

Abstract
The serologic hallmark of primary biliary cirrhosis (PBC) is the presence of antimitochondrial autoantibodies (AMA) directed against the E2 subunit of PDC-E2. The PBC-related autoepitope of PDC-E2 contains lipoic acid, and previous work has demonstrated that mimics of lipoic acid following immunization of mice lead to a PBC-like disease. Furthermore, approximately one third of patients who have ingested excessive amounts of acetaminophen (paracetamol) develop AMA of the same specificity as patients with PBC. Quantitative structure-activity relationship (QSAR) data indicates that acetaminophen metabolites are particularly immunoreactive with AMA, and we submit that in genetically susceptible hosts, electrophilic modification of lipoic acid in PDC-E2 by acetaminophen or similar drugs can facilitate a loss of tolerance and lead to the development of PBC.
The Natural History and Genetics of Primary Biliary Cirrhosis
Primary biliary cirrhosis (PBC) is a middle-age onset, liver-specific autoimmune disease that has an average incidence of 2.7 cases per 100,000 in a well defined US population [1], but epidemiological studies suggest that the incidence of PBC is increasing [2]. There is variation in the rates of disease between geographic locations [2, 3], and PBC is more prevalent in Northern Europe and North America and less common in Eastern Asia, Africa, and Australia [4, 5]. It predominantly affects women with a female:male patient ratio of 9:1 [6].
PBC is characterized by the presence of high titer antimitochondrial autoantibodies (AMA) and the immune-mediated progressive destruction of biliary epithelial cells (BEC) of small bile ducts, eventually leading to cholestasis, fibrosis, and, potentially, liver cirrhosis [6]. Approximately 50–60% of patients are asymptomatic at diagnosis. The disease has a long latency period [7, 8], followed by the development of symptoms that usually include fatigue, pruritus hyperpigmentation of the skin, and, later, bleeding varices, edema, or ascites [9]. The prognosis of patients diagnosed with PBC has improved significantly over the past two decades, perhaps because patients are being diagnosed earlier.
The female predominance among individuals with PBC suggests that there are significant genetic components in this disease, supported by the high frequency of X chromosome monosomy in patients with PBC [10]. Reports from recent genetic studies demonstrate that in addition to the HLA locus, several loci are associated with susceptibility to PBC: interleukin (IL) 12-related pathways, SPIB, IRF5-TNPO3, and 17q12-2. New candidate genes identified by genome wide association studies include STAT4, DENND1B, CD80, IL7R, CXCR5, TNFRSF1A, CLEC16A, and NFKB1 [11–14]. Data on the familial clustering of PBC demonstrates that first-degree relatives of PBC patients have an increased risk of developing disease and most often these familial clusters involve mother-daughter pairs, consistent with the female preponderance of the disease [15, 16]. Furthermore, twin studies have demonstrated a high concordance for PBC in monozygotic twins and a low concordance among dizygotic twins [17]. These clusters provide evidence for a genetic basis underlying PBC. In addition, clusters of unrelated individuals suggest that environmental factors also play a role in the development of the disease [18], and environmental components including chemicals [19–21] and bacteria [21–23] have been implicated in initiating the disease.
Immunological Features
AMAs are present in over 95% of patients with PBC and are the most specific diagnostic antibody marker of PBC [24, 25]. The autoantigens of AMA have been identified as the E2 subunits of the 2-oxo-acid dehydrogenase complexes (2OADC-E2), including the E2 subunits of the pyruvate dehydrogenase complex (PDC-E2), branched chain 2-oxo acid dehydrogenase complex (BCOADC-E2), and 2-oxo-glutarate dehydrogenase complex (OGDC-E2) [26–30]. The AMA target antigens are all localized to the inner mitochondrial matrix and catalyze the oxidative decarboxylation of 2-oxo-acid acid substrates [31]. The E2 enzymes have a common structure consisting of a single N-terminal catalytic domain that contains at least one binding site (two in the case of mammalian PDC-E2) for the covalently attached lipoic acid cofactor. Previous studies have demonstrated that the epitopes most often recognized by AMA are all within the lipoyl binding domains of these target antigens [32]. Notably, AMA can be detected in individuals long before the manifestation of liver pathology and remain at high titer in patients with PBC even after liver transplantation [7, 8].
In patients with PBC, T helper (CD4+TCR αβ+) and CD8+ T cells are present in portal tracts around damaged bile ducts, strongly suggesting the participation of cellular immune mechanisms in biliary damage [6]. PDC-E2 autoreactive CD4 T cells are present in peripheral blood and liver [33], and there is a specific 100- to 150-fold increase in the number of PDC-E2-specific CD4 T cells in the hilar lymph nodes and liver versus peripheral blood in patients with PBC [34]. The PDC-E2 autoepitope for both CD4 and HLA class I-restricted CD8 T cells, overlaps with the B cell epitope, which spans the lipoyl domain [35]. Similar to CD4 autoreactive T cells, there is a 10-fold higher frequency of CD8 T cells specific for PDC-E2 within the liver versus peripheral blood. Moreover, the precursor frequency of PDC-E2-specific autoreactive CD8 T cells is significantly higher in the early, rather than the late stage, of the disease [35]. Recent reports substantiate the significance of innate immunity, including monocytes, toll-like receptors, and natural killer cells, in the development of PBC [36–39]. The multi-lineage response to the immunodominant mitochondrial autoantigen PDC-E2 in PBC suggest that a loss of tolerance to PDC-E2 is the initiating event in the development of clinical biliary pathology [6].
Susceptibility of the Lipoyl Domain to Chemical Modification
The uniqueness of AMA epitope specificity to the lipoyl domains of the 2OADC-E2 enzymes in patients with PBC suggests that the lipoic acid domain is likely a lynchpin to the etiology of PBC. In particular, the immune reactivity of AMA is directed against a conformational epitope that includes a highly conserved lysine-lipoyl conjugate and the signature amino acid motifs Glu-Thr-Asp-Lys-Ala, Glu-Thr-Asp-Lys-(Thr), and (Gln-Ser)-Asp-Lys-Ala in PDC-E2, OGDC-E2, and BCOADC-E2, respectively [27–29]. High-resolution structural analysis and modeling studies of the PDC-E2 lipoyl domains from both prokaryotes and eukaryotes demonstrates that lipoic acid is covalently attached to the ε group of lysine (K) via an amide bond and is prominently displayed on the outer surface of PDC-E2. More importantly, the ability of lipoic acid to rotate by means of its “swinging arms” with respect to the bulk of the PDC-E2 molecule allows access to its dithiolane ring for reduction acylation [40, 41]. Although the change in conformation and the existence of multiple conformations of the lipoyl domain during reductive acetylation is important in catalyzing acyl transfer [40], it also renders PDC-E2 susceptible to aberrant chemical modification.
The strikingly high titer of AMA with epitope specificity located within the lipoyl domains of 2OADC-E2 enzymes, the required conformational changes of the lipoyl domain during acyl transfer, and the susceptibility of these domains to chemical modification have led us to postulate that chemical modification of the lipoyl domain of PDC-E2 by xenobiotics is sufficient to break self-tolerance. Indeed, using quantitative structure-activity relationship (QSAR) analysis on a microarray platform, we have demonstrated that when the lipoyl domain of PDC-E2 is modified with specific synthetic small molecule lipoyl mimics, the ensuing structures are very specifically bound by antibodies in PBC sera, often at levels higher than the native PDC-E2 molecule [20, 42, 43]. These mimics include compounds that are widely used in the environment including perfumes, lipstick, and many common food flavorings [20, 43]. Furthermore, animals immunized with selected AMA-positive xenobiotics produce AMA and display liver pathology similar to PBC [44–46].
QSAR analysis on a focused panel of lipoic acid mimics in which the lipoyl di-S bond are modified provided evidence suggesting that direct alteration of the lipoyl ring – that is, disruption of the S-S linkage – renders the lipoic acid “activated” and receptive for xenobiotic modification and subsequent AMA recognition [42]. The compounds 6,8-bis(acetylthio)octanoic acid (SAc), 8-(acetylthio)octanoic acid (OASAc), and 6,8-bis(propionylthio)octanoic acid (SCOEt) had very high levels of reactivity against sera from AMA-positive PBC patients; levels that were even higher than the reactivity against lipoic-acid-conjugated PDC-E2 peptide (Figure 1). Structural analysis of these three compounds suggests that they are similar to the products of naturally occurring physiological reactions, and these thioester modified analogs can arise from acylation of reduced lipoic acid. That is, these compounds contain a reduced lipoic acid with the addition of one (OASAc) or two (SAc) acetyl groups or two propionyl groups (SCOEt). Interestingly, the oxidative states of lipoic acid affect the immunogenicity of PDC-E2 [47]. The lipoic-acid-conjugated PDC-E2 is more immunogenic in a reduced form [48], which corresponds to the ring-open form of lipoic acid. Altogether, these findings suggest that xenobiotic modification of the lipoyl S-S linkage during lipoyl electron transfer could break tolerance to PDC-E2 and elicit AMA.
Figure 1.
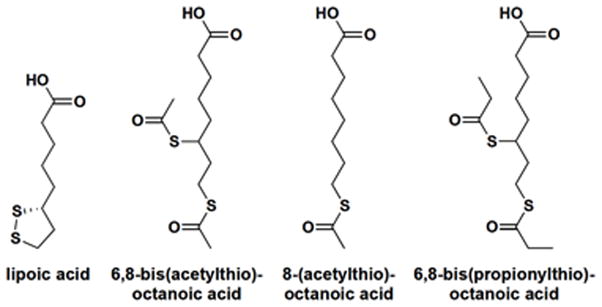
Molecular structures of lipoic acid, 6,8-bis(acetylthio)octanoic acid (SAc), 8-(acetylthio)octanoic acid (OASAc), and 6,8-bis(propionylthio)octanoic acid (SCOEt). Note that these compounds contain a reduced lipoic acid to which one (OASAc) or two (SAc) acetyl groups or two propionyl groups (SCOEt) have been added.
AMAs are Present in Sera of Patients with Acetaminophen-Induced Liver Injuries
In the United States, most liver injury is the consequence of drug overdose. The liver is at risk for drug-induced injury because many environmental chemicals are metabolized primarily by hepatocytes [49]. The pathogenesis of most drug-induced liver injuries is initiated by the metabolic conversion of xenobiotics into reactive intermediate species, such as electrophilic compounds or free radicals, that can potentially alter the structure and function of cellular macromolecules to form neo-antigens. Furthermore, reactive intermediate species could lead to oxidative stress, deregulation of cell signaling pathways, the dysfunction of biomolecules, organelle malfunction, and eventual cell death [50]. Although it is not clear how xenobiotics or the modified cellular proteins initiate autoimmunity in PBC, analysis of serum samples from subjects with acute liver failure indicate that a severe liver oxidant injury can lead to AMA production [51]. Characterization of 217 serum samples from 69 patients with acute liver failure (ALF) collected up to 24 months post-ALF were compared with controls for titer and reactivity with 2OADC-E2. AMAs were detected in 28/69 (40.6%) ALF patients and reactivity was found against all of the major mitochondrial autoantigens. The strikingly high frequency of AMAs in patients with ALF supports the thesis that oxidative stress-induced liver damage may lead to AMA production. In particular, we note that AMA with the same antigen and epitope specificity as in patients with PBC was found in almost 35% of individuals poisoned by ingesting excessive amounts of acetyl-para-aminophenol (commonly known as acetaminophen or paracetamol and abbreviated here as APAP), suggesting that the PDC-E2 lipoyl domain is likely a target of APAP-induced reactive oxygen species. Similar observations have been reported by Bernal et al. in a cohort of ALF patients in the United Kingdom [52], suggesting that autoantibodies can be generated in ALF in which self-proteins are released upon liver damage. This finding is significant because toxic doses of APAP produces reactive oxygen species, nitrogen species, and metabolites [53, 54] that could result in mitochondrial damage and liver injury, as evidenced by the elevation of serum alanine amino transferase and P450 dependent centrilobular damage [55, 56].
On the other hand, we also note that the production of AMAs do not persist in ALF overdose individuals. The intervention of immune regulatory mechanisms such as B regulatory cells, T regulatory cells, and miRNAs [57, 58] after the initial AMAs production may modulate the production of AMAs. Similar to the multi-hit hypothesis postulated in the pathogenesis of alcohol- and obesity-induced fatty liver diseases [59], it is likely that the initial triggering of AMAs by APAP is only a priming step. In PBC susceptible individuals, the initial priming by electrophilic drugs such as APAP is necessary to provide the signals required for the perpetuation of AMAs and progression.
Mechanism of APAP Metabolism and Protein Modification
APAP is the most widely used non-prescription drug in the United States, and at the recommended therapeutic dosage of 1,000 mg per single dose (and up to 4,000 mg per day for adults) 85% of the ingested APAP is metabolized in the liver to non-toxic compounds via conjugation of the aromatic ring to sulfate or glucuronic acid. The remaining 15% is converted into a highly-electrophilic metabolite, N-acetyl-p-benzoquinoneimine (NAPQI), by isozymes of microsomal cytochrome P450 [60]. In the presence of the reduced form of glutathione (GSH), NAPQI can either be covalently linked to GSH via Michael’s addition to the aromatic ring or reduced back to APAP [61]. The predominant method of NAPQI detoxification occurs through the former mechanism, leading to depletion of the intracellular glutathione pool [62]. However, in the presence of excess APAP, or when levels of microsomal P450 increase, hepatic GSH is more extensively depleted and cannot efficiently neutralize the increased levels of NAPQI. The reactive NAPQI that accumulates in the absence of GSH can then react with nucleophilic sites such as cysteine and lysine residues on cellular proteins and related cofactors [63].
Previous data [31] suggested that glutathiolation would decrease the antigenicity of PDC-E2, but because of cellular glutathione depletion very little glutathione would be available for such covalent protection of PDC-E2. Glutathione depletion could lead to the production of possible neo-antigens through modification of native PDC-E2 by high levels of reactive NAPQI or other electrophilic agents. We reason that in PBC such electrophilic modification on lipoic-acid-conjugated PDC-E2 will inhibit the physiological function of PDC-E2 and subsequently lead to disruption of ATP synthesis, cell death, and the release of either unmasked PDC-E2 or neoantigens formed by xenobiotic-modified PDC-E2. The exposure of this chemically modified self-protein to the immune system of genetically susceptible individuals could lead to the breakdown of self-tolerance to native PDC-E2 by molecular mimicry and epitope spreading (Figure 2). Thus, in genetically susceptible individuals, prolonged exposure to electrophilic agents such as acetaminophen may initiate and/or enhance the breakdown of self-tolerance to PDC-E2 and eventually lead to PBC.
Figure 2.

Acetyl-para-aminophenol (acetaminophen/paracetamol; APAP) metabolism and proposed mechanism of APAP-mediated breaking of immune tolerance.
APAP is metabolized in the liver to nontoxic compounds via conjugation of the aromatic ring to sulfate or glucuronic acid. Approximately 15% is converted into a highly electrophilic metabolite, N-acetyl-p-benzoquinoneimine (NAPQI) by microsomal cytochrome P450 oxidation. NAPQI can either be covalently linked to reduced glutathione (GSH) or reduced back to APAP. In the presence of excess APAP, or when microsomal P450 is increased, hepatic GSH is depleted to a greater extent and is insufficient to neutralize NAPQI. Consequently, reactive NAPQI accumulates and can form adducts with cellular proteins, leading to disruption of cellular functions, generation of neo-antigens, and loss of tolerance.
Significance of AMA in the Pathophysiology of bile duct epithelial cells in PBC
Despite the fact that mitochondrial targets are proteins ubiquitously expressed in all nucleated cells, the immunopathology of PBC is characterized by the presence of CD4+ and CD8+ T cell infiltrates in liver and the specific destruction of small bile duct epithelial cells (BEC) [6]. This apparent paradox suggests there are unique immunopathological characteristics of BEC, the target tissue. Previous work suggested that AMA preferentially recognize PDC-E2 with reduced sulfhydryl groups, and hence blocking these sites would likely eliminate the epitope site responsible for generating autoantibodies [48]. This concept was further supported by studies characterizing the autoantibody recognition site and the effects of apoptosis on the immunogenicity of PDC-E2 [47]. The authors demonstrated that after apoptosis HeLa, Jurkat T, and Caco-2 cells presented a form of PDC-E2 that is undetectable by AMA. This loss of recognition of apoptotic cell-derived PDC-E2 by AMA cannot be attributed to disappearance or degradation of the enzyme, but rather to a reversible structural change in the protein. By contrast, AMA recognition of PDC-E2 could be detected in samples of apoptotic BEC. This immunoreactivity can be eliminated by the addition of oxidized glutathione to sodium dodecyl sulfate – treated cholangiocyte cell lysates, which renders PDC-E2 nonantigenic when probed with AMA. Conversely, HeLa cells pretreated with buthionine sulfoximine (a glutathione synthetase inhibitor) before apoptotic stimulus were reactive with patient sera, demonstrating that the antigen could be “resurrected” by depleting glutathione. The authors concluded that the persistent recognition of PDC-E2 derived from cholangiocytes is caused by a failure in these cells to covalently link PDC-E2 to glutathione during the course of apoptosis, suggesting that the depletion of GSH by NAPQI may be important for PDC-E2 immunogenicity in the context of APAP toxicity (Figure 3).
Figure 3.

Role of N-acetyl-p-benzoquinoneimine (NAPQI) in primary biliary cirrhosis (PBC). APAP (acetyl-para-aminophenol) is converted to NAPQI by P450 in the liver. Under conditions of glutathione depletion and bile duct epithelial cells (BEC) apoptosis, the native PDC-E2 remains immunologically intact and becomes accessible to modification by electrophilic drugs, such as NAPQI, thereby generating neoantigens. Through crossreactivity and antigen spreading tolerance is broken, subsequently leading to BEC damage.
Apoptosis of BEC has been proposed as a potential source of neo-antigens responsible for activating autoreactive lymphocytes [64]. It is important to note that when BECs, but not control cells, undergo apoptosis, PDC-E2 remains immunologically intact [47, 65]. More importantly, when monocyte-derived macrophages from PBC patients were incubated with apoptotic bodies from BECs in the presence of AMAs, there was intense proinflammatory cytokine production, and this production was inhibited by anti-CD16, an antibody against the Fc receptor [66]. In light of the fact that AMA can be induced in patients with APAP overdose, these recent findings on apoptotic BECs further emphasize a unique role of AMAs in the etiopathogenesis of PBC.
Conclusions and Future Directions
The identification of AMA reactive compounds such as6,8-bis(acetylthio)octanoic acid, 8-(acetylthio)octanoic acid, and 6,8-bis(propionylthio)octanoic acid, which structurally resemble lipoic acid with a disrupted disulfide bond, and the presence of AMA in acetaminophen overdose patients argue strongly that electrophilic drugs could modify the lipoic acid domain of PDC-E2 and break tolerance in genetically susceptible hosts. We are cognizant that the breaking of tolerance is crucial in the initiation of PBC. Future understanding of the biochemical processes involved in an electrophilic attack of PDC-E2 and the identification of such adducts will provide clues to the molecular mechanisms in the development of PBC. Finally, neoantigen generation and breaking of tolerance through modification of cellular proteins by electrophilic drugs may not be confined to PBC and has implication for other autoimmune diseases.
Acknowledgments
This work is supported in part by National Institutes of Health grants DK39588 and DK067003. The authors would like to thank Mr. Thomas Kenny and Ms. Nikki Phipps for assistance in preparation of the manuscript.
Abbreviations
- AMA
antimitochondrial antibodies
- APAP
acetaminophen
- PDC-E2
pyruvate dehydrogenase complex
- BCOADC-E2
branched chain 2-oxo acid dehydrogenase complex
- OGDC-E2
2-oxo-glutarate dehydrogenase complex
- PBC
primary biliary cirrhosis
- BEC
biliary epithelial cells
- QSAR
quantitative structure-activity relationship
- SAc
6,8-bis(acetylthio)octanoic acid
- OASAc
8-(acetylthio)octanoic acid
- SCOEt
6,8-bis(propionylthio)octanoic acid
- NAPQI
N-acetyl-p-benzoquinoneimine
- GSH
glutathione
- ALF
acute liver failure
Glossary
- Ascites
accumulation of fluid in the peritoneal cavity, commonly due to cirrhosis and severe liver disease
- Bleeding varices
Engorged veins are called varices; they are fragile and can bleed easily because veins are not designed to handle high internal pressures
- Cholangiocytes
epithelial cells of the bile duct
- Epitope spreading
the shifting of an immune reaction from its primary antigenic binding site to other targets. The new targets need not to be structurally similar to the primary target
- Hyper pigmentation of the skin
darkening in the natural color of the skin
- Molecular mimicry
a theoretical mechanism by which the immune system attacks self-antigens that are structurally similar to foreign antigens; it is due to the cross-activation of immunological response
- Portal tracts
collections of branches of the portal vein, hepatic artery, and the biliary ducts bound together in perivascular connective tissues throughout the liver
- Pruritus
an unpleasant sensation that elicits the desire to scratch the skin
- Quantitative structure-activity relationship (QSAR)
A regression model commonly used in biological sciences and chemistry to predict the properties of chemicals and its biological activity
- Xenobiotics
chemical substances foreign to an organism because they are not normally produced or expected to be present in the organism’s system
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kim WR, et al. Epidemiology and natural history of primary biliary cirrhosis in a US community. Gastroenterology. 2000;119:1631–1636. doi: 10.1053/gast.2000.20197. [DOI] [PubMed] [Google Scholar]
- 2.Myers RP, et al. Epidemiology and natural history of primary biliary cirrhosis in a Canadian health region: a population-based study. Hepatology. 2009;50:1884–1892. doi: 10.1002/hep.23210. [DOI] [PubMed] [Google Scholar]
- 3.Rautiainen H, et al. Prevalence and incidence of primary biliary cirrhosis are increasing in Finland. Scandinavian journal of gastroenterology. 2007;42:1347–1353. doi: 10.1080/00365520701396034. [DOI] [PubMed] [Google Scholar]
- 4.Farrell GC. Primary biliary cirrhosis in Asians: less common than in Europeans, but just as depressing. Journal of gastroenterology and hepatology. 2008;23:508–511. doi: 10.1111/j.1440-1746.2008.05379.x. [DOI] [PubMed] [Google Scholar]
- 5.Invernizzi P. Geoepidemiology of autoimmune liver diseases. Journal of autoimmunity. 2010;34:J300–306. doi: 10.1016/j.jaut.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Gershwin ME, Mackay IR. The causes of primary biliary cirrhosis: Convenient and inconvenient truths. Hepatology. 2008;47:737–745. doi: 10.1002/hep.22042. [DOI] [PubMed] [Google Scholar]
- 7.Benson GD, et al. Serial analysis of antimitochondrial antibody in patients with primary biliary cirrhosis. Clinical & developmental immunology. 2004;11:129–133. doi: 10.1080/10446670410001722113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayo MJ. Natural history of primary biliary cirrhosis. Clinics in liver disease. 2008;12:277–288. viii. doi: 10.1016/j.cld.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 9.Prince MI, et al. Asymptomatic primary biliary cirrhosis: clinical features, prognosis, and symptom progression in a large population based cohort. Gut. 2004;53:865–870. doi: 10.1136/gut.2003.023937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Invernizzi P, et al. Frequency of monosomy X in women with primary biliary cirrhosis. Lancet. 2004;363:533–535. doi: 10.1016/S0140-6736(04)15541-4. [DOI] [PubMed] [Google Scholar]
- 11.Liu X, et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nature genetics. 2010;42:658–660. doi: 10.1038/ng.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mells GF, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nature genetics. 2011;43:329–332. doi: 10.1038/ng.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirschfield GM, et al. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cirrhosis. Nature genetics. 2010;42:655–657. doi: 10.1038/ng.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirschfield GM, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. The New England journal of medicine. 2009;360:2544–2555. doi: 10.1056/NEJMoa0810440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bach N, Schaffner F. Familial primary biliary cirrhosis. Journal of hepatology. 1994;20:698–701. doi: 10.1016/s0168-8278(05)80137-0. [DOI] [PubMed] [Google Scholar]
- 16.Lazaridis KN, et al. Increased prevalence of antimitochondrial antibodies in first-degree relatives of patients with primary biliary cirrhosis. Hepatology. 2007;46:785–792. doi: 10.1002/hep.21749. [DOI] [PubMed] [Google Scholar]
- 17.Selmi C, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology. 2004;127:485–492. doi: 10.1053/j.gastro.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 18.Smyk D, et al. Primary biliary cirrhosis: family stories. Autoimmune diseases. 2011;2011:189585. doi: 10.4061/2011/189585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ala A, et al. Increased prevalence of primary biliary cirrhosis near Superfund toxic waste sites. Hepatology. 2006;43:525–531. doi: 10.1002/hep.21076. [DOI] [PubMed] [Google Scholar]
- 20.Amano K, et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. Journal of immunology. 2005;174:5874–5883. doi: 10.4049/jimmunol.174.9.5874. [DOI] [PubMed] [Google Scholar]
- 21.Selmi C, et al. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium. Hepatology. 2003;38:1250–1257. doi: 10.1053/jhep.2003.50446. [DOI] [PubMed] [Google Scholar]
- 22.Leung PS, et al. Is there a relation between Chlamydia infection and primary biliary cirrhosis? Clinical & developmental immunology. 2003;10:227–233. doi: 10.1080/10446670310001642429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang Y, et al. Smoking, family history and urinary tract infection are associated with primary biliary cirrhosis: A meta-analysis. Hepatology research : the official journal of the Japan Society of Hepatology. 2011;41:572–578. doi: 10.1111/j.1872-034X.2011.00806.x. [DOI] [PubMed] [Google Scholar]
- 24.Oertelt S, et al. A sensitive bead assay for antimitochondrial antibodies: Chipping away at AMA-negative primary biliary cirrhosis. Hepatology. 2007;45:659–665. doi: 10.1002/hep.21583. [DOI] [PubMed] [Google Scholar]
- 25.Selmi C, et al. Primary biliary cirrhosis and Sjogren’s syndrome: Autoimmune epithelitis. Journal of autoimmunity. 2012;39:34–42. doi: 10.1016/j.jaut.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gershwin ME, et al. Identification and specificity of a cDNA encoding the 70 kd mitochondrial antigen recognized in primary biliary cirrhosis. Journal of immunology. 1987;138:3525–3531. [PubMed] [Google Scholar]
- 27.Leung PS, et al. Autoantibodies to BCOADC-E2 in patients with primary biliary cirrhosis recognize a conformational epitope. Hepatology. 1995;22:505–513. [PubMed] [Google Scholar]
- 28.Moteki S, et al. Epitope mapping and reactivity of autoantibodies to the E2 component of 2-oxoglutarate dehydrogenase complex in primary biliary cirrhosis using recombinant 2-oxoglutarate dehydrogenase complex. Hepatology. 1996;23:436–444. doi: 10.1002/hep.510230307. [DOI] [PubMed] [Google Scholar]
- 29.Van de Water J, et al. The autoepitope of the 74-kD mitochondrial autoantigen of primary biliary cirrhosis corresponds to the functional site of dihydrolipoamide acetyltransferase. The Journal of experimental medicine. 1988;167:1791–1799. doi: 10.1084/jem.167.6.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu H, et al. PBC screen: an IgG/IgA dual isotype ELISA detecting multiple mitochondrial and nuclear autoantibodies specific for primary biliary cirrhosis. Journal of autoimmunity. 2010;35:436–442. doi: 10.1016/j.jaut.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Mao TK, et al. Sidechain biology and the immunogenicity of PDC-E2, the major autoantigen of primary biliary cirrhosis. Hepatology. 2004;40:1241–1248. doi: 10.1002/hep.20491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moteki S, et al. Use of a designer triple expression hybrid clone for three different lipoyl domain for the detection of antimitochondrial autoantibodies. Hepatology. 1996;24:97–103. doi: 10.1002/hep.510240117. [DOI] [PubMed] [Google Scholar]
- 33.Zhang W, et al. T cell clonal expansions detected in patients with primary biliary cirrhosis express CX3CR1. Journal of autoimmunity. 2011;37:71–78. doi: 10.1016/j.jaut.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimoda S, et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. The Journal of clinical investigation. 1998;102:1831–1840. doi: 10.1172/JCI4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumura S, et al. Comprehensive mapping of HLA-A0201-restricted CD8 T-cell epitopes on PDC-E2 in primary biliary cirrhosis. Hepatology. 2002;36:1125–1134. doi: 10.1053/jhep.2002.36161. [DOI] [PubMed] [Google Scholar]
- 36.Mao TK, et al. Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology. 2005;42:802–808. doi: 10.1002/hep.20859. [DOI] [PubMed] [Google Scholar]
- 37.Moritoki Y, et al. AMA production in primary biliary cirrhosis is promoted by the TLR9 ligand CpG and suppressed by potassium channel blockers. Hepatology. 2007;45:314–322. doi: 10.1002/hep.21522. [DOI] [PubMed] [Google Scholar]
- 38.Shimoda S, et al. CD4 T-cell autoreactivity to the mitochondrial autoantigen PDC-E2 in AMA-negative primary biliary cirrhosis. Journal of autoimmunity. 2008;31:110–115. doi: 10.1016/j.jaut.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 39.You Z, et al. The immunopathology of liver granulomas in primary biliary cirrhosis. Journal of autoimmunity. 2012 doi: 10.1016/j.jaut.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones DD, et al. Restricted motion of the lipoyl-lysine swinging arm in the pyruvate dehydrogenase complex of Escherichia coli. Biochemistry. 2000;39:8448–8459. doi: 10.1021/bi992978i. [DOI] [PubMed] [Google Scholar]
- 41.Vijayakrishnan S, et al. Solution structure and characterisation of the human pyruvate dehydrogenase complex core assembly. Journal of molecular biology. 2010;399:71–93. doi: 10.1016/j.jmb.2010.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naiyanetr P, et al. Electrophile-modified lipoic derivatives of PDC-E2 elicits anti-mitochondrial antibody reactivity. Journal of autoimmunity. 2011;37:209–216. doi: 10.1016/j.jaut.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rieger R, et al. Identification of 2-nonynoic acid, a cosmetic component, as a potential trigger of primary biliary cirrhosis. Journal of autoimmunity. 2006;27:7–16. doi: 10.1016/j.jaut.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 44.Leung PS, et al. Induction of primary biliary cirrhosis in guinea pigs following chemical xenobiotic immunization. Journal of immunology. 2007;179:2651–2657. doi: 10.4049/jimmunol.179.4.2651. [DOI] [PubMed] [Google Scholar]
- 45.Leung PS, et al. Immunization with a xenobiotic 6-bromohexanoate bovine serum albumin conjugate induces antimitochondrial antibodies. Journal of immunology. 2003;170:5326–5332. doi: 10.4049/jimmunol.170.10.5326. [DOI] [PubMed] [Google Scholar]
- 46.Wakabayashi K, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology. 2008;48:531–540. doi: 10.1002/hep.22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Odin JA, et al. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. The Journal of clinical investigation. 2001;108:223–232. doi: 10.1172/JCI10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mendel-Hartvig I, et al. Primary biliary cirrhosis: further biochemical and immunological characterization of mitochondrial antigens. Clinical and experimental immunology. 1985;62:371–379. [PMC free article] [PubMed] [Google Scholar]
- 49.Sevior DK, et al. Hepatocytes: the powerhouse of biotransformation. The international journal of biochemistry & cell biology. 2012;44:257–261. doi: 10.1016/j.biocel.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 50.Gu X, Manautou JE. Molecular mechanisms underlying chemical liver injury. Expert reviews in molecular medicine. 2012;14:e4. doi: 10.1017/S1462399411002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leung PS, et al. Antimitochondrial antibodies in acute liver failure: implications for primary biliary cirrhosis. Hepatology. 2007;46:1436–1442. doi: 10.1002/hep.21828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernal W, et al. Disease-specific autoantibodies in patients with acute liver failure: the King’s College London Experience. Hepatology. 2008;47:1096–1097. doi: 10.1002/hep.22179. author reply 1097. [DOI] [PubMed] [Google Scholar]
- 53.Ferret PJ, et al. Detoxification of reactive oxygen species by a nonpeptidyl mimic of superoxide dismutase cures acetaminophen-induced acute liver failure in the mouse. Hepatology. 2001;33:1173–1180. doi: 10.1053/jhep.2001.24267. [DOI] [PubMed] [Google Scholar]
- 54.Lores Arnaiz S, et al. Oxidative stress by acute acetaminophen administration in mouse liver. Free radical biology & medicine. 1995;19:303–310. doi: 10.1016/0891-5849(95)00023-q. [DOI] [PubMed] [Google Scholar]
- 55.Hinson JA, et al. Acetaminophen-induced hepatotoxicity. Life sciences. 1981;29:107–116. doi: 10.1016/0024-3205(81)90278-2. [DOI] [PubMed] [Google Scholar]
- 56.Hinson JA, et al. Mechanism of paracetamol toxicity. Lancet. 1990;335:732. doi: 10.1016/0140-6736(90)90851-u. [DOI] [PubMed] [Google Scholar]
- 57.Bernuzzi F, et al. Phenotypical and functional alterations of CD8 regulatory T cells in primary biliary cirrhosis. Journal of autoimmunity. 2010;35:176–180. doi: 10.1016/j.jaut.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Padgett KA, et al. Primary biliary cirrhosis is associated with altered hepatic microRNA expression. Journal of autoimmunity. 2009;32:246–253. doi: 10.1016/j.jaut.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mantena SK, et al. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free radical biology & medicine. 2008;44:1259–1272. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harvison PJ, et al. Cytochrome P-450 isozyme selectivity in the oxidation of acetaminophen. Chemical research in toxicology. 1988;1:47–52. doi: 10.1021/tx00001a009. [DOI] [PubMed] [Google Scholar]
- 61.Moldeus P. Paracetamol metabolism and toxicity in isolated hepatocytes from rat and mouse. Biochemical pharmacology. 1978;27:2859–2863. doi: 10.1016/0006-2952(78)90201-0. [DOI] [PubMed] [Google Scholar]
- 62.David Josephy P. The molecular toxicology of acetaminophen. Drug metabolism reviews. 2005;37:581–594. doi: 10.1080/03602530500205200. [DOI] [PubMed] [Google Scholar]
- 63.Larson AM. Acetaminophen hepatotoxicity. Clinics in liver disease. 2007;11:525–548. vi. doi: 10.1016/j.cld.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 64.Lleo A, et al. Etiopathogenesis of primary biliary cirrhosis. World journal of gastroenterology : WJG. 2008;14:3328–3337. doi: 10.3748/wjg.14.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lleo A, et al. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology. 2009;49:871–879. doi: 10.1002/hep.22736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lleo A, et al. Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology. 2010;52:987–998. doi: 10.1002/hep.23783. [DOI] [PMC free article] [PubMed] [Google Scholar]