

Abstract
New polyomaviruses are continually being identified, and it is likely that links between this virus family and disease will continue to emerge. Unfortunately, a specific treatment for polyomavirus-associated disease is lacking. Because polyomaviruses express large Tumor Antigen, TAg, we hypothesized that small molecule inhibitors of the essential ATPase activity of TAg would inhibit viral replication. Using a new screening platform, we identified inhibitors of TAg's ATPase activity. Lead compounds were moved into a secondary assay, and ultimately two FDA approved compounds, bithionol and hexachlorophene, were identified as the most potent TAg inhibitors known to date. Both compounds inhibited Simian Virus 40 replication as assessed by plaque assay and quantitative PCR. Moreover, these compounds inhibited BK virus, which causes BKV Associated Nephropathy. In neither case was host cell viability compromised at these concentrations. Our data indicate that directed screening for TAg inhibitors is a viable method to identify polyomavirus inhibitors, and that bithionol and hexachlorophene represent lead compounds that may be further modified and/or ultimately used to combat diseases associated with polyomavirus infection.
Keywords: polyomavirus, bithionol, hexachlorophene, T antigen, molecular chaperone, high throughput screen
1. Introduction
Polyomaviruses are double stranded DNA viruses that replicate in non-dividing cells, and in humans these viruses persist as a life-long infection (Imperiale and Major, 2007). The majority of the population is infected with at least one of the human polyomaviruses (Kean et al., 2009). In most cases, polyomavirus infection does not result in disease, but replication can proceed unabatedly in immunosuppressed patients and results in several diseases (Jiang et al., 2009). For example, reactivation of BK virus can lead to BK Virus Associated Nephropathy (BKVAN), or hemorrhagic cystitis (Hirsch and Randhawa, 2009). JC virus can cause progressive multifocal leukoencephalopathy in AIDS patients and in patients receiving specific treatments for Multiple Sclerosis (Safak and Khalili, 2003).
Six new polyomaviruses were recently identified: The KI and WU viruses were isolated from patients with respiratory infections (Allander et al., 2007; Gaynor et al., 2007); Merkel Cell Polyomavirus was recognized in Merkel Cell Carcinomas (Feng et al., 2008; Shuda et al., 2008); Human Polyomaviruses 6 and 7 were identified on the skin of healthy individuals (Schowalter et al., 2010); Trichodysplasia spinulosa virus was found in dysplastic hair follicle cells from immunocompromised individuals (van der Meijden et al., 2010); and a human virus that resembled the monkey lymphotropic virus was sequenced from immunosuppressed patients (Scuda et al., 2011). Early evidence suggests that these viruses may be reactivated in immunosuppressed individuals (Sharp et al., 2009). Given the recent and rapid increase in the number of new polyomaviruses, it is likely that additional members of this virus family linked to other diseases will emerge, particularly as the immunocompromised population expands (Kunisaki and Janoff, 2009) and because polyomaviruses are adept at replicating at low levels in non-dividing cells (Fanning et al., 2009). Unfortunately, the current treatments for polyomavirus related diseases are not specific, inadequate and/or have undesirable side effects (Josephson et al., 2006b).
In addition to the message encoding capsid proteins, polyomaviruses express one essential transcript, which encodes the conserved, multidomain transforming factor, the large tumor (T) antigen (TAg) (Cantalupo et al., 2005; Pipas, 1992). TAg triggers cell cycle progression by binding to Rb and p53, as well as a variety of other factors that regulate host cells (Pipas, 2009). To initiate viral DNA synthesis, TAg binds to the viral origin of replication and serves as the replicative helicase via its AAA+ ATPase domain (Li and Kelly, 1984). In addition, TAg harbors an essential “J domain” that stimulates the ATPase activity of the Hsp70 family of molecular chaperones (Campbell et al., 1997; Fewell et al., 2002; Kelley and Georgopoulos, 1997; Srinivasan et al., 1997). The TAg-Hsc70 complex is hypothesized to help release the transcription factor E2F from Rb (Sullivan et al., 2000), which is required for cell cycle activation. These properties are essential for viral replication and are conserved amongst most TAgs (Cantalupo et al., 2005). The early transcript also expresses a protein known as small t Antigen (tAg) that performs activities important for infection but is not required for replication of the viral genome (Sablina and Hahn, 2008).
Of the eight known human polyomaviruses, replication systems only exist for BK and JC virus. In contrast to the slow replication of these viruses, the related Simian Virus 40 (SV40) replicates well in monkey kidney cell lines and expresses a highly conserved TAg. Most of our current understanding of TAg emerged from studies on SV40 (Pipas, 2009).
Because the TAgs are conserved, and because humans lack a TAg homologue, we suggested that this protein would provide a therapeutic target to prevent polyomavirus replication. We further hypothesized that compounds that inhibit the ATPase activity of TAg, which is required for viral replication, might be identified. As a first proof-of-principle, Wright et al. determined that MAL2-11B, which slows the ATPase activity of TAg and the ability of the TAg J-domain to stimulate Hsp70 ATPase activity, inhibits the replication of SV40 and BKV (Wright et al., 2009). However, MAL2-11B is not suitable as a therapeutic due to its low solubility, poor permeability, and low antiviral potency. Moreover, MAL2-11B was not identified in a screen using TAg as a probe, but was a structural analogue of compounds that alter the ability of J domain-containing proteins to activate Hsp70 (Fewell et al., 2004; Wright et al., 2008). Thus, this agent was neither identified nor optimized as a TAg-specific target. In a second attempt to identify anti-polyomavirus agents based on TAg ATPase inhibition, a screen was performed that surveyed compounds that inhibited TAg ATPase activity and were provided by the Molecular Libraries Screening Centers Network. From these efforts—and via subsequent chemical synthesis—a lead compound was identified (Seguin et al., 2012). Unfortunately, the compound, bisphenol A (BPA), is cytotoxic and is therefore also unsuitable as a current therapeutic for polyomavirus-associated diseases.
To identify more potent, less toxic, and selective polyomavirus inhibitors, we now report on a novel high throughput screen with purified SV40 TAg. The screen took advantage of a new platform to assess modulators of ATP-hydrolyzing enzymes. In addition, to optimize our chances that potential therapeutics might be isolated, the library included MAL2-11B-like compounds as well as compounds developed and approved for other uses. As hoped, we isolated TAg ATPase inhibitors that were significantly more potent than MAL2-11B and less toxic than BPA. A preliminary structure-activity relationship (SAR) identified the key chemical features of the molecules that gave rise to their effects. The resulting lead compounds also inhibited the replication of SV40 and BKV in cultured cells with an effect dose-50% (ED50) that was >10-fold higher than the dose at which toxicity was apparent, thus providing a satisfactory therapeutic index (TI). Because the lead compounds are approved by the FDA for other applications, our efforts set the stage for the development and testing of new anti-polyomavirus agents.
2. Materials and methods
2.1 Reagents and chemical methods
The MicroSource MS2000 library contains ~2200 bioactive compounds with a minimum of 95% purity. The collection includes drug components (50%), natural products (30%) and other bioactive compounds (20%) (Miyata et al., 2010).
Bithionol (CAS 97-18-7) and hexachlorophene (CAS 70-30-4) were purchased from Sigma Aldrich, and the compounds ST024951 (1), ST029256 (2), ST033374 (3) ST033385 (4), ST057728 (5) and ST5308743 (6) were purchased from Timtec (Newark DE). Compound 5250892 (7) was obtained from ChemBridge (San Diego, CA), and resorcinol sulfoxide (8) and R128546 (9) were purchased from Sigma-Aldrich. The purities of these bisphenol-like compounds were confirmed by HPLC/UV, and by mass spectrometry when ionization was possible. Cidofovir was kindly provided by Dr. Abhay Vats (University of Pittsburgh). Eight known dihydropyrimidines were synthesized as described (Wisen et al., 2008), and their structures were confirmed using high resolution mass spectrometry and 1H NMR.
SV40 TAg was purified essentially as reported (Wright et al., 2009). In brief, Sf9 cells were infected with a baculovirus engineered for the expression of wild type TAg for 45 h. The cells were harvested and lysed, and the lysate was clarified by high-speed centrifugation. The resulting supernatant was loaded on to an immunoaffinity column containing the monoclonal TAg antibody pAB419 (Harlow et al., 1981) covalently linked to Protein G Sepharose Fast-Flow beads. The column was washed, and fractions highly enriched for TAg were eluted at pH 11. After the pH was adjusted to neutrality, the protein was dialyzed against 10mM Tris-Cl, pH 8.0, 1mM EDTA, 100mM NaCl, 10% glycerol, and 1mM DTT. TAg-containing fractions and the purity of the final preparation were verified by Coomassie Brilliant Blue staining of SDS-polyacrylamide gels and by western blot analysis. TAg was stored at −80°C until use.
2.2 A high throughput screen to identify inhibitors of TAg ATPase activity
A collection of ~150 dihydropyrimidines was screened in 96-well plates. Briefly, a stock solution of TAg was prepared in assay buffer (100 mM Tris-Cl, pH 7.4, 20 mM KCl, and 6 mM MgCl2, 0.01 % Triton X-100) to yield a final concentration of 2.5 nM (0.20 μg/μL). An aliquot of this solution (14 μL) was added to each well of a clear 96-well plate and 1 μL of either the assay compound (final concentration 200 μM) or DMSO was added. The reaction was initiated with 10 μL of 2.5 mM ATP. The plates were then incubated for 3 h at 37 °C. After incubation, each well received 80 μL of the quinaldine red reagent (Miyata et al., 2010), which was quenched by the addition of 10 μL of a 32 % sodium citrate solution. However, screening of the larger MS2000 collection necessitated development of a higher density format platform. Toward this goal, a high throughput screen to identify inhibitors of the ATPase activity of TAg was performed as described (Miyata et al., 2010), but with several modifications. First, 5uL of the a stock solution of TAg (final concentration 2.5 nM) was added to each well of opaque, white, low-volume, nonsterile, polystyrene 384-well plates (Thermo Fisher Scientific, Inc). It was critical to use these plates because assay sensitivity was strictly dependent on fluorescence quenching between the polystyrene plates and the quinaldine red agent (data not shown). To each well, 0.2 μL of either the compound (at ~5 mM in DMSO) or DMSO was added. We found that assay performance decreased above 5% DMSO. The reaction was then initiated with 2 μL of 3.2 mM ATP, and the plates were incubated for 3 h at 37 °C. After incubation, each well received 15 μL of the quinaldine red reagent, which was ultimately quenched by the addition of 2 μL of a 32% sodium citrate solution. The plates were incubated for an additional 15 min before fluorescence intensity (excitation 430 nm, emission 530 nm) was measured on a PHERAstar plate reader. The Z factor (Zhang et al., 1999) was calculated to be 0.70. The coefficient of variation ranged from 6 to 10% and the signal-to-noise was ~50.
2.3 Biochemical and kinetic assays to assess TAg activity
A steady state ATPase assay with purified SV40 TAg was performed as reported previously (Wright et al., 2009). In short, TAg was preincubated with the indicated compound for 15 min on ice, and upon addition of α32P-ATP the reaction was moved to 30°C. At the indicated times, aliquots were removed, the reaction was quenched, and the ATP and generated ADP species were resolved by thin layer chromatography. The resulting data were analyzed on a Fujifilm BAS-2500 phosphoimager and quantified using ImageGuage software (Fuji Film Science Lab). The data obtained represent the results from at least 3 independent replicates. IC50 values were determined by titrating compounds into this assay such that the volume of DMSO was constant, and data were standardized relative to a DMSO control. A sigmoid, 3-parameter line regression of the resulting data was performed using SigmaPlot (Systat Software, San Jose, CA).
A variation of this assay was used to obtain the desired kinetic constants via a Lineweaver-Burk plot. In these assays, ATP was titrated from a final concentration of 7.14 μM to 50 μM and in the presence or absence of the indicated compound, and the rates of the reactions were determined as described above. Each assay was performed at least three times. These data were plotted as a double reciprocal, and the Km and Vmax values were determined from a line fit. The differences in the obtained data sets were determined by ANCOVA, performed by SAS (SAS Institute, Cary, NC). Pairwise comparisons were performed for both the slope (used to determine the Km) and intercept (used to determine the Vmax) of the line determined by SAS, and the p values for these comparisons are reported.
Limited proteolysis was based on a previously described protocol with some modification (Weisshart et al., 2004). TAg was purified as above. Reactions were assembled with 3 μg TAg, 30 μM compound in DMSO and 4 mM nucleotide (either ATP or AMP-PNP), then incubated for 20 min on ice prior to addition of protease. A denatured TAg control sample included 3.5 μL of TCA sample buffer (80 mM Tris HCl pH 8, 8 mM EDTA, 120 mM DTT, 3.5% SDS, 0.29% glycerol, 0.08% Tris base and 0.01% bromophenol blue) in place of DMSO or nucleotide, and was heated at 75 °C for 15 min and cooled before addition of protease. Reactions were incubated at 37 °C for 5 min with 1.8 ng of Proteinase K (Sigma) diluted in 10 mM Tris pH8.0, 1.0 mM EDTA, 100 mM NaCl and 10% glycerol; the final volume was 25 μL. The reaction was stopped with 5 μL of 100% trichloroacetic acid (Fischer Scientific) and incubated for 10 min on ice. Reactions were centrifuged at 13,000×g at 4 °C for 10 min and the supernatants were removed. The pellets were resuspended in TCA sample buffer, and then separated by 12.5% SDS-PAGE. The proteins were visualized with Coomassie Brilliant Blue.
2.4 Viral replication and cell culture assays
CV1 cells were grown in MEM + 10% FBS and pen/strep and Vero cells (kindly provided by Dr. Bruce McClane, University of Pittsburgh) were grown in DMEM + 10% FBS + pen/strep at 37°C in a 5% CO2 incubator. SV40 stocks were prepared by plating CV1 cells into 24-well dishes and infecting the cells with SV40 at a multiplicity of infection (MOI) of 2 for 2 h. Next, the media was removed and replaced with media containing the desired compound or an equivalent volume of DMSO. Two biological replicates corresponding to each treatment were performed. The media was refreshed at 24 hours post infection (hpi), and again supplemented with either DMSO or the indicated compound. At 48 hpi, when the viral replication cycle of SV40 is nearly complete, the cells were frozen and thawed 3 times to provide a viral stock. This stock was titred by plaque assay using at least 3 technical replicates, based on a previously reported protocol (Murata et al., 2008). In brief, CV1 cells were grown on 6 cm dishes to near confluence and dilutions of the viral stock were plated onto the monolayer for 2 h, and then replaced by a 4mL overlay of media in 0.9% Noble agar. On 3 and 6 days post infection (dpi), an additional agar overlay was made. At 9 dpi, the agar was removed and the monolayer was stained with crystal viol et. Plaques were counted by eye, and viral mediated cell clearing was confirmed by light microscope.
A quantitative DNA replication assay for SV40 was performed as previously described (Huryn et al., 2011; Li and Kelly, 1984; Randhawa et al., 2005a). CV1 cells at ~90% confluency were infected with SV40 at an MOI of 6, and after a 2 h infection the media was removed and replaced with media containing the desired compound or an appropriate volume of DMSO. These growth conditions were used to mimic normal viral infection in non-dividing cells. The media containing the compound was refreshed daily and at 48 hpi the viral DNA was harvested by free-thawing the cells (at −20 °C) in media three times. The DNA from the resulting cell lysates was stored at −20°C and was then quantified by quantitative PCR (qPCR) as outlined below.
To obtain larger quantities of SV40 DNA and to directly visualize the viral DNA (see Figure S2), CV1 cells were grown to 90% confluency in 10 cm culture dishes, and were infected with SV40 at an MOI of 6. After 2 h of infection, the virus-containing media was replaced with fresh media containing the appropriate drug concentrations or the appropriate volume of DMSO. Dilutions were prepared in DMSO for bisphenol and hexachlorophene and each dilution was examined in cells in triplicate. After 24 hpi, the media was replaced with fresh media containing the appropriate drug dilutions. At 48 hpi, the cells were collected and viral DNA was extracted using the modified Qiagen miniprep protocol (Cantalupo et al., 2005). Specifically, cells were washed in PBS and then 250 μL of buffer P1 was added to each well. Next, 250 μL of P2 was immediately added to lyse the cells. When cells were visibly lysed by examination by light microscope, the lysate was removed and incubated with 500 μg Proteinase K at 55 °C for 1 h. The lysate was neutralized with N3 Buffer, and mixed gently. Samples were centrifuged for 10 min at 15,000g at 4 °C, and the resulting supernatant was loaded onto the supplied mini-column and centrifuged for 1 min at 15,000g at room temp. The column was washed in 700 μL PE, and briefly centrifuged to remove residual ethanol. Finally, 50 μL of TE pre-warmed to 50 °C was added to the column and equilibrated for 5 min before a final 1 min elution with the 50μl of TE buffer at room temperature was performed. The concentration of the purified viral DNA was determined using a nanodrop (Nanodrop 2000, Thermo Scientific), and 10 μl of the purified DNA was incubated with BamHI for 2 h at 37°C to linearize the SV40 DNA. Next, the total restriction digestion reaction was run on 0.8% agarose gel, alongside a 1 kb DNA ladder. Gel images were taken at identical resolutions and the viral DNA products were quantified using Image J software.
To quantify the replication of BKV, Vero cells at ~90% confluency in 12-well dishes were infected with BKV, kindly provided by Dr. Michael Imperiale (University of Michigan), for 1 h at an estimated MOI of 0.01. (BKV does not replicate as quickly or efficiently as SV40, as a result less viral stock was available for use in BKV infections than SV40 infections.) The infectious media was removed and replaced with 1 mL media containing the indicated compound or DMSO. Media containing the compound or the DMSO control was refreshed daily for 5 d in contrast to 2 d for SV40, which reflects the much slower replication cycle of BKV. In any event, these time periods represent less than one full round of viral replication. Ultimately, the cultures were lysed 5 dpi by three freeze-thaw cycles at −20 °C. This viral stock was further homogenized by pipetting before quantification by pPCR (see below).
qPCR was performed to quantify the amount of SV40 or BKV DNA and to assess the extent of viral replication. These DNA stocks, obtained above, were serially diluted for qPCR assays as described (Huryn et al., 2011). To perform the qPCR assay, a mix of primers (see below) and Taqman probe (IDT) were assembled with PerfeCta qPCR SuperMix, ROX (QuantaBiosciences) in MicroAmp plates (ABI) on a 7300 Real-Time PCR System. Absolute quantification was performed using pSVB3 (Pipas et al., 1983) as a reference for SV40 and pBKV (Dalrymple and Beemon, 1990) as a reference for BKV. Each of these plasmids contains the entire viral genome cloned in to a BamH1 site. A mix of primers that target the VP1 protein and Taqman probe (IDT DNA) (for SV40 5' (FAM)/ATCAGGAACCCAGCACTCCACT/(IOWA BLACK) 3', primer 1- 5' GATGAACACTGACCACAAGG 3', primer 2- 5' GCACATTTTCCCCACCT 3') were used to detect SV40 (Wright et al., 2009). The primers for BKV detection were reported elsewhere (Huryn et al., 2011).
To assess compound toxicity, an MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay was performed (Cory et al., 1991; Minguez et al., 2003). CV1 cells were plated in 96 well dishes and grown to 90% confluency. The desired concentration of compounds was added to warm MEM and plated onto cells in at least three technical replicates. At 24 h, the media was aspirated and the indicated compound dissolved in media was re-added. At 48 h, the media was again aspirated but was replaced with diluted MTS reagent (Promega) in MEM lacking phenol red. The plate was returned to the 37 °C incubator for 3 h. Data were collected on a BioRad iMark Microplate Reader (Hercules, CA) at 490 nm. The data were compared to both a DMSO (negative) and 1 M staurosporine (positive) control. The LD50 was determined by sigmoid 3-parameter line regression of the data using SigmaPlot (Systat Software, San Jose, CA).
2.4 In silico chemical similarity searches
Similarity searches were conducted in an in-house database MScreen (http://mscreen.lsi.umich.edu/) using each scaffold against the external library set consisting of structures from Asinex, ChemBridge, ChemDiv, MayBridge, NCI, PubChem, Sigma, and TimTec. All structures within 70% similarity were retrieved for each chemical scaffold. The combined results were imported into the software program DVS and fingerprints using the 166 public MACCS keys were computed for the structures. Fingerprints were not successfully computed for all of the structures. Tanimoto distances were computed for each chemical scaffold against all of the retrieved structures. The results were then imported into Benchware DataMiner along with the structures and the original scaffolds. To aid in sorting, each original scaffold was given a Tanimoto distance of “−1” to itself.
3. Results
3.1 Identification of compounds that inhibit TAg ATP hydrolysis
We hypothesized that inhibitors of the conserved and essential SV40 TAg ATPase activity would be specific inhibitors of polyomaviruses, since each virus encodes a highly conserved TAg. Toward this goal, we adapted a procedure originally developed to identify inhibitors of Hsp70 ATPase activity (Chang et al., 2008; Miyata et al., 2010). This method measures the enzymatic release of inorganic phosphate from ATP using the quinaldine red (QR) reagent. First, a library of approximately 150 dihydropyrimidines was screened to identify close structural derivatives of MAL2-11B that may be better TAg inhibitors (Wisen et al., 2008; Wright et al., 2009). Compounds in this collection were screened at 200 μM in 96-well plates, as described in the Materials and methods. From this process, thirteen compounds were identified that inhibited > 35% of the ATPase activity of TAg. Based on the fact that it lacks endogenous ATPase activity, the compounds are predicted to have no effect on small TAg. The activities of these compounds were then confirmed by re-screening them against TAg in duplicate. To complement these inhibitors and to identify alternative chemical scaffolds, we next screened the commercially available Spectrum Collection (MicroSource Discovery Inc., Groton, Conn), which contains 2,240 known bioactives and drugs approved by the FDA for a range of indications. However, because of the larger size of this library, a higher throughput (e.g. 384-well plates) version of the QR assay was required. Therefore, we conducted pilot experiments with TAg to understand whether a miniaturized version would still give robust screening performance. We found that the signal was linear for >3 hours when the concentration of TAg was at least 2.5 nM (0.20 μg/μL), the levels of ATP were 1 mM, and the reaction was performed at 37 °C (Figure 1A). TAg also yielded a dose-dependent increase in signal intensity (Figure 1B). Moreover, the assay exhibited good screening parameters under these conditions, with an average Z factor between 0.6 and 0.7. Encouraged by these findings, we screened the MS2000 collection in low volume, 384-well microtiter plates to identify potential inhibitors. These compounds were screened at 100 μM and 33 chemicals (1.4% of the collection) were identified as potential inhibitors because they reduced ATPase activity by > 20% (Figure 1C). Of these compounds, the 20 with the best inhibitory activity were confirmed in duplicate. Together, these combined screening efforts resulted in the identification of 13 dihydropyrimidines and 20 other compounds that appeared to directly inhibit TAg ATPase activity.
Figure 1.

Development of a high throughput screen for TAg. A. The signal is linear for at least 3 hours at 0.20 μg/well (2.5 nM) of TAg, 1 mM ATP and 37 °C. Phosphate levels were measured using quinaldine red (QR) and converted to pmoles using a set of sodium phosphate standards. B. Relationship between TAg concentration and raw phosphate signal (OD620). To conserve protein, 0.20 μg per well (2.5 nM) was chosen for screening. Results are the average of triplicate wells and the error bars represent the standard deviation (SD). C. The results from screening the MS2000 collection. TAg was screened in 384-well plates, as described in the Materials and methods. The red line is 3 standard deviations of the negative control. The purple line is the Z factor, calculated by plate. The controls (positive (red): no enzyme; negative (blue): DMSO) were present on each plate. D. Chemical structures of the active compounds from the MS2000 collection that were confirmed in duplicate and then screened in the steady-state, radioactive ATPase assay (see Figure 2). E. The structure of MAL2-11B is shown for comparison.
Next, we wanted to test these compounds in an independent, steady state ATPase assay. This assay relies on 32P radioactivity instead of the colorimetric QR reagent. Unlike the QR assay, the steady state assay allows us to measure kinetics constants and is conducted under conditions in which the enzyme is stable; thus, it is expected to identify false positives. From the list of active compounds, we re-synthesized 8 of the most potent dihydroyrimidines and purchased 12 of the commercially available compounds from the MS2000 list. These compounds included a number of flavones, polyphenols and bisphenols, such as quercitin, baicalein, and chloranil (Figure 1D). We established that only three of these compounds, bithionol, chloranil and hexachlorophene, were significantly more potent inhibitors than MAL2-11B when used at a final concentration of 100 μM (Figure 2A). The remainder of the compounds were either false positives or their activities were not significantly better than MAL2-11B. Because chloranil caused TAg to aggregate under the utilized assay conditions (Figure S1), we focused our efforts on bithionol and hexachlorophene (Figure 2B). Titrations of bithionol and hexachlorophene into the steady state ATPase assay revealed apparent inhibitory concentration-50% (IC50) values of 3.3±0.34 and 1.7±0.17 μM, respectively (Figure 2C). Thus, the potencies of bithionol and hexachlorophene were greater than those of any other reported TAg inhibitor, including MAL2-11B, BPA, ellagic acid, and spiperone (Goodwin et al., 2009; Seguin et al., 2012; Wright et al., 2009).
Figure 2.
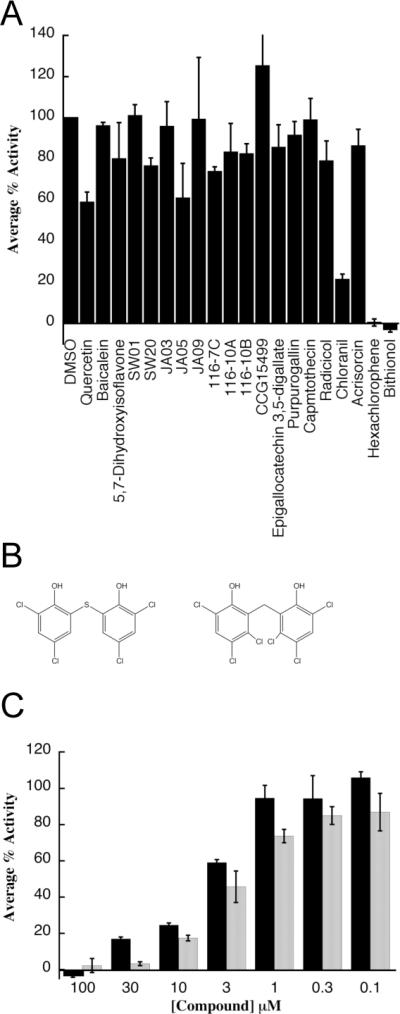
A high throughput screen identifies three inhibitors of TAg ATPase activity. A. The indicated compounds were tested in triplicate at a final concentration of 100 μM in a steady state ATPase assay and the data plotted relative to the DMSO control, which is set at “100.” B. The structures of bithionol and hexachlorophene are shown. C. Results from triplicate steady state ATPase assays are shown, +/− SD, for bithionol, grey bars, and hexachlorophene, black bars, at the indicated concentrations. Data were again plotted relative to the DMSO control, which was set at “100.”
3.2 A structure-activity relationship for novel TAg ATPase inhibitors
Because the structures of bithionol and hexachlorophene are related, we hypothesized that the directed screening of other related compounds would help us establish a structure-activity relationship (SAR) for the inhibition of TAg ATPase activity. To this end, an in silico search was performed and 60 compounds were identified that were either structurally similar to these compounds or could be employed to test specific hypotheses regarding the chemical groups that mediate the inhibitory activity on TAg. A subset of these compounds was then selected based on commercial availability (Figure 3A), and these were screened in the steady state ATPase assay at a final concentration of 30 μM (Figure 3B). This concentration was chosen based on the fact that bithionol and hexachlorophene inhibited the ATPase activity of TAg by ~80% (Figure 2C). We found that only two of the tested compounds inhibited TAg ATPase activity, and these agents, (6,6'-thiobis(2-bromo-4-chlorophenol) and 6,6'-methylenebis(2-bromo-4-chlorophenol)), are most structurally similar to bithionol. Based on the data in Figure 3B, we also conclude that some essential features for TAg inhibition include a bisphenol-like moiety, flexibility at the biraryl linker group, and the presence of substituents at positions 2 and 4 on the phenols; however, bulky groups at these positions appeared to disrupt inhibitory activity.
Figure 3.

A structure-activity relationship for the action of select bisphenols and bisphenol-like compounds for inhibition of TAg ATPase activity. A. An in silico search for bisphenol-related structures based on Tanimoto distances was performed (see Materials and methods), and from these results nine compounds were selected for further analysis of their effects on TAg ATPase activity. B. The indicated compounds were tested in a steady-state ATPase assay, as in Figure 2, at a final concentration of 30 μM. The results are shown relative to a DMSO control (−), in triplicate, +/− SD. Data were standardized relative to the DMSO control, which was set at “100.”
3.3 Bithionol and hexachlorophene inhibit SV40 replication
Bithionol and hexachlorophene were derived from the Spectrum 2000 library as FDA-approved compounds for their bacteriostatic and antiparasitic properties. In fact, both compounds were used as topical antiseptics before other alternatives became available. In addition, bithionol has successfully been used to treat outbreaks of the parasite Paragonimus westermani in humans, and can be taken orally at a concentration of 40 mg/kg every other day (Kim, 1970). To our knowledge, neither compound has been examined for its ability to treat a viral infection.
To examine whether the compounds inhibit polyomavirus replication, the activities of bithionol and hexachlorophene were first tested in an SV40 plaque assay. CV1 monkey kidney cells were infected with SV40 and treated with bithionol and hexachlorophene at a final concentration of 30 μM. As a control, we also retested MAL2-11B at a final concentration of 100 μM. It is important to note that in these and all subsequent viral replication assays (see below), the compounds were added after viral infection, so that only effects on viral replication (and not viral entry) would be assessed. Next, a crude viral stock was obtained and titred by plaque assay. We first observed that MAL2-11B reduced the number of plaques by nearly 60% (Figure 4A), which is consistent with previously published results (Wright et al., 2009). We also discovered that bithionol and hexachlorophene inhibited viral replication nearly 100-fold relative to the DMSO control, indicating that both compounds are significantly more potent inhibitors of SV40 propagation than MAL2-11B. A more significant dose dependence on the effects of bithionol and hexachlorophene on SV40 replication was then performed, but in this assay total DNA was harvested and linearized with HinDIII in order to visualize the viral DNA (see Materials and methods; Figure S2). By quantifying these data we obtained EC50s for bithionol and hexachlorophene of 2.2±0.51 μM and 3.2±0.55 μM, respectively.
Figure 4.
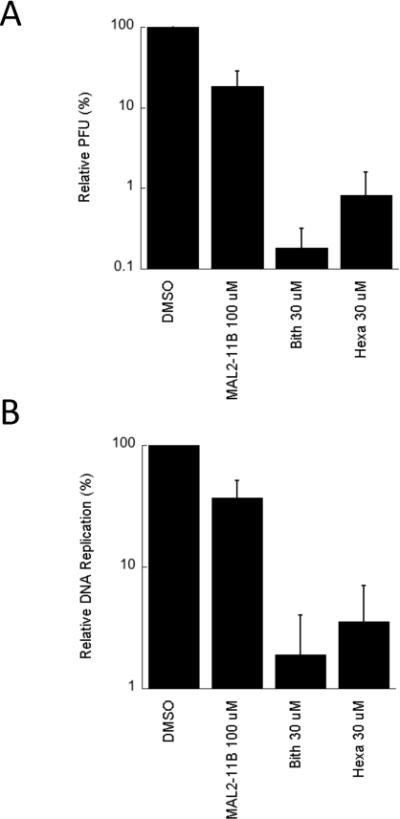
Bithionol and hexachlorophene inhibit SV40 replication in CV1 monkey kidney cells. A. SV40 replication was assessed in the presence of MAL2-11B, bithionol (“Bith”) or hexachlorophene (“Hexa”) for 2 days, after which the replication-competent virus was titred by plaque assay. The data represent three separate infections, each measured in at least two technical replicates, +/− SD. Data were standardized relative to the DMSO control, which was set at “100%.” B. Replication of viral DNA in a parallel experiment was quantified by qPCR under identical conditions to those used in part A. The data represent the means of 12 independent experiments, +/− SE. Please note that in A and B MAL2-11B was used at a final concentration of 100 μM, whereas bithionol and hexachlorophene were used at a final concentration of 30 μM.
To confirm these results and to provide a quantitative method to rapidly test TAg inhibitors, a qPCR assay was developed to measure SV40 DNA replication in our cell culture system. This assay is based on previous work to detect the human polyomavirus BKV in patient samples (Randhawa et al., 2005a; Randhawa et al., 2005b). The assay takes advantage of the small size of the polyomavirus genome (~5 kb), which can be purified like a plasmid or isolated in sufficient quantities in a supernatant fraction after subjecting the cells to freeze-thaw cycles. In brief, cells were infected as described for the plaque assay and processed, and the viral DNA was titred by absolute quantification in a qPCR assay (see Materials and methods). Finally, the data were compared to an untreated, infected control.
As anticipated, we again found that MAL2-11B at a final concentration of 100 μM inhibited DNA replication in culture by ~60% (Figure 4B). Further, both bithionol and hexachlorophene inhibited DNA replication ~100-fold at 30 μM (Figure 4B). This result is completely consistent with the results of the plaque assay, thus validating the protocol. Although not all viral DNA replicated during infection leads to infectious viral particles (Rigby and Berg, 1978), we observed a clear correlation between the outcome of the plaque assay and the qPCR assay. More generally, the data support our hypothesis that a directed isolation of TAg ATPase inhibitors can be used to identify compounds that inhibit polyomavirus replication.
One possible mechanism to explain the inhibition of viral replication is that the compounds compromise host cell viability. Indeed, because of the bisphenol-like moiety, bithionol and hexachlorophene are somewhat structurally related to BPA, which is quite toxic in some cell types (Seguin et al., 2012). To exclude this scenario, a viability assay was performed with each compound in CV1 cells. The data were also compared to a DMSO negative control and to a positive control, the toxic agent staurosporine. Importantly, the compounds were added and the cells were treated under conditions that were identical to those used when the effects of the compounds on SV40 replication were assessed (i.e., ~90% confluency). It is also important to note that theses assays were purposely performed on cells that were nearly confluent and were not in log-phase, as these viruses normally reside and replicate in non-proliferative cells (see Introduction). In this assay, the MI50 (metabolic index-50) is defined as the concentration of compound that yields one-half the signal compared to the negative control. The MI50 values obtained for bithionol and hexachlorophene were 68 and 55 μM, respectively (95% confidence ranges based on the sigmoidal fit of 41–60 and 29–71 μM, respectively), which is significantly less toxic than BPA, as measured under similar conditions (Seguin et al., 2012). This analysis also leads to the derivation of the Therapeutic Index (TI) (Goodman et al., 2006) for the compounds, which is defined as the MI50/EC50. For bithionol and hexachlorophene the TIs were 31 and 17, respectively. These values indicate a relatively high degree of specificity, and of note, some commonly used drugs (e.g., digoxin) have a TI of only ~2. We therefore conclude that the antiviral effects of bithionol and hexachlorophene are independent of their toxicities, and represent improved compounds compared to BPA (also see Discussion).
In order to characterize how bithionol and hexachlorophene inhibit the ATPase activity of TAg, ATP was titrated into the steady state ATPase assay while the concentration of inhibitor was held constant. The data were analyzed by double reciprocal (i.e., Lineweaver-Burk) plots. To verify that meaningful kinetic information could be obtained, we also assessed the ability of ADP to function as a bona fide competitive inhibitor for TAg's ATPase activity. Figure 5 presents an example of the analysis lacking or containing ADP at a constant, final concentration of 150 μM. As expected, the competitive inhibitor ADP increased the Km for ATP whereas the Vmax stayed relatively constant, which is typical of competitive inhibition (Segel, 1976). When bithionol and hexachlorophene were examined at a final concentration of 3 μM, which inhibits TAg ATPase activity by ~50%, there were statistically significant increases in the Km values for ATP (Figure 5 and Table 1). Effects on the Vmax values were also apparent but these were not significantly different from the DMSO control (Table 1). The simplest interpretation of these data is that bithionol and hexachlorophene act as competitive inhibitors for ATP. However, because of the significant differences in the structures of ATP and these two bisphenols, it is also possible that the compounds bind another site, which alters TAg conformation (see Discussion section). It is also important to note that the compounds do not cause a global unfolding of TAg, as evidenced by the use of a limited proteolysis assay (Weisshart et al., 1996) (Figure S3).
Figure 5.
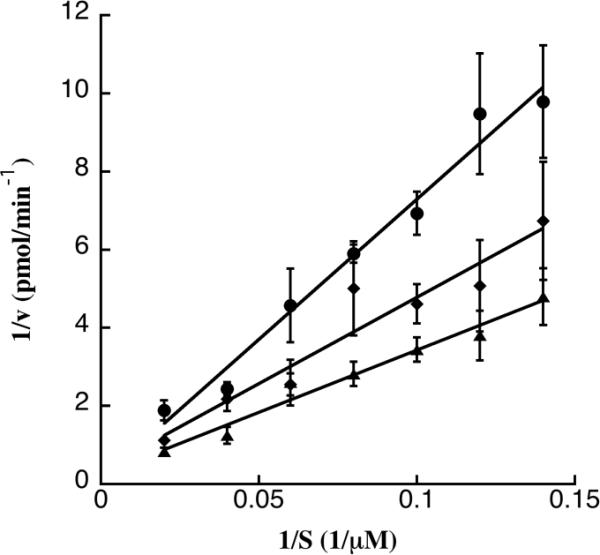
A Lineweaver-Burk analysis of TAg was performed over a range of ATP concentrations and was used to determine the Km and Vmax. In this figure, the effect of ADP at a final concentration of 150 μM was examined as a competitive inhibitor of a range (final concentrations of 5–50 μM ATP) of substrate (S) concentrations. Data represent values obtained either in the presence of DMSO (triangles), 150μM ADP (circles) or 3 μM hexachlorophene (diamonds). Assays were performed at least in triplicate, and are shown as means of the data, +/− SD.
Table 1.
Bithionol and hexachlorophene affect the Km for ATP in TAg. Kinetic parameters (Km and Vmax) were determined by Lineweaver-Burk analysis of TAg in the presence of 150 μM ADP or 3 μM bithionol or hexachlorophene over a range of ATP concentrations, as shown in Figure 4. ANCOVA was used to determine the p values for each pair-wise comparison.
| Km (μM) | Vmax (pmol/min) | |
|---|---|---|
| TAg + DMSO | 110 | 3.5 |
| TAg + ADP | 200** | 2.9‡ |
| TAg + Bithionol | 590** | 10‡ |
| TAg + Hexachlorophene | 290* | 6.6‡ |
TAg + DMSO versus TAg + ADP or bithionol,
p<0.01;
TAg + DMSO versus TAg + hexachlorophene,
p<0.05;
TAg + DMSO versus TAg + ADP, bithionol or hexachlorophene,
p>0.5
3.4 Bithionol and hexachlorophene inhibit BKV replication
We next examined whether bithionol and hexachlorophene inhibit the replication of a clinically relevant polyomavirus, BKV. For this assay, a low titre infection was performed in Vero cells, and the DNA was quantified by qPCR, as performed for SV40 (Figure 4B). The low titre infection is a result of the limited replication of BKV in vitro. Data were compared to a DMSO control, MAL2-11B and to cidofovir, which has been shown to inhibit BKV replication in vitro and has been used clinically (Farasati et al., 2005; Johnston et al., 2010). (A full range of cidofovir concentrations and the resulting effects on SV40 replication are shown in Figure S4.) The results of the experiment showed that bithionol and hexachlorophene inhibit BKV replication by ~70% at a final concentration of 30 μM, and this effect is comparable to the impact of MAL2-11B or cidofovir on BKV when used at much higher final concentrations (100 and 350 μM, respectively; Figure 6). Interestingly, hexachlorophene maintained its potent inhibition at 10 μM, but bithionol was somewhat less effective at this lower concentration (Figure 6A). A model to describe this phenomenon is provided in the Discussion.
Figure 6.

Both bithionol and hexachlorophene inhibit BKV DNA replication in Vero monkey kidney cells. A. The replication of BKV DNA was assessed after 5 d of infection with MAL2-11B, cidofovir, bithionol or hexachlorophene, and the levels of viral DNA were quantified by qPCR as described in the Materials and methods. The data were standardized to the DMSO control, which is set to “100” +/− SE. Data represent the means of 12 experiments from four independent infections. Please note that MAL2-11B was used at a final concentration of 100 μM and cidofovir at 350 μM, whereas bithionol and hexachlorophene were used at a final concentration of 10 or 30 μM. B. The combination of bithionol and cidofovir does not further inhibit SV40 DNA replication in CV1 monkey kidney cells. Replication of viral DNA was quantified by qPCR as described in the Materials and methods. The data represent the means of 8 independent experiments, +/− SE. Each compound was tested as indicated at a final concentration of 10 μM. The combination of 10 μM bithionol and 10 μM cidofovir was not significantly different from the effect of 10 μM cidofovir alone.
Together, our data indicate that the directed screening for TAg ATPase inhibitors can be used to identify inhibitors of polyomavirus infection. Among these inhibitors, two compounds were identified that are more potent than clinically evaluated anti-virals and are less toxic and have improved pharmacological properties than other isolated TAg inhibitors (also see below). Our data also indicate that second-round screening of agents via qPCR assay for SV40 replication is a valid route to isolate inhibitors of BKV replication, which is a significantly more laborious virus to propagate and analyze.
4. Discussion
Only recently have there been significant efforts to identify polyomavirus inhibitors. One previous screening strategy to identify polyomavirus inhibitors took advantage of indirect read-outs for viral infection, and led to the identification of ellagic acid and spiperone as inhibitors of TAg expression in SV40, BKV, and JCV infected cells (Goodwin et al., 2009). A preliminary analysis of these compounds indicated that they may block viral entry. In these previous studies, target and off-target effects could not be ascertained. In contrast, we took advantage of our knowledge of polyomaviruses biology to identify druggable components of the viral lifecycle. We then created an assay to target this activity. The assay co-opts the fact that TAg's ATPase activity is essential for viral replication (Castellino et al., 1997; Fanning et al., 2009), and that humans lack a TAg homologue. The data presented in this manuscript indicate the success of this targeted, mechanism-based approach. Based on the excellent Z factor and signal-to-noise for the targeted screen (0.7 and 50, respectively), the specificity and structural similarities of the lead compounds, and the fact that the estimated therapeutic index for these compounds is >10, we are also confident that more potent and efficacious inhibitors can be obtained via subsequent efforts with more diverse libraries.
Our work was initiated to address the paucity of available therapeutics for polyomavirus infection. Previous studies to identify compounds that inhibit polyomavirus led to the discovery of cidofovir, leflunomide (Farasati et al., 2005), and the quinolones (Ali et al., 2007). These agents have been used with some success in the clinic. Cidofovir inhibits BKV in vitro and is commonly used as a treatment for BKVAN (Farasati et al., 2005), but high creatine levels, which are typical in nephropathy, contraindicate its use. In addition, there has been no controlled clinical study to suggest that the lower dosage levels used in BKVAN actually compromise viral replication beyond the impact of reducing immunosuppression (Hirsch and Randhawa, 2009). In our studies, the bisphenols at a final concentration of 100 μM inhibited BKV replication to approximately the same extent as a clinically used compound, cidofovir, at a final concentration of 350 μM (Figure 6A). Similarly, the immunosuppressant used to treat rheumatoid arthritis, leflunomide, has been shown to decrease BKV replication in vitro but has not been rigorously examined in clinical trials (Faguer et al., 2007; Josephson et al., 2006a; Josephson et al., 2006b). A third class of compounds used for the treatment of BKVAN is the quinolones, including ciprofloxacin and ofloxacin. While these antibiotics have shown promise both in vitro and in the clinic (Leung et al., 2005; Randhawa, 2005), many patients cannot tolerate them, and they exhibit relatively low potency (Ali et al., 2007). In addition, we previously uncovered MAL2-11B as an inhibitor of SV40 and BKV replication, but this agent was neither obtained via directed screening efforts nor optimized as an anti-viral agent (Wright et al., 2009). Not surprisingly, then, the low solubility and efficacy of this compound requires further refinement if the MAL2-11B scaffold is to be pursued as an anti-viral lead structure. We also uncovered BPA as a TAg ATPase inhibitor but we found that this toxic agent led to cell death at approximately the same concentration that inhibition of TAg was evident (Seguin et al., 2012). Thus, this compound was also unsuitable for development as a potential anti-viral agent.
Even though the potencies of the compounds described in this report are comparable to MAL2-11B, various physicochemical parameters and drug-like structural filters can be used to highlight the differences between MAL2-11B, bithionol, and hexachlorophene. For example, the total polar surface area (TPSA) for the three compounds is 95.9, 40.5, and 40.5, respectively, and their molecular weights and numbers of rotatable bonds are 512.6, 356.1, and 406.9, and 12, 2, and 2. Therefore, as anticipated, bithionol and hexachlorophene are far better than MAL2-11B in satisfying the standard structural filters for drug-likeliness. Even though all three compounds have clogP's >5, MAL2-11B does not pass either lead likeliness, Lipinski, or Veber filters, whereas bithionol passes lead likeliness and Veber filters, as well as the Lipinski rule of 5 (3 of 4), and hexachlorophene passes the Veber filter and the Lipinski rule of 5 (3 of 4) (Lipinski et al., 2001). In support that the effects of the bisphenols do not stem from cytotoxicity, we noted that the ability of bithionol and cidofovir, both at 10 μM, to inhibit SV40 replication were not additive (Figure 6B). Cidofovir-mediated inhibition of SV40 was previously observed, and as shown in Figure S4, we have titrated this effect (Andrei et al., 1997). Cidofovir is a cytosine analog and a general polymerase inhibitor. If bithionol non-specifically inhibited viral replication, then one would instead predict an additive or synergistic effect of cidofovir and bithionol on replication. Overall, based on their physicochemical properties and relative specificities, bithionol and hexachlorophene can be considered promising candidates for the development of active pharmaceuticals.
As described above, bithionol and hexachlorophene were included in the library used for our screen by virtue of the fact that they are FDA approved for other applications. Both compounds have been used as topical anti-microbials, but it is known that overexposure can be toxic (Kimbrough, 1973; Powell and Lampert, 1973). Under conditions in which the potential neurotoxic effects of these agents are not as problematic, bithionol and hexachlorophene also have veterinary applications as anti-parasitic and anti-fluke agents. In fact, these compounds may be of use for the veterinary treatment of Avian Polyomavirus, which can cause acute death in a wide range of psittacine birds (Katoh et al., 2010). Further, in geographical regions where fluke outbreaks in humans are more problematic, bithionol has been used to successfully clear infections in humans after oral administration (Bacq et al., 1991; Kim, 1970; Lee and Kim, 2006; Price et al., 1993; Seo et al., 1982).
The correlation between SV40 inhibition and BKV inhibition confirms our hypothesis that the isolation of SV40 TAg ATPase and SV40 replication inhibitors can lead to the identification of inhibitors of other polyomaviruses. It was interesting that the relative potencies of hexachlorophene and bithionol on purified TAg activity and on SV40 and BKV replication are within the same range, although the compounds were somewhat less potent in the cell-based replication assays. Indeed, one might expect effects to be much more potent in vitro, particularly as compounds may be metabolized to inactive species when incubated with cells. Thus, it is worth noting that the compounds needed to be added with each media change in the viral assays (see Materials and methods); the reason for this was that the combined effects of viral replication and the compounds led to a significant number of cells that rounded-up, an observation that suggests that a toxic byproduct of the compounds is produced and needs to be removed. It was also curious that bithionol and hexachlorophene inhibit BKV to a different extent when used at a final concentration of 10 μM, although their effects on the ATPase activity of TAg and on SV40 were essentially identical. Notably, however, this phenomenon has been observed previously (Wright et al., 2009). It may reflect bithionol's differential interactions with the SV40 and BKV TAgs, which—while highly conserved (74% identity)—are certainly not identical. It is also worthwhile to note that while bithionol and hexachlorophene are structurally similar, they may not interact identically with TAg. Indeed, their effects on the Km for ATP in TAg are unique (Table 1). A recent report also indicated that these agents inhibit glutamate dehydrogenase, but interestingly they bound somewhat differently to this hexameric enzyme when analyzed structurally (Li et al., 2009). Hexachlorophene binds the center core of the hexamer, while bithionol binds at the two-fold axis between pairs of subunits. Thus, it is possible that bithionol and hexachlorophene also interact differently with TAg, perhaps even at a site that is distinct from the ATP biding pocket. Indeed, even though the effects of bithionol and hexachlorophene on the Vmax of TAg were not statistically significant (Table 1), it is possible that the compounds have pleiotropic effects on TAg: A primary binding site might be at or near the nucleotide pocket, as observed by competitive inhibition of the NADPH binding enzyme 3-oxoacyl-ACP reductase (Wickramasinghe et al., 2006), whereas secondary binding may occur at the hexameric interfaces in TAg. Nevertheless, we note that the addition of both bithionol and hexachlorophene to TAg in the in vitro assay did not lead to an additive or synergistic effect, suggesting that the compounds do bind to a similar site on TAg (data not shown). Together, specific questions regarding the mechanisms of action of these compounds can best be resolved via a structural analysis of bisphenol binding to TAg, an effort that is underway. Regardless, it is important to emphasize that whatever off-target effects these agents may possess, the magnitude of these interactions is insufficient to alter cell viability anywhere near the effective concentration needed to inhibit SV40 or BKV replication.
Overall, the experiments reported in this manuscript document three important findings. First, we were able to design a high throughput screen to identify inhibitors of the SV40 TAg. This method was suitable for screening in 384-well plates and it may be co-opted in the future to screen larger and more diverse libraries. Second, we identified two FDA-approved compounds that inhibit both TAg ATPase activity and the replication of two polyomaviruses, SV40 and BKV. Third, we developed and verified the efficacy of qPCR assays that can be employed to perform secondary and tertiary screens to more rapidly verify the efficacies of polyomavirus inhibitors in future efforts. The recent rise in the number of new polyomaviruses and the fact that the population of immunocompromised individuals is increasing indicate a dire need for new antiviral therapies. Our work not only provides a novel strategy to identify potent and specific polyomavirus inhibitors, but the two most promising candidates from this effort may be re-formulated and tested for their effects in the clinic.
Supplementary Material
Highlights
Polyomaviruses are linked to an increasing number of diseases
To date, specific therapeutics that target this class of viruses are not known
A high-throughput screen was performed with a polyomavirus-encoded target
Two FDA-approved compounds were identified that inhibit polyomavirus growth
A structure activity relationship establishes key features of the compounds
Acknowledgements
We gratefully acknowledge Paul Cantalupo, Abhilasha Rathi and other members of the Pipas laboratory for helpful discussion and reagents. We also thank Marnin Wolfe and Chris Guerriero for help with the statistical analyses, and Kayla Lloyd for technical support. J.E.G. acknowledges the experimental assistance of Martha Larsen and Paul Kirchhoff at the University of Michigan's Center for Chemical Genomics. This work was supported by National Institutes of Health grants MH84077 and DK78307 (“The Pittsburgh Center for Kidney Research”) to J.L.B, P50 GM067082 (the University of Pittsburgh Chemical Methodologies and Library Development Center) to P.W., and NS059690 to J.E.G from the National Institutes of Health. A.W.I. acknowledges funding from the Howard Hughes Medical Institute fellowship program and the Beckman Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript.The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali SH, Chandraker A, DeCaprio JA. Inhibition of Simian virus 40 large T antigen helicase activity by fluoroquinolones. Antivir Ther. 2007;12:1–6. [PubMed] [Google Scholar]
- Allander T, Andreasson K, Gupta S, Bjerkner A, Bogdanovic G, Persson MA, Dalianis T, Ramqvist T, Andersson B. Identification of a third human polyomavirus. J Virol. 2007;81:4130–4136. doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei G, Snoeck R, Vandeputte M, De Clercq E. Activities of various compounds against murine and primate polyomaviruses. Antimicrobial agents and chemotherapy. 1997;41:587–593. doi: 10.1128/aac.41.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacq Y, Besnier JM, Duong TH, Pavie G, Metman EH, Choutet P. Successful treatment of acute fascioliasis with bithionol. Hepatology. 1991;14:1066–1069. [PubMed] [Google Scholar]
- Campbell KS, Mullane KP, Aksoy IA, Stubdal H, Zalvide J, Pipas JM, Silver PA, Roberts TM, Schaffhausen BS, DeCaprio JA. DnaJ/hsp40 chaperone domain of SV40 large T antigen promotes efficient viral DNA replication. Genes Dev. 1997;11:1098–1110. doi: 10.1101/gad.11.9.1098. [DOI] [PubMed] [Google Scholar]
- Cantalupo P, Doering A, Sullivan CS, Pal A, Peden KW, Lewis AM, Pipas JM. Complete nucleotide sequence of polyomavirus SA12. J Virol. 2005;79:13094–13104. doi: 10.1128/JVI.79.20.13094-13104.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellino AM, Cantalupo P, Marks IM, Vartikar JV, Peden KW, Pipas JM. trans-Dominant and non-trans-dominant mutant simian virus 40 large T antigens show distinct responses to ATP. J Virol. 1997;71:7549–7559. doi: 10.1128/jvi.71.10.7549-7559.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Bertelsen EB, Wisen S, Larsen EM, Zuiderweg ER, Gestwicki JE. High-throughput screen for small molecules that modulate the ATPase activity of the molecular chaperone DnaK. Anal Biochem. 2008;372:167–176. doi: 10.1016/j.ab.2007.08.020. [DOI] [PubMed] [Google Scholar]
- Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- Dalrymple SA, Beemon KL. BK virus T antigens induce kidney carcinomas and thymoproliferative disorders in transgenic mice. J Virol. 1990;64:1182–1191. doi: 10.1128/jvi.64.3.1182-1191.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faguer S, Hirsch HH, Kamar N, Guilbeau-Frugier C, Ribes D, Guitard J, Esposito L, Cointault O, Modesto A, Lavit M, Mengelle C, Rostaing L. Leflunomide treatment for polyomavirus BK-associated nephropathy after kidney transplantation. Transpl Int. 2007;20:962–969. doi: 10.1111/j.1432-2277.2007.00523.x. [DOI] [PubMed] [Google Scholar]
- Fanning E, Zhao X, Jiang X. Polyomavirus Life Cycle. In: Damania B, Pipas JM, editors. DNA Tumor Viruses. Springer New York; New York, NY: 2009. [Google Scholar]
- Farasati NA, Shapiro R, Vats A, Randhawa P. Effect of leflunomide and cidofovir on replication of BK virus in an in vitro culture system. Transplantation. 2005;79:116–118. doi: 10.1097/01.tp.0000149338.97084.5f. [DOI] [PubMed] [Google Scholar]
- Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewell SW, Pipas JM, Brodsky JL. Mutagenesis of a functional chimeric gene in yeast identifies mutations in the simian virus 40 large T antigen J domain. Proc Natl Acad Sci U S A. 2002;99:2002–2007. doi: 10.1073/pnas.042670999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, Day BW, Brodsky JL. Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J Biol Chem. 2004;279:51131–51140. doi: 10.1074/jbc.M404857200. [DOI] [PubMed] [Google Scholar]
- Gaynor AM, Nissen MD, Whiley DM, Mackay IM, Lambert SB, Wu G, Brennan DC, Storch GA, Sloots TP, Wang D. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007;3:e64. doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman LS, Gilman A, Brunton LL, Lazo JS, Parker KL. Goodman & Gilman's the pharmacological basis of therapeutics. 11th ed. McGraw-Hill; New York: 2006. [Google Scholar]
- Goodwin EC, Atwood WJ, DiMaio D. High-throughput cell-based screen for chemicals that inhibit infection by simian virus 40 and human polyomaviruses. J Virol. 2009;83:5630–5639. doi: 10.1128/JVI.00203-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E, Crawford LV, Pim DC, Williamson NM. Monoclonal antibodies specific for simian virus 40 tumor antigens. J Virol. 1981;39:861–869. doi: 10.1128/jvi.39.3.861-869.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch HH, Randhawa P. BK virus in solid organ transplant recipients. Am J Transplant. 2009;9(Suppl 4):S136–146. doi: 10.1111/j.1600-6143.2009.02904.x. [DOI] [PubMed] [Google Scholar]
- Huryn DM, Brodsky JL, Brummond KM, Chambers PG, Eyer B, Ireland AW, Kawasumi M, Laporte MG, Lloyd K, Manteau B, Nghiem P, Quade B, Seguin SP, Wipf P. Chemical methodology as a source of small-molecule checkpoint inhibitors and heat shock protein 70 (Hsp70) modulators. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:6757–6762. doi: 10.1073/pnas.1015251108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperiale MJ, Major EO. Polyomaviruses. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Field's Virology. 5th ed. Lippincott Williams & Wilkins; Philadelphia: 2007. [Google Scholar]
- Jiang M, Abend JR, Johnson SF, Imperiale MJ. The role of polyomaviruses in human disease. Virology. 2009;384:266–273. doi: 10.1016/j.virol.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston O, Jaswal D, Gill JS, Doucette S, Fergusson DA, Knoll GA. Treatment of polyomavirus infection in kidney transplant recipients: a systematic review. Transplantation. 2010;89:1057–1070. doi: 10.1097/TP.0b013e3181d0e15e. [DOI] [PubMed] [Google Scholar]
- Josephson MA, Gillen D, Javaid B, Kadambi P, Meehan S, Foster P, Harland R, Thistlethwaite RJ, Garfinkel M, Atwood W, Jordan J, Sadhu M, Millis MJ, Williams J. Treatment of renal allograft polyoma BK virus infection with leflunomide. Transplantation. 2006a;81:704–710. doi: 10.1097/01.tp.0000181149.76113.50. [DOI] [PubMed] [Google Scholar]
- Josephson MA, Williams JW, Chandraker A, Randhawa PS. Polyomavirus-associated nephropathy: update on antiviral strategies. Transpl Infect Dis. 2006b;8:95–101. doi: 10.1111/j.1399-3062.2006.00150.x. [DOI] [PubMed] [Google Scholar]
- Katoh H, Ogawa H, Ohya K, Fukushi H. A review of DNA viral infections in psittacine birds. J Vet Med Sci. 2010;72:1099–1106. doi: 10.1292/jvms.10-0022. [DOI] [PubMed] [Google Scholar]
- Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley WL, Georgopoulos C. The T/t common exon of simian virus 40, JC, and BK polyomavirus T antigens can functionally replace the J-domain of the Escherichia coli DnaJ molecular chaperone. Proc Natl Acad Sci U S A. 1997;94:3679–3684. doi: 10.1073/pnas.94.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS. Treatment of Paragonimus westermani infections with bithionol. Am J Trop Med Hyg. 1970;19:940–942. doi: 10.4269/ajtmh.1970.19.940. [DOI] [PubMed] [Google Scholar]
- Kimbrough RD. Review of the toxicity of hexachlorophene, including its neurotoxicity. J Clin Pharmacol. 1973;13:439–444. doi: 10.1002/j.1552-4604.1973.tb00196.x. [DOI] [PubMed] [Google Scholar]
- Kunisaki KM, Janoff EN. Influenza in immunosuppressed populations: a review of infection frequency, morbidity, mortality, and vaccine responses. Lancet Infect Dis. 2009;9:493–504. doi: 10.1016/S1473-3099(09)70175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee OJ, Kim TH. Indirect evidence of ectopic pancreatic fascioliasis in a human. J Gastroenterol Hepatol. 2006;21:1631–1633. doi: 10.1111/j.1440-1746.2006.03185.x. [DOI] [PubMed] [Google Scholar]
- Leung AY, Chan MT, Yuen KY, Cheng VC, Chan KH, Wong CL, Liang R, Lie AK, Kwong YL. Ciprofloxacin decreased polyoma BK virus load in patients who underwent allogeneic hematopoietic stem cell transplantation. Clin Infect Dis. 2005;40:528–537. doi: 10.1086/427291. [DOI] [PubMed] [Google Scholar]
- Li JJ, Kelly TJ. Simian virus 40 DNA replication in vitro. Proc Natl Acad Sci U S A. 1984;81:6973–6977. doi: 10.1073/pnas.81.22.6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Smith CJ, Walker MT, Smith TJ. Novel inhibitors complexed with glutamate dehydrogenase: allosteric regulation by control of protein dynamics. J Biol Chem. 2009;284:22988–23000. doi: 10.1074/jbc.M109.020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Minguez JM, Kim SY, Giuliano KA, Balachandran R, Madiraju C, Day BW, Curran DP. Synthesis and biological assessment of simplified analogues of the potent microtubule stabilizer (+)-discodermolide. Bioorg Med Chem. 2003;11:3335–3357. doi: 10.1016/s0968-0896(03)00186-x. [DOI] [PubMed] [Google Scholar]
- Miyata Y, Chang L, Bainor A, McQuade TJ, Walczak CP, Zhang Y, Larsen MJ, Kirchhoff P, Gestwicki JE. High-throughput screen for Escherichia coli heat shock protein 70 (Hsp70/DnaK): ATPase assay in low volume by exploiting energy transfer. J Biomol Screen. 2010;15:1211–1219. doi: 10.1177/1087057110380571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata H, Peden K, Lewis AM., Jr. Identification of a mutation in the SV40 capsid protein VP1 that influences plaque morphology, vacuolization, and receptor usage. Virology. 2008;370:343–351. doi: 10.1016/j.virol.2007.08.040. [DOI] [PubMed] [Google Scholar]
- Pipas JM. Common and unique features of T antigens encoded by the polyomavirus group. J Virol. 1992;66:3979–3985. doi: 10.1128/jvi.66.7.3979-3985.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipas JM. SV40: Cell transformation and tumorigenesis. Virology. 2009;384:294–303. doi: 10.1016/j.virol.2008.11.024. [DOI] [PubMed] [Google Scholar]
- Pipas JM, Peden KW, Nathans D. Mutational analysis of simian virus 40 T antigen: isolation and characterization of mutants with deletions in the T-antigen gene. Mol Cell Biol. 1983;3:203–213. doi: 10.1128/mcb.3.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell HC, Lampert PW. Bithionol: a possible substitute for hexachlorophene. Pediatrics. 1973;52:859–861. [PubMed] [Google Scholar]
- Price TA, Tuazon CU, Simon GL. Fascioliasis: case reports and review. Clin Infect Dis. 1993;17:426–430. doi: 10.1093/clinids/17.3.426. [DOI] [PubMed] [Google Scholar]
- Randhawa P, Shapiro R, Vats A. Quantitation of DNA of polyomaviruses BK and JC in human kidneys. J Infect Dis. 2005a;192:504–509. doi: 10.1086/431522. [DOI] [PubMed] [Google Scholar]
- Randhawa P, Vats A, Shapiro R. Monitoring for polyomavirus BK And JC in urine: comparison of quantitative polymerase chain reaction with urine cytology. Transplantation. 2005b;79:984–986. doi: 10.1097/01.tp.0000157573.90090.fd. [DOI] [PubMed] [Google Scholar]
- Randhawa PS. Anti-BK virus activity of ciprofloxacin and related antibiotics. Clin Infect Dis. 2005;41:1366–1367. doi: 10.1086/497080. [DOI] [PubMed] [Google Scholar]
- Rigby PW, Berg P. Does simian virus 40 DNA integrate into cellular DNA during productive infection? J Virol. 1978;28:475–489. doi: 10.1128/jvi.28.2.475-489.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablina AA, Hahn WC. SV40 small T antigen and PP2A phosphatase in cell transformation. Cancer Metastasis Rev. 2008;27:137–146. doi: 10.1007/s10555-008-9116-0. [DOI] [PubMed] [Google Scholar]
- Safak M, Khalili K. An overview: Human polyomavirus JC virus and its associated disorders. J Neurovirol. 2003;9(Suppl 1):3–9. doi: 10.1080/13550280390195360. [DOI] [PubMed] [Google Scholar]
- Schowalter RM, Pastrana DV, Pumphrey KA, Moyer AL, Buck CB. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe. 2010;7:509–515. doi: 10.1016/j.chom.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuda N, Hofmann J, Calvignac-Spencer S, Ruprecht K, Liman P, Kuhn J, Hengel H, Ehlers B. A novel human polyomavirus closely related to the African green monkey-derived lymphotropic polyomavirus (LPV) J Virol. 2011 doi: 10.1128/JVI.02602-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segel IH. Biochemical calculations : how to solve mathematical problems in general biochemistry. 2d ed. Wiley; New York: 1976. [Google Scholar]
- Seguin SP, Evans CW, Nebane-Akah M, McKellip S, Ananthan S, Tower NA, Sosa M, Rasmussen L, White EL, Maki BE, Matharu DS, Golden JE, Aube J, Brodsky JL, Noah JW. High-throughput screening identifies a bisphenol inhibitor of SV40 large T antigen ATPase activity. Journal of biomolecular screening. 2012;17:194–203. doi: 10.1177/1087057111421630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo BS, Lee SH, Hong ST, Hong SJ, Kim CY, Lee HY. Studies On Intestinal Trematodes In Korea: V. A Human Case Infected By Fibricola Seoulensis (Trematoda: Diplostomatidae) Korean Journal of Parasitology. 1982;20:93–99. doi: 10.3347/kjp.1982.20.2.93. [DOI] [PubMed] [Google Scholar]
- Sharp CP, Norja P, Anthony I, Bell JE, Simmonds P. Reactivation and Mutation of Newly Discovered WU, KI, and Merkel Cell Carcinoma Polyomaviruses in Immunosuppressed Individuals. J Infect Dis. 2009;199:398–404. doi: 10.1086/596062. [DOI] [PubMed] [Google Scholar]
- Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, Chang Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci U S A. 2008;105:16272–16277. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan A, McClellan AJ, Vartikar J, Marks I, Cantalupo P, Li Y, Whyte P, Rundell K, Brodsky JL, Pipas JM. The amino-terminal transforming region of simian virus 40 large T and small t antigens functions as a J domain. Mol Cell Biol. 1997;17:4761–4773. doi: 10.1128/mcb.17.8.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Cantalupo P, Pipas JM. The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol Cell Biol. 2000;20:6233–6243. doi: 10.1128/mcb.20.17.6233-6243.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meijden E, Janssens RW, Lauber C, Bouwes Bavinck JN, Gorbalenya AE, Feltkamp MC. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog. 2010;6:e1001024. doi: 10.1371/journal.ppat.1001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisshart K, Bradley MK, Weiner BM, Schneider C, Moarefi I, Fanning E, Arthur AK. An N-terminal deletion mutant of simian virus 40 (SV40) large T antigen oligomerizes incorrectly on SV40 DNA but retains the ability to bind to DNA polymerase alpha and replicate SV40 DNA in vitro. J Virol. 1996;70:3509–3516. doi: 10.1128/jvi.70.6.3509-3516.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisshart K, Friedl S, Taneja P, Nasheuer HP, Schlott B, Grosse F, Fanning E. Partial proteolysis of simian virus 40 T antigen reveals intramolecular contacts between domains and conformation changes upon hexamer assembly. J Biol Chem. 2004;279:38943–38951. doi: 10.1074/jbc.M406159200. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe SR, Inglis KA, Urch JE, Muller S, van Aalten DM, Fairlamb AH. Kinetic, inhibition and structural studies on 3-oxoacyl-ACP reductase from Plasmodium falciparum, a key enzyme in fatty acid biosynthesis. Biochem J. 2006;393:447–457. doi: 10.1042/BJ20050832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisen S, Androsavich J, Evans CG, Chang L, Gestwicki JE. Chemical modulators of heat shock protein 70 (Hsp70) by sequential, microwave-accelerated reactions on solid phase. Bioorg Med Chem Lett. 2008;18:60–65. doi: 10.1016/j.bmcl.2007.11.027. [DOI] [PubMed] [Google Scholar]
- Wright CM, Chovatiya RJ, Jameson NE, Turner DM, Zhu G, Werner S, Huryn DM, Pipas JM, Day BW, Wipf P, Brodsky JL. Pyrimidinone-peptoid hybrid molecules with distinct effects on molecular chaperone function and cell proliferation. Bioorg Med Chem. 2008;16:3291–3301. doi: 10.1016/j.bmc.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CM, Seguin SP, Fewell SW, Zhang H, Ishwad C, Vats A, Lingwood CA, Wipf P, Fanning E, Pipas JM, Brodsky JL. Inhibition of Simian Virus 40 replication by targeting the molecular chaperone function and ATPase activity of T antigen. Virus research. 2009;141:71–80. doi: 10.1016/j.virusres.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.