

The indole 3-position is highly electron-rich and typically functions as the primary nucleophilic site to react with a large array of electrophiles, leading to various functionalized indoles.[1] The reversal of this prime reactivity, i.e., making the indole 3-position electrophilic, would be of significant synthetic utility and provide a complementary strategy to access derivatives[2] otherwise difficult to prepare conventionally. This reactivity umpolung[3] of indole has, however, only been realized in limited cases.[4]
For the past few years we have engaged in extensive studies of gold-catalyzed intra-[5] and intermolecular[6] alkyne oxidations using oxygen-delivering oxidants,[7] where reactive α-oxo gold carbene intermediates are presumably generated[8] and responsible for the diverse reaction outcomes. Lately we extended this strategy to the use of nitrene precursors as oxidants, providing access to reactive α-imino gold carbenes (Scheme 1A);[9] however, the chemistry has so far been limited to ynamides,[10] which are activated alkynes. In our effort to expand the scope of this type of gold-catalyzed nitrene transfer,[11] we decided to use an azido group as a nitrene precursor, which was inspired by previous studies of gold-[12] and platinum-catalyzed[13] pyrrole synthesis. We reasoned that closely and rigidly positioned C-C triple bonds in ortho-azidoarylalkynes might facilitate an intramolecular nitrene transfer from the azido group to the C-C triple bond. Importantly, the thus-formed gold carbene B would serve as an electrophilic indole equivalent, as depicted in its resonance form C, therefore realizing reactivity umpolung of the indole 3-position (Scheme 1B).[14]
Scheme 1.

Gold-catalyzed nitrene transfer: realizing reactivity umpolung at the indole 3-position.
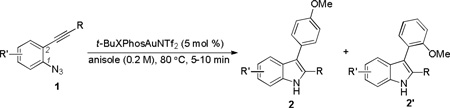
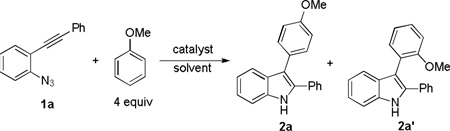
We started by using ortho-azidophenylalkyne 1a as the substrate and anisole as the nucleophile, and the initial reaction was run in toluene using Ph3PAuNTf2[15] as the catalyst. To our delight, the desired indole regioisomers 2a and 2a’ were indeed formed (entry 1), confirming that the azido group could function as a nitrene precursor and a gold carbene of type B might be indeed formed; moreover, this proposed reactive intermediate seemingly reacted mainly via its cationic resonance form C[16] as no Büchner reaction,[17] i.e., the formation of cycloheptatriene products, which is characteristic of carbene chemistry, occurred. The regioselectivity on the anisole ring is consistent with an electrophilic aromatic substitution mechanism. To our surprise, the majority of the putative gold intermediate B/C reacted with the solvent toluene, yielding a mixture of regioisomers (p-3/o-3/m-3 = 41%/11%/8%). Although the concentration of toluene is ~190 time of that of anisole, anisole is much more nucleophilic than toluene.[18] These results indicate that intermediate C is strongly electrophilic and hence less selective. This conclusion is consistent with the ratio of p-3 vs. m-3 (~5), lower than that in the case of nitration (>10),[19] suggesting that C might be even more electrophilic than NO2+. Since products of type 2a, 2a’, and 3 are also good nucleophiles, we anticipated that it is essential to use excess intended nucleophiles in order to minimizing their competing reactions with highly electrophilic C.
Other solvents were screened in order to minimize solvent participation (entries 2–4). While benzene also interfered the desired reaction (entry 2), neither DCE (entry 3) nor chlorobenzene (entry 4) did; a better reaction yield was realized in DCE. Examining different gold catalysts (entries 5–10) at a beneficiary higher reaction temperature (comparing entries 3 and 5) revealed that IPrAuNTf2 (entry 6) gave the best yield and bulky t-BuXPhosAuNTf2 gave the best para/ortho ratio (entry 8). We also run the reaction using anisole as the solvent at a higher concentration (0.2 M). Somewhat to our surprise, the reaction became dramatically faster and finished in 5 min at 80 °C; moreover, the yield was excellent. Perhaps even more surprising is that the para/ortho ratio decreased as the reaction temperature got lower (comparing entries 11–13). This may suggest the involvement of another reaction mechanism. As shown in Scheme 2, at a higher temperature (e.g., 80 °C), the formation of B/C should be facilitated, but at a lower temperature (e.g., −20 °C) its precursor, i.e. A, may persist and play an increasing role in the reaction by reacting with nucleophiles via an SN2’ process; since the Au-C bond length in B/C should be shorter than the Au-C bond in A, one might expect that the more the reaction goes through B/C the more regioselective as the bulky ligand (i.e., t- BuXPhos) can be more sterically imposing.[20]
Scheme 2.

Rationale for the inversed relationship between regioselectivities and reaction temperatures.
The scope of o-azidoarylalkynes was subsequently examined by first varying the alkyne substituent. Using anisole as the solvent, primary alkyl groups such as n-butyl (Table 2, entry 1) and phenethyl (Table 2, entry 2) reacted smoothly; a lower yield was obtained with 1d containing a benzyl ether (Table 2, entry 3); cyclic secondary alkyl groups (Table 2, entries 4–6) as well as a tert-butyl group (Table 2, entry 7) all led to good yields; interestingly, substrate 1i with a terminal alkyne also worked (Table 2, entry 8); aryl groups with either a p-MeO (Table 2, entry 10) or a p-methoxycarbonyl group (Table 2, entry 11) were readily tolerated, and the corresponding indoles were formed in good to excellent yields. In all the cases, the regioselectivities correlated well with the substituent steric size, and the best 2/2’ ratio was realized with the tert-butyl alkyne 1h. In addition, substrates with substituted benzene ring also reacted with good efficiencies, thus offering highly substituted indole products (entries 11 and 12).
Table 2.
The scope of o-azidoarylalkyne substrates.[a]
 | |||||
|---|---|---|---|---|---|
| entry | 1 (R) | R’ | 2 | 2/2’ | yield[b] |
| 1 | 1b (n-butyl) | H | 2b | 7/1 | 74% |
| 2 | 1c (PhCH2CH2) | H | 2c | 5/1 | 83% |
| 3 | 1d (BnOCH2) | H | 2d | 5/1 | 51%[c] |
| 4 | 1e (cyclopropyl) | H | 2e | 8/1 | 76% |
| 5 | 1f (cyclopentyl) | H | 2f | 15/1 | 75% |
| 6 | 1g (cyclohexyl) | H | 2g | 16/1 | 82% |
| 7 | 1h (t-butyl) | H | 2h | 25/1 | 75% |
| 8 | 1i (H) | H | 2i | 3/1 | 91% |
| 9 | 1j (p-MeOPh) | H | 2j | 7/1 | 76% |
| 10 | 1k (p-MeO2CPh) | H | 2k | 7/1 | 95% |
| 11 | 1l (n-butyl) | 4,6-Me2 | 2l | 8/1 | 82% |
| 12[d] | 1m (n-butyl) | 3,5-Cl2 | 2m | 7/1 | 78% |
Vial reaction.
The combined isolated yield of 2 and 2’.
About 4% of 7 was formed.
Reaction time: 1 h.
The applicability of this chemistry to other nucleophiles was then probed using the conditions in Table 1, Entry 6, and the successful examples are shown in Table 3. As expected, p-xylene, when used as solvent, served as a suitable nucleophile for this chemistry (entry 1). In the case of naphthalene, the α-position is preferred due to its stronger nucleophilicity (entry 2). More activated benzene rings (entries 3 and 4) gave good yields of desired products, and a good selectivity (10:1) was observed with 1,3-dimethoxybenzene, reflecting the more congested nature of the 2-position. In the case of N-methylpyrrole, apparently the electronic and the steric factors were working against each other; consequently, no regioselectivity was observed (entry 5); nevertheless, the overall efficiency was excellent. Increasing the steric hindrance at the pyrrole 2-position by installing a TIPS group on the ring nitrogen indeed made the reaction occurred selectively at the 3-position (entry 6). When N-benzylindole was used as the nucleophile, to our surprise, at least three inseparable regioisomers were formed, suggesting that its benzene ring participated in this reaction as the nucleophilic site. To our delight, with strongly electron-withdrawing nitro group on the indole benzene ring, the substitution selectively occurred on the 3-position, affording nonsymmetrical 3,3’-biindoles with good yields (entries 7 and 8). The use of the more polar azidoalkyne substrate 1k facilitated purification of the products. With the indole 2-position substituted and its benzene ring again deactivated, the electrophilic substitution proceeded expectedly at the 3-position to yield hindered biindole 5i. Notably, the 3,3-biindole structure has been found in natural products[21] and compounds of medical interest,[22] and their syntheses often require multiple steps.[23] Dimedone methyl ether could react as a nucleophile as well albeit accompanied with in-situ hydrolysis and in a relatively low reaction yield (entry 10). Besides carbon nucleophiles, alcohols could react with intermediates of type C (entries 11 and 12). The moderate yields were to some extent due to
 |
(1) |
 |
(2) |
the susceptibility of the products towards aerobic oxidation. Interestingly, with allyl alcohol as the nucleophile, the product underwent one-pot Claisen rearrangement, yielding indol-3-one 5m in a serviceable yield (entry 13). Notably, no desired product was observed when using N-methyltosylamide as the nucleophile.
Table 1.
Initial discovery and condition optimizations.[a]
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | solvent | conditions | yield[b] | 2a/2a’ |
| 1 | Ph3PAuNTf2 | toluene | 60 °C, 11 h | 20%[c] | 2.4 |
| 2 | Ph3PAuNTf2 | benzene | 60 °C, 11 h | 40%[d] | 2.4 |
| 3 | Ph3PAuNTf2 | DCE | 60 °C, 11 h | 52% | 1.9 |
| 4 | Ph3PAuNTf2 | PhCl | 60 °C, 11 h | 44% | 2.1 |
| 5 | Ph3PAuNTf2 | DCE | 80 °C, 11 h | 60% | 1.9 |
| 6 | IPrAuNTf2 | DCE | 80 °C, 11 h | 81% | 4.3 |
| 7 | Cy-JohnPhosAuNTf2 | DCE | 80 °C, 11 h | 60% | 2.7 |
| 8 | t-BuXPhosAuNTf2 | DCE | 80 °C, 11 h | 66% | 8.6 |
| 9 | (ArO)3PAuNTf2[e] | DCE | 80 °C, 11 h | 73% | 2.0 |
| 10 | AuCl3 | DCE | 80 °C, 11 h | <20% | - |
| 11 | t-BuXPhosAuNTf2 | anisole (0.2 M) | 80 °C, 5 min | >95%[f] | 6.5 |
| 12 | t-BuXPhosAuNTf2 | anisole (0.2 M) | 40 °C, 1.5 h | >95% | 5.2 |
| 13 | t-BuXPhosAuNTf2 | anisole (0.2 M) | −20 °C, 12 h | >95% | 3.9 |
[1] = 0.05 M.
Estimated by 1H NMR using diethyl phthalate as the internal reference.
Regioisomers of 3 were formed due to reaction with solvent toluene.
2,3-diphenylindole (4) was formed in 10% yield.
Ar = 2,4-di-tert-butylphenyl.
Isolated yield: 89%.
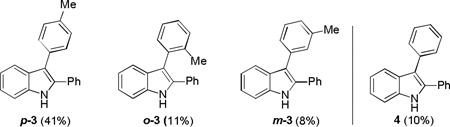
Table 3.
The scope of different nucleophiles.[a]
 | ||||
|---|---|---|---|---|
| entry | NuH | time | 5[b] | yield[c] |
| 1 | p-xylene[d] | 5 h |  |
70% |
| 2 | 3 h |  |
78% | |
| 3 | 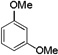 |
3 h | 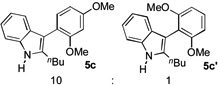 |
84% |
| 4 | 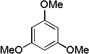 |
3 h | 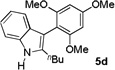 |
84% |
| 5 | 8 h | 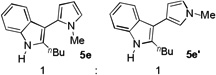 |
91% | |
| 6[e] | 12 h | 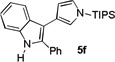 |
69%[f] | |
| 7 | 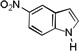 |
3 h | 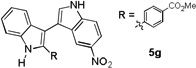 |
83% |
| 8 | 3 h | 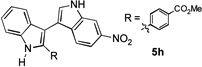 |
81% | |
| 9 | 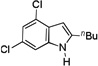 |
3 h | 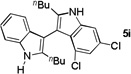 |
64% |
| 10 | 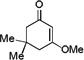 |
1 h | 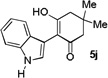 |
49% |
| 11 | n-BuOH | 15 h | 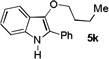 |
66% |
| 12 | n-BuOH | 12 h | 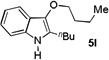 |
62% |
| 13 | allyl alcohol | 10 h | 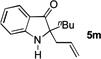 |
65% |
[1] = 0.1 M.
Regioisomers not separated. Ratio determined by 1H NMR.
Isolated yield.
Used as solvent.
10 mol % of IPrAuNTf2 used.
The regioselectivity is >9:1, and the yield is for the major isomer.
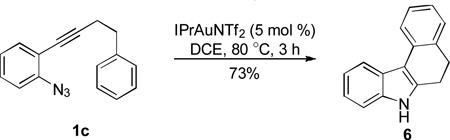
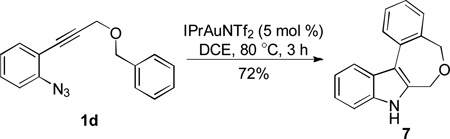
This reactivity umpolung of indole was briefly tested in intramolecular scenarios. With azidoalkyne 1c, in the absence of an external nucleophile such as anisole (e.g., in Table 2, entry 2), the tethered benzene ring reacted as the nucleophile, efficiently trapping the electrophilic indole 3-position and thereby forming tetracyclic product 6 in a good yield (Eq. 1). To our surprise, even a seven-membered ring could be readily formed via this intramolecular trapping (Eq. 2); moreover, this reaction appeared to be rather facile as the tetracyclic product 7 was formed in ca. 4% yield even when anisole was used as the solvent (Table 2, entry 3), again implicating the high electrophilicity of the intermediate B/C.
In summary, we have developed a new approach to achieving reactivity umpolung of indole at the 3-position via gold catalysis. By using an ortho-azido group to deliver a nitrene intramolecularly, an arylalkyne can be converted into a gold carbene intermediate that contains the indole skeleton but highly electrophilic at the 3-position. The reaction of this electrophilic indole intermediate with variously nucleophiles provides a novel and expedient synthesis of a range of functional indoles.
Experimental Section
General procedure for the gold-catalyzed formation of indole 5
o-azidoarylalkyne 1 (0.30 mmol) and IPrAuNTf2 (12.9 mg, 0.015 mmol) were added to a solution of a nucleophile (1.2 mmol) in DCE (3 mL) at room temperature. The reaction mixture was heated at 80 °C, and the progress of the reaction was monitored by TLC. The reaction typically took 3 h. Upon completion, the mixture was concentrated and the residue was purified by silica gel flash chromatography (eluent: hexanes/ethyl acetate) to afford the desired products.
Supplementary Material
Acknowledgments
The authors thank NIGMS (R01 GM084254) and UCSB for generous financial support and Dr. Guang Wu for helping with X-ray crystallography. LZ is a Sloan fellow.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For selected reviews on indole synthesis, see: a) Cacchi S, Fabrizi G. Chem. Rev. 2011;111:PR215. doi: 10.1021/cr100403z. Bandini M, Eichholzer A. Angew. Chem., Int. Ed. 2009;48:9608. doi: 10.1002/anie.200901843.
- 2.These derivatives require the coupling of indoles with nucleophiles (NuH), which has been realized via oxidative processes. For selected examples, see: a) Haro Td, Nevado C. J. Am. Chem. Soc. 2010;132:1512. doi: 10.1021/ja909726h. Jeevanandam A, Srinivasan PC. Synth. Commun. 1995;25:3427. Dwight TA, Rue NR, Charyk D, Josselyn R, DeBoef B. Org. Lett. 2007;9:3137. doi: 10.1021/ol071308z. Stuart DR, Fagnou K. Science. 2007;316:1172. doi: 10.1126/science.1141956. Stuart DR, Villemure E, Fagnou K. J. Am. Chem. Soc. 2007;129:12072. doi: 10.1021/ja0745862. He C-Y, Fan S, Zhang X. J. Am. Chem. Soc. 2010;132:12850. doi: 10.1021/ja106046p. Potavathri S, Pereira KC, Gorelsky SI, Pike A, LeBris AP, DeBoef B. J. Am. Chem. Soc. 2010;132:14676. doi: 10.1021/ja107159b.
- 3.a) Seebach D. Angew. Chem., Int. Ed. 1979;18:239. [Google Scholar]; b) Seeback D, Kolb M. Chem. Ind. (London) 1974:687. [Google Scholar]
- 4.for a review, see: Joule JA. Sci. Synth. 2000;10:541.
- 5.a) Cui L, Zhang L. Chem. Commun. 2010 doi: 10.1039/c001314e. Accepted; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cui L, Li C, Zhang L. Angew. Chem., Int. Ed. 2010;49:9178. doi: 10.1002/anie.201004712. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cui L, Zhang G, Peng Y, Zhang L. Org. Lett. 2009;11:1225. doi: 10.1021/ol900027h. [DOI] [PubMed] [Google Scholar]; d) Cui L, Peng Y, Zhang L. J. Am. Chem. Soc. 2009;131:8394. doi: 10.1021/ja903531g. [DOI] [PubMed] [Google Scholar]; e) Li G, Zhang L. Angew. Chem., Int. Ed. 2007;46:5156. doi: 10.1002/anie.200701449. [DOI] [PubMed] [Google Scholar]
- 6.a) Ye L, He W, Zhang L. J. Am. Chem. Soc. 2010;132:8550. doi: 10.1021/ja1033952. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ye L, Cui L, Zhang G, Zhang L. J. Am. Chem. Soc. 2010;132:3258. doi: 10.1021/ja100041e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ye L, He W, Zhang L. Angew. Chem., Int. Ed. 2011;50:3236. doi: 10.1002/anie.201007624. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lu B, Li C, Zhang L. J. Am. Chem. Soc. 2010;132:14070. doi: 10.1021/ja1072614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For works done by other research groups, see: a) Shapiro ND, Toste FD. J. Am. Chem. Soc. 2007;129:4160. doi: 10.1021/ja070789e. Yeom HS, Lee JE, Shin S. Angew. Chem., Int. Ed. 2008;47:7040. doi: 10.1002/anie.200802802. Yeom HS, Lee Y, Lee JE, Shin S. Org. Biomol. Chem. 2009;7:4744. doi: 10.1039/b910757f. Davies PW, Albrecht SJC. Angew. Chem., Int. Ed. 2009;48:8372. doi: 10.1002/anie.200904309. Yeom HS, Lee Y, Jeong J, So E, Hwang S, Lee JE, Lee SS, Shin S. Angew. Chem., Int. Ed. 2010;49:1611. doi: 10.1002/anie.200906346. Jadhav AM, Bhunia S, Liao H-Y, Liu R-S. J. Am. Chem. Soc. 2011;133:1769. doi: 10.1021/ja110514s. Yeom H-S, So E, Shin S. Chem. Eur. J. 2011:1764. doi: 10.1002/chem.201002863. Davies PW, Cremonesi A, Martin N. Chem. Commun. 2011;47:379. doi: 10.1039/c0cc02736g.
- 8.For studies that exclude the formation of α-oxo gold carbene intermediates, see: a) Xu C-F, Xu M, Jia Y-X, Li C-Y. Org. Lett. 2011;13:1556. doi: 10.1021/ol200270t. Li C-W, Pati K, Lin G-Y, Sohel SMA, Hung H-H, Liu R-S. Angew. Chem., Int. Ed. 2010;49:9891. doi: 10.1002/anie.201004647. Cuenca AB, Montserrai S, Hossain KM, Mancha G, Lledos A, Medio-Simon M, Ujaque G, Asensio G. Org. Lett. 2009;11:4906. doi: 10.1021/ol9020578.
- 9.Li C, Zhang L. Org. Lett. 2011:1738. doi: 10.1021/ol2002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For a review on ynamides see: De KKA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP. Chem. Rev. 2010;110:5064. doi: 10.1021/cr100003s.
- 11.a) Li Z, Capretto DA, Rahaman RO, He C. J. Am. Chem. Soc. 2007;129:12058. doi: 10.1021/ja0724137. [DOI] [PubMed] [Google Scholar]; b) Li Z, Ding X, He C. J. Org. Chem. 2006;71:5876. doi: 10.1021/jo060016t. [DOI] [PubMed] [Google Scholar]
- 12.Gorin DJ, Davis NR, Toste FD. J. Am. Chem. Soc. 2005;127:11260. doi: 10.1021/ja053804t. [DOI] [PubMed] [Google Scholar]
- 13.Hiroya K, Matsumoto S, Ashikawa M, Ogiwara K, Sakamoto T. Org. Lett. 2006;8:5349. doi: 10.1021/ol062249c. [DOI] [PubMed] [Google Scholar]
- 14.This work is prompted by the submission of a closely related work by Professor Fabien Gagosz. For the reference, see: Wetzel A, Gagosz F. Angew. Chem., Int. Ed. 2011;50:7354. doi: 10.1002/anie.201102707.
- 15.Mézailles N, Ricard L, Gagosz F. Org. Lett. 2005;7:4133. doi: 10.1021/ol0515917. [DOI] [PubMed] [Google Scholar]
- 16.Seidel G, Mynott R, Fürstner A. Angew. Chem., Int. Ed. 2009;48:2510. doi: 10.1002/anie.200806059. [DOI] [PubMed] [Google Scholar]
- 17.Doyle MP, McKervey MA, Ye T. Modern catalytic methods for organic synthesis with diazo compounds : from cyclopropanes to ylides. New York: Wiley; 1998. [Google Scholar]
- 18.According to the Mayr-Patz equation (H. Mayr, M. Patz, Angew. Chem., Int. Ed. 1994, 33, 938): logk20 °C = sN(N + E) (N: nucleophilicity parameter, E: electrophilicity parameter, sN: nucleophile-specific sensitivity parameter), with 4,4’-bis(p-methoxy)benzhydrylium ion as the electrophile (E = 0), anisole [N/sN(anisole): −1.18/1.20] (H. Mayr, B. Kempf, A. R. Ofial, Acc. Chem. Res. 2002, 36, 66) would react 3 × 104 times faster than toluene [N/sN(toluene): −4.47/1.32] (H. Mayr, T. Bug, M. F. Gotta, N. Hering, B. Irrgang, B. Janker, B. Kempf, R. Loos, A. R. Ofial, G. Remennikov, H. Schimmel, J. Am. Chem. Soc. 2001, 123, 9500).
- 19.a) Fang D, Shi Q-R, Cheng J, Gong K, Liu Z-L. Appl. Catal., A. 2008;345:158. [Google Scholar]; b) Yin W-P, Shi M. J. Chem. Res. 2006:549. [Google Scholar]; c) Prakash GKS, Mathew T, Marinez ER, Esteves PM, Rasul G, Olah GA. J. Org. Chem. 2006;71:3952. doi: 10.1021/jo0604181. [DOI] [PubMed] [Google Scholar]; d) Stock LM, Brown HC. In: Advances in Physical Organic Chemistry, Vol. Volume 1. Gold V, editor. Academic Press; 1963. p. 35. [Google Scholar]
- 20.Due to the cationic nature of B/C, it is difficult to probe the reaction mechanism via typical carbene chemistries such as cyclopropanation. In fact, no cyclopropanation would be observed in the presence of styrene.
- 21.a) Shi D, Han L, Sun J, Li S, Wang S, Yang Y, Fan X, Shi J. Chin. Chem. Lett. 2005;16:770. [Google Scholar]; b) Hodder AR, Capon RJ. J. Nat. Prod. 1991;54:1661. [Google Scholar]
- 22.Carter MD, Weaver DF, Jacobo SMH, Lu E, Gao F. PCT Int. Appl. 2008 WO 2008058402 A1 20080522. [Google Scholar]
- 23.Ramesh C, Kavala V, Kuo C-W, Raju BR, Yao C-F. Eur. J. Org. Chem. 2010;2010:3796. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.