hyperactivation has been reported in SAD patients during conditions of social threat (50–51), as well as during non-threatening tasks involving human, as compared to non-human or computer-simulated, interaction (52). Moreover, the adjacent fusiform gyrus—part of the parahippocampal cortex (and included in the detected clusters)— is cardinal in facial recognition (53) and processing of facial expression(51, 54). A recent fMRI study reported that when asked to passively view socially threatening stimuli, persons with SAD had higher BOLD signal increases in bilateral PHG than controls; however, if asked to try to actively regulate their negative responses to the same stimuli, the SAD group had decreased responses in fusiform (50). These differences were not replicated if social threat was replaced with physical threat. In the present study, PHG and fusiform GM differences were observed only in the SAD group (Table 4a). GM volume in the PD group was not only significantly lower than in the SAD group, as shown in Table 4b, but lower than in the controls as well. Whilst no other study to our knowledge has directly contrasted these two anxiety disorders at the morphological level, a number of reports (55–57) have reported reduced PHG volume among panic patients. Our data, coupled with the functional literature on SAD, suggest that increased PHG activity may serve as a marker for social-based anxiety constructs. Incidentally, lower caudate volume, the only other regional abnormality associated with PD in the aforementioned PD meta-analysis(55), was also replicated in our PD group (Table 4a).
Grey Matter Decreases Associated with SAD
The temporal pole, that is, the anterior region of the superior temporal cortex corresponding to Brodmann’s Area 38, had significantly lower GM volume in both samples of SAD subjects. The anterior temporal cortex has been implicated in the processing of abstract conceptual knowledge, but BA38— and particularly its right hemisphere— may more specifically index social concepts (58–59). Studies in healthy subjects have reported activation in this region during social competition and perception of others’ mental states(52). Conversely, lesions and degeneration of BA 38 have led to changes in the ability to characterize social attributes of behavior (58, 60). Among persons with SAD, public speaking(15) and anticipation thereof (61) have been associated with decreased blood flow to the temporal pole. Interestingly, selectively increased surface area in the left temporal pole has been reported in Williams Syndrome (WS), a rare genetic disorder that in terms of its behavioral phenotype seems the opposite of SAD (e.g., hyper-sociability, lack of fear of interacting with strangers)(62). That, as well as one other report (63) also showed decreased PHG volume in WS patients, again the opposite of what we find here with SAD. Though WS and SAD may be etiologically different disorders, the common regional focus of abnormalities suggests that the above regions may mediate common constructs of social cognition, with different neuroanatomical aberrations leading to different clinical syndromes.
The SAD group also had lower GM in the lateral orbitofrontal cortex (~BA11,47). OFC regulates expression of emotion, and assigns positive and negative stimulus response contingencies(64–65). Concordantly, GM disturbances (particularly in the left hemisphere) have been linked to multiple anxiety and mood problems(18). The OFC also receives direct reciprocal input from the amygdala(47), a central mediator of the fear response, and in persons with SAD, the uncinate fasiculus— the white matter tract connecting OFC to temporal cortex— is compromised(66–67). Although our study does not address temporal sequence, these disturbances are likely to begin early in life, as infants with high-reactive and inhibited temperament— which are risk factors for later onset of SAD(68–69) — show reduced cortical thickness in similar left OFC regions when imaged in adulthood, even if they did not go on to develop the full disorder(70).
Finally, two other interesting but inconsistently observed regional differences deserve comment. First, significantly lower GM among SAD cases was identified in the primary motor and sensory cortex in sample 1 only. Although the right hemispheric precentral gyrus is thought to control motor function, it also has been associated with self-face recognition(48) and imitations of facial expressions(49) that could hold implications for social anxiety. Second, multiple clusters were observed for the control>SAD contrast in the middle cingulate in sample 1 (Table 2). Although not directly mirrored in sample 2, within the SAD cases of sample 2, increased severity of social anxiety was associated with lower cingulate GM. Though it is unlikely that these patterns are specific to SAD (they were observed in the PD group, and similar abnormalities have been reported for other anxiety and mood disorders (21)) the overall inverse relationships with GM-SAD diagnosis (sample 1) and GM-severity (sample 2) are broadly consistent with functional models positing anxiety as a failure of the frontal cortex to down-regulate limbic activation (71).
Limitations
The reported findings should be interpreted within the context of the following limitations. First, the study is cross-sectional and does not therefore speak to the causal relationship between brain structure and diseased state, as the identified GM differences could either predispose to, or be a result of the disorder. Disentangling causal from compensatory pathways would require more complex epidemiological approaches(72)— e.g., selecting subjects who are at high-risk (by virtue of family history, presence of a prodrome, etc) but asymptomatic at recruitment, and then tracking brain changes longitudinally as a subset go on to develop the syndrome. Related, the GM differences should also not be used to make diagnostic inferences, as they are based on overall group differences and do not account for important individual brain, behavioral, and environmental variations that shape whether a given subjects will have a diagnosis(73). Second, standard methodological limitations to VBM, particularly its vulnerability to normalization and smoothing errors(37, 74) apply here as well. Third, it is possible that gender-related variance partially contributed to observed group differences in sample 1, where anxiety groups had a greater proportion of female subjects. We did, however, adjust for gender in all models, and furthermore, the main findings were replicated in the second sample, which was matched on gender, as well as in the final combined sample. Fourth, although other lifetime anxiety diagnoses did not contribute to the observed results, we cannot rule out that other behavioral traits that differed across the two groups but do not directly index SAD, contributed to group differences. Finally, we only compared SAD with PD. Different patterns might have been observed with different comparison group: for example, specific phobia may have yielded largely overlapping coordinates, but PTSD diverging significantly, particularly within frontal regulatory regions(71). Related, we only included SAD subjects without a history of PD, and vice versa. This could have biased selection toward milder or less generalizable cases. Alternative approaches would be to include a 3rd group with both SAD and PD, or to permit all comorbidity and then model the variance statistically. Both approaches, however, would have necessitated a substantially larger sample.
Conclusions
This report contributes to the currently limited literature on the neurobiology of SAD by identifying structural deficits that may predispose to functional abnormalities. The rigorous ascertainment criteria, retest in an independent sample, and comparison with PD strengthen both the reliability and the interpretability of our findings. The confirmation in a second sample is particularly valuable in an imaging context, given the preponderance of failures to replicate original reports(75), and should be considered in future designs whenever possible. Finally, because the results include regions (e.g., cerebellum, temporal pole) that are not among the primary nodes of fear circuitry, the validity of these regions, as well as their specific roles in mediating constructs of SAD will require further investigation.
Supplementary Material
Acknowledgments
Sample 1 collection and ascertainment was funded by a NARSAD Young Investigator Grant (Talati, P.I.); some cases were recruited from an ongoing National Institute of Mental Health (NIMH) Program Project Grant P01 MH60970 (Rene Hen, Ph.D., Overall P.I.; Weissman, Ph.D.; Project P.I.). Brain scans were funded by the Program for Imaging and Cognitive Sciences at Columbia University (Hirsch, P.I.). Sample 2 was funded by a NIMH separate grant R21 MH077976 (Schneier, P.I).
Dr. Talati is funded by a 5 year K01 Award (1 K01 DA029598) from the National Institute of Drug Abuse; Spiro Pantazatos is funded by an F31 award (F31MH088104-02) from the NIMH.
The authors express their gratitude to Christine Platzek, M.S., and Sandhya Rawal, B.A. candidate, for assistance with subject recruitment and data analysis respectively.
Footnotes
Financial Disclosures
In the past two years, Dr. Weissman has received royalties from the Oxford University Press, Perseus Press, the American Psychiatric Association Press, and MultiHealth Systems. Dr. Schneier has received an honorarium from GlaxoSmithKline for speaking at two conferences, and royalties from Cambridge University Press and UpToDate. None of these present a conflict of interest with the present study. Drs. Talati, Pantazatos, and Hirsch report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Diagnostic and Statistical Manual of Psychiatric Disorders. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 2.Delgado MR, Nearing KI, Ledoux JE, Phelps EA. Neural circuitry underlying the regulation of conditioned fear and its relation to extinction. Neuron. 2008;59(5):829–38. doi: 10.1016/j.neuron.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bienvenu OJ, Samuels JF, Wuyek LA, Liang KY, Wang Y, Grados MA, et al. Is obsessive-compulsive disorder an anxiety disorder, and what, if any, are spectrum conditions? A family study perspective. Psychol Med. 2011:1–13. doi: 10.1017/S0033291711000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stein DJ, Craske MG, Friedman MJ, Phillips KA. Meta-structure issues for the DSM-5: how do anxiety disorders, obsessive-compulsive and related disorders, post-traumatic disorders, and dissociative disorders fit together? Curr Psychiatry Rep. 2011;13(4):248–50. doi: 10.1007/s11920-011-0207-1. [DOI] [PubMed] [Google Scholar]
- 5.Jefferys D. Social phobia. The most common anxiety disorder. Aust Fam Physician. 1997;26(9):1061, 4–7. [PubMed] [Google Scholar]
- 6.Weissman MM, Bland RC, Canino GJ, Greenwald S, Lee CK, Newman SC, et al. The cross-national epidemiology of social phobia: a preliminary report. Int Clin Psychopharmacol. 1996;11 (Suppl 3):9–14. doi: 10.1097/00004850-199606003-00003. [DOI] [PubMed] [Google Scholar]
- 7.Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617–27. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Filho AS, Hetem LA, Ferrari MC, Trzesniak C, Martin-Santos R, Borduqui T, et al. Social anxiety disorder: what are we losing with the current diagnostic criteria? Acta Psychiatr Scand. 2010;121(3):216–26. doi: 10.1111/j.1600-0447.2009.01459.x. [DOI] [PubMed] [Google Scholar]
- 9.Schneier FR. Clinical practice. Social anxiety disorder. N Engl J Med. 2006;355(10):1029–36. doi: 10.1056/NEJMcp060145. [DOI] [PubMed] [Google Scholar]
- 10.Stein MB, Chartier MJ, Hazen AL, Kozak MV, Tancer ME, Lander S, et al. A direct-interview family study of generalized social phobia. Am J Psychiatry. 1998;155(1):90–7. doi: 10.1176/ajp.155.1.90. [DOI] [PubMed] [Google Scholar]
- 11.Freitas-Ferrari MC, Hallak JE, Trzesniak C, Filho AS, Machado-de-Sousa JP, Chagas MH, et al. Neuroimaging in social anxiety disorder: a systematic review of the literature. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(4):565–80. doi: 10.1016/j.pnpbp.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 12.Pietrini F, Godini L, Lazzeretti L, Benni L, Pracucci C, Talamba GA, et al. Neuroimaging and neurobiology of social anxiety. Riv Psichiatr. 2010;45(6):349–60. [PubMed] [Google Scholar]
- 13.Safren SA, Heimberg RG, Horner KJ, Juster HR, Schneier FR, Liebowitz MR. Factor structure of social fears: The Liebowitz Social Anxiety Scale. J Anxiety Disord. 1999;13(3):253–70. doi: 10.1016/s0887-6185(99)00003-1. [DOI] [PubMed] [Google Scholar]
- 14.Lorberbaum JP, Kose S, Johnson MR, Arana GW, Sullivan LK, Hamner MB, et al. Neural correlates of speech anticipatory anxiety in generalized social phobia. Neuroreport. 2004;15(18):2701–5. [PubMed] [Google Scholar]
- 15.Tillfors M, Furmark T, Marteinsdottir I, Fischer H, Pissiota A, Langstrom B, et al. Cerebral blood flow in subjects with social phobia during stressful speaking tasks: a PET study. Am J Psychiatry. 2001;158(8):1220–6. doi: 10.1176/appi.ajp.158.8.1220. [DOI] [PubMed] [Google Scholar]
- 16.Schneier FR, Pomplun M, Sy M, Hirsch J. Neural response to eye contact and paroxetine treatment in generalized social anxiety disorder. Psychiatry Res. 2011 doi: 10.1016/j.pscychresns.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrews-Hanna JR, Reidler JS, Sepulcre J, Poulin R, Buckner RL. Functional-anatomic fractionation of the brain’s default network. Neuron. 2010;65(4):550–62. doi: 10.1016/j.neuron.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitfield-Gabrieli S, Moran JM, Nieto-Castanon A, Triantafyllou C, Saxe R, Gabrieli JD. Associations and dissociations between default and self-reference networks in the human brain. Neuroimage. 2011;55(1):225–32. doi: 10.1016/j.neuroimage.2010.11.048. [DOI] [PubMed] [Google Scholar]
- 19.Ferrari MC, Busatto GF, McGuire PK, Crippa JA. Structural magnetic resonance imaging in anxiety disorders: an update of research findings. Rev Bras Psiquiatr. 2008;30(3):251–64. doi: 10.1590/s1516-44462008000300013. [DOI] [PubMed] [Google Scholar]
- 20.Potts NL, Davidson JR, Krishnan KR, Doraiswamy PM. Magnetic resonance imaging in social phobia. Psychiatry Res. 1994;52(1):35–42. doi: 10.1016/0165-1781(94)90118-x. [DOI] [PubMed] [Google Scholar]
- 21.Radua J, van den Heuvel OA, Surguladze S, Mataix-Cols D. Meta-analytical comparison of voxel-based morphometry studies in obsessive-compulsive disorder vs other anxiety disorders. Arch Gen Psychiatry. 2010;67(7):701–11. doi: 10.1001/archgenpsychiatry.2010.70. [DOI] [PubMed] [Google Scholar]
- 22.van Tol MJ, van der Wee NJ, van den Heuvel OA, Nielen MM, Demenescu LR, Aleman A, et al. Regional brain volume in depression and anxiety disorders. Arch Gen Psychiatry. 2010;67(10):1002–11. doi: 10.1001/archgenpsychiatry.2010.121. [DOI] [PubMed] [Google Scholar]
- 23.Cassimjee N, Fouche JP, Burnett M, Lochner C, Warwick J, Dupont P, et al. Changes in regional brain volumes in social anxiety disorder following 12 weeks of treatment with escitalopram. Metab Brain Dis. 2010;25(4):369–74. doi: 10.1007/s11011-010-9218-6. [DOI] [PubMed] [Google Scholar]
- 24.Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet. 2006;368(9540):1023–32. doi: 10.1016/S0140-6736(06)69418-X. [DOI] [PubMed] [Google Scholar]
- 25.Weissman MM. Family genetic studies of panic disorder. J Psychiatr Res. 1993;27 (Suppl 1):69–78. doi: 10.1016/0022-3956(93)90018-w. [DOI] [PubMed] [Google Scholar]
- 26.Schneier FR, Johnson J, Hornig CD, Liebowitz MR, Weissman MM. Social phobia. Comorbidity and morbidity in an epidemiologic sample. Arch Gen Psychiatry. 1992;49(4):282–8. doi: 10.1001/archpsyc.1992.01820040034004. [DOI] [PubMed] [Google Scholar]
- 27.Kendler KS, Walters EE, Neale MC, Kessler RC, Heath AC, Eaves LJ. The structure of the genetic and environmental risk factors for six major psychiatric disorders in women. Phobia, generalized anxiety disorder, panic disorder, bulimia, major depression, and alcoholism. Arch Gen Psychiatry. 1995;52(5):374–83. doi: 10.1001/archpsyc.1995.03950170048007. [DOI] [PubMed] [Google Scholar]
- 28.Goldstein RB, Wickramaratne PJ, Horwath E, Weissman MM. Familial aggregation and phenomenology of ‘early’-onset (at or before age 20 years) panic disorder. Arch Gen Psychiatry. 1997;54(3):271–8. doi: 10.1001/archpsyc.1997.01830150097014. [DOI] [PubMed] [Google Scholar]
- 29.Talati A, Ponniah K, Strug LJ, Hodge SE, Fyer AJ, Weissman MM. Panic disorder, social anxiety disorder, and a possible medical syndrome previously linked to chromosome 13. Biol Psychiatry. 2008;63(6):594–601. doi: 10.1016/j.biopsych.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fyer A, Endicott J, Mannuzza S, Klein DF. Anxiety Disorders Clinic. New York State Psychiatric Institute; New York: 1985. Schedule for affective disorders and schizophrenia-lifetime version, modified for the study of anxiety disorders (SADS-LA) [DOI] [PubMed] [Google Scholar]
- 31.Weissman MM, Wickramaratne P, Adams P, Wolk S, Verdeli H, Olfson M. Brief screening for family psychiatric history: the family history screen. Arch Gen Psychiatry. 2000;57(7):675–82. doi: 10.1001/archpsyc.57.7.675. [DOI] [PubMed] [Google Scholar]
- 32.Leckman JF, Sholomskas D, Thompson WD, Belanger A, Weissman MM. Best estimate of lifetime psychiatric diagnosis: a methodological study. Arch Gen Psychiatry. 1982;39(8):879–83. doi: 10.1001/archpsyc.1982.04290080001001. [DOI] [PubMed] [Google Scholar]
- 33.Spielberger C, Gorsuch RL, Lushene RE. Manual for the State-Trait Anxiety Inventory. Palo Alto, CA: Consulting Psychologists Press; 1970. [Google Scholar]
- 34.First MB, Spitzer RL, Gibbon M, Willians JBW. Structured Clinical Interview for DSM-IV Axis 1 Disorders. Washington, DC: American Psychiatric Press; 1997. [Google Scholar]
- 35.Liebowitz MR. Social phobia. Mod Probl Pharmacopsychiatry. 1987;22:141–73. doi: 10.1159/000414022. [DOI] [PubMed] [Google Scholar]
- 36.Ashburner J, Friston KJ. Voxel-based morphometry--the methods. Neuroimage. 2000;11(6 Pt 1):805–21. doi: 10.1006/nimg.2000.0582. [DOI] [PubMed] [Google Scholar]
- 37.Ashburner J, Friston KJ. Why voxel-based morphometry should be used. Neuroimage. 2001;14(6):1238–43. doi: 10.1006/nimg.2001.0961. [DOI] [PubMed] [Google Scholar]
- 38.Ashburner J, Friston KJ. Unified segmentation. Neuroimage. 2005;26(3):839–51. doi: 10.1016/j.neuroimage.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 39.Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage. 2007;38(1):95–113. doi: 10.1016/j.neuroimage.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 40.Keller SS, Wilke M, Wieshmann UC, Sluming VA, Roberts N. Comparison of standard and optimized voxel-based morphometry for analysis of brain changes associated with temporal lobe epilepsy. Neuroimage. 2004;23(3):860–8. doi: 10.1016/j.neuroimage.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 41.Mikl M, Marecek R, Hlustik P, Pavlicova M, Drastich A, Chlebus P, et al. Effects of spatial smoothing on fMRI group inferences. Magn Reson Imaging. 2008;26(4):490–503. doi: 10.1016/j.mri.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 42.Lazaro L, Castro-Fornieles J, Cullell C, Andres S, Falcon C, Calvo R, et al. A voxel-based morphometric MRI study of stabilized obsessive-compulsive adolescent patients. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(8):1863–9. doi: 10.1016/j.pnpbp.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 43.Yoo HK, Kim MJ, Kim SJ, Sung YH, Sim ME, Lee YS, et al. Putaminal gray matter volume decrease in panic disorder: an optimized voxel-based morphometry study. Eur J Neurosci. 2005;22(8):2089–94. doi: 10.1111/j.1460-9568.2005.04394.x. [DOI] [PubMed] [Google Scholar]
- 44.Henley SM, Ridgway GR, Scahill RI, Kloppel S, Tabrizi SJ, Fox NC, et al. Pitfalls in the use of voxel-based morphometry as a biomarker: examples from huntington disease. AJNR Am J Neuroradiol. 2010;31(4):711–9. doi: 10.3174/ajnr.A1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silver M, Montana G, Nichols TE. False positives in neuroimaging genetics using voxel-based morphometry data. Neuroimage. 2011;54(2):992–1000. doi: 10.1016/j.neuroimage.2010.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayasaka S, Phan KL, Liberzon I, Worsley KJ, Nichols TE. Nonstationary cluster-size inference with random field and permutation methods. Neuroimage. 2004;22(2):676–87. doi: 10.1016/j.neuroimage.2004.01.041. [DOI] [PubMed] [Google Scholar]
- 47.Warwick JM, Carey P, Jordaan GP, Dupont P, Stein DJ. Resting brain perfusion in social anxiety disorder: a voxel-wise whole brain comparison with healthy control subjects. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1251–6. doi: 10.1016/j.pnpbp.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 48.Kilts CD, Kelsey JE, Knight B, Ely TD, Bowman FD, Gross RE, et al. The neural correlates of social anxiety disorder and response to pharmacotherapy. Neuropsychopharmacology. 2006;31(10):2243–53. doi: 10.1038/sj.npp.1301053. [DOI] [PubMed] [Google Scholar]
- 49.Baldacara L, Borgio JG, Lacerda AL, Jackowski AP. Cerebellum and psychiatric disorders. Rev Bras Psiquiatr. 2008;30(3):281–9. doi: 10.1590/s1516-44462008000300016. [DOI] [PubMed] [Google Scholar]
- 50.Goldin PR, Manber T, Hakimi S, Canli T, Gross JJ. Neural bases of social anxiety disorder: emotional reactivity and cognitive regulation during social and physical threat. Arch Gen Psychiatry. 2009;66(2):170–80. doi: 10.1001/archgenpsychiatry.2008.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Straube T, Kolassa IT, Glauer M, Mentzel HJ, Miltner WH. Effect of task conditions on brain responses to threatening faces in social phobics: an event-related functional magnetic resonance imaging study. Biol Psychiatry. 2004;56(12):921–30. doi: 10.1016/j.biopsych.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 52.Polosan M, Baciu M, Cousin E, Perrone M, Pichat C, Bougerol T. An fMRI study of the social competition in healthy subjects. Brain Cogn. 2011;77(3):401–11. doi: 10.1016/j.bandc.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 53.Fairhall SL, Ishai A. Effective connectivity within the distributed cortical network for face perception. Cereb Cortex. 2007;17(10):2400–6. doi: 10.1093/cercor/bhl148. [DOI] [PubMed] [Google Scholar]
- 54.Gentili C, Gobbini MI, Ricciardi E, Vanello N, Pietrini P, Haxby JV, et al. Differential modulation of neural activity throughout the distributed neural system for face perception in patients with Social Phobia and healthy subjects. Brain Res Bull. 2008;77(5):286–92. doi: 10.1016/j.brainresbull.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 55.Lai CH. Gray matter deficits in panic disorder: a pilot study of meta-analysis. J Clin Psychopharmacol. 2011;31(3):287–93. doi: 10.1097/JCP.0b013e31821a1045. [DOI] [PubMed] [Google Scholar]
- 56.Massana G, Serra-Grabulosa JM, Salgado-Pineda P, Gasto C, Junque C, Massana J, et al. Parahippocampal gray matter density in panic disorder: a voxel-based morphometric study. Am J Psychiatry. 2003;160(3):566–8. doi: 10.1176/appi.ajp.160.3.566. [DOI] [PubMed] [Google Scholar]
- 57.Uchida RR, Del-Ben CM, Busatto GF, Duran FL, Guimaraes FS, Crippa JA, et al. Regional gray matter abnormalities in panic disorder: a voxel-based morphometry study. Psychiatry Res. 2008;163(1):21–9. doi: 10.1016/j.pscychresns.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 58.Zahn R, Moll J, Krueger F, Huey ED, Garrido G, Grafman J. Social concepts are represented in the superior anterior temporal cortex. Proc Natl Acad Sci U S A. 2007;104(15):6430–5. doi: 10.1073/pnas.0607061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zahn R, Moll J, Paiva M, Garrido G, Krueger F, Huey ED, et al. The neural basis of human social values: evidence from functional MRI. Cereb Cortex. 2009;19(2):276–83. doi: 10.1093/cercor/bhn080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu W, Miller BL, Kramer JH, Rankin K, Wyss-Coray C, Gearhart R, et al. Behavioral disorders in the frontal and temporal variants of frontotemporal dementia. Neurology. 2004;62(5):742–8. doi: 10.1212/01.wnl.0000113729.77161.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tillfors M, Furmark T, Marteinsdottir I, Fredrikson M. Cerebral blood flow during anticipation of public speaking in social phobia: a PET study. Biol Psychiatry. 2002;52(11):1113–9. doi: 10.1016/s0006-3223(02)01396-3. [DOI] [PubMed] [Google Scholar]
- 62.Meda SA, Pryweller JR, Thornton-Wells TA. Regional brain differences in cortical thickness, surface area and subcortical volume in individuals with Williams syndrome. PLoS One. 2012;7(2):e31913. doi: 10.1371/journal.pone.0031913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reiss AL, Eckert MA, Rose FE, Karchemskiy A, Kesler S, Chang M, et al. An experiment of nature: brain anatomy parallels cognition and behavior in Williams syndrome. J Neurosci. 2004;24(21):5009–15. doi: 10.1523/JNEUROSCI.5272-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blackmon K, Barr WB, Carlson C, Devinsky O, Dubois J, Pogash D, et al. Structural evidence for involvement of a left amygdala-orbitofrontal network in subclinical anxiety. Psychiatry Res. 2011;194(3):296–303. doi: 10.1016/j.pscychresns.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Milad MR, Rauch SL. The role of the orbitofrontal cortex in anxiety disorders. Ann N Y Acad Sci. 2007;1121:546–61. doi: 10.1196/annals.1401.006. [DOI] [PubMed] [Google Scholar]
- 66.Baur V, Bruhl AB, Herwig U, Eberle T, Rufer M, Delsignore A, et al. Evidence of frontotemporal structural hypoconnectivity in social anxiety disorder: A quantitative fiber tractography study. Hum Brain Mapp. 2011 doi: 10.1002/hbm.21447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baur V, Hanggi J, Rufer M, Delsignore A, Jancke L, Herwig U, et al. White matter alterations in social anxiety disorder. J Psychiatr Res. 2011;45(10):1366–72. doi: 10.1016/j.jpsychires.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 68.Biederman J, Hirshfeld-Becker DR, Rosenbaum JF, Herot C, Friedman D, Snidman N, et al. Further evidence of association between behavioral inhibition and social anxiety in children. Am J Psychiatry. 2001;158(10):1673–9. doi: 10.1176/appi.ajp.158.10.1673. [DOI] [PubMed] [Google Scholar]
- 69.Schwartz CE, Snidman N, Kagan J. Adolescent social anxiety as an outcome of inhibited temperament in childhood. J Am Acad Child Adolesc Psychiatry. 1999;38(8):1008–15. doi: 10.1097/00004583-199908000-00017. [DOI] [PubMed] [Google Scholar]
- 70.Schwartz CE, Kunwar PS, Greve DN, Moran LR, Viner JC, Covino JM, et al. Structural differences in adult orbital and ventromedial prefrontal cortex predicted by infant temperament at 4 months of age. Arch Gen Psychiatry. 2010;67(1):78–84. doi: 10.1001/archgenpsychiatry.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. 2007;164(10):1476–88. doi: 10.1176/appi.ajp.2007.07030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weissman MM, Brown AS, Talati A. Translational epidemiology in psychiatry: linking population to clinical and basic sciences. Arch Gen Psychiatry. 2011;68(6):600–8. doi: 10.1001/archgenpsychiatry.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davatzikos C. Why voxel-based morphometric analysis should be used with great caution when characterizing group differences. Neuroimage. 2004;23(1):17–20. doi: 10.1016/j.neuroimage.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 74.Crum WR, Griffin LD, Hill DL, Hawkes DJ. Zen and the art of medical image registration: correspondence, homology, and quality. Neuroimage. 2003;20(3):1425–37. doi: 10.1016/j.neuroimage.2003.07.014. [DOI] [PubMed] [Google Scholar]
- 75.Ioannidis JP. Excess significance bias in the literature on brain volume abnormalities. Arch Gen Psychiatry. 2011;68(8):773–80. doi: 10.1001/archgenpsychiatry.2011.28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.