

Abstract
The use of bis-boronic acid for the direct synthesis of boronic acids has greatly facilitated the two-step, one-pot borylation/Suzuki cross-coupling reaction between aryl- and heteroaryl halides. Making use of Buchwald’s second generation XPhos preformed catalyst, high yields of cross-coupled products were obtained for most substrates. The method also allows an efficient two-step, one-pot synthesis, providing access to three distinct cross-coupled products after column chromatography. The method also provides a rapid and convenient route to teraryl compounds.
INTRODUCTION
The Suzuki-Miyaura cross-coupling of boronic acids with organic halides is one of the most widely applied methods in current synthetic organic chemistry.1, 2 Since the first report of the palladium-catalyzed cross-coupling between an aryl halide and an arylboronic acid by Suzuki and Miyaura in 1981,3 it has emerged as a functional group tolerant method, providing reliable and efficient access to structurally diverse biaryl motifs. 4 It is for these reasons that it remains one of the most important methods of choice for C-C bond formation in industrial and academic groups alike. Fueled by the commercial availability of numerous organic halides, boronic acids, and the constant development of improved catalyst systems,5, 6 intense research efforts continue in this area.
With all of the advances, the Suzuki-Miyaura cross-coupling reaction still suffers a major limitation in that it relies upon the direct use of boronic acids. Although many boronic acids are commercially available, they can be very expensive and decompose with storage over time, often resulting in the necessity to use at least 1.2 equivalents (with regard to the organic halide) in a typical Suzuki-Miyaura cross-coupling reaction. 7, 8 Additionally, if the boronic acid is not commercially available, its synthesis is required, adding additional, often lengthy, steps to the research process.9–18
Over the last 15 years, progress has been made to circumvent some of the limitations of the Suzuki-Miyaura cross-coupling reaction with the advent of one-pot Miyaura borylation/Suzuki cross-coupling reactions. The first reported system by Miyaura in 1997 utilized two organotriflates and bis(pinacolato)diboron (B2Pin2) in refluxing dioxane. The method required the synthesis of excess boronate and a second addition of catalyst to facilitate the cross-coupling in high yield. However, it demonstrated for the first time that the need to isolate or purchase a boronic acid could be eliminated. Instead, the method allows the coupling of two aryl triflates in a simple and efficient manner.12
Since Miyaura’s seminal paper in 1997, other groups have followed with improvements on the process. In 2000 and 2002 Baudoin et al. extended the method to include aryl iodides and bromides with the more atom economical pinacolborane (H-BPin). This improved method eliminated the need to add additional catalyst in the second step but required 5 mol % of a palladium catalyst and dioxane at elevated temperatures, was limited in scope, and still required an excess of the boronate ester coupling partner.19, 20
The next major advance came in 2007 when Buchwald et al. successfully demonstrated the first general Miyaura borylation of aryl chlorides. In the same paper, they went on to show that the method could be extended to a one-pot Miyaura borylation/Suzuki cross-coupling reaction between two aryl chlorides. The one-pot method made use of B2Pin2 with efficient catalyst loads of 1 mol %, but required the use of excess boronate ester in refluxing dioxane.21
More recently, Wang et al. demonstrated that aryl- and heteroaryl bromides and chlorides could be used efficiently in the one-pot process when utilizing B2Pin2. However, the catalyst system developed is currently not commercially available and still requires the use of refluxing dioxane. In addition, there are no examples of aryl chlorides undergoing the borylation reaction, and therefore they can only be used in the second step. Further, the method still requires that the boron coupling component be synthesized in excess.22
Overall, the one-pot Miyaura borylation/Suzuki cross-coupling is a very efficient method. The fact that boronic acids no longer have to be purchased or isolated, coupled with the ease of synthetic strategy employing just two aryl halide (or pseudohalide) coupling partners, make this method very attractive. However, although significant progress has been made, many disadvantages remain: 1) Current protocols make use of the atom inefficient bis(pinacolato)diboron or its derivatives to make the boronate ester. 2) The synthesis of excess boronate ester is required with respect to the second aryl halide. 3) High temperatures are utilized in solvents that are not environmentally sound (dioxane, DMF, toluene). 4) High catalyst loads are employed or a second loading of catalyst is required to facilitate the Suzuki reaction in the second step. 5) Some catalyst systems are used that are not commercially available.
RESULTS AND DISCUSSION
Recently, we described the full scope of the borylation of aryl and heteroaryl electrophiles (bromides, chlorides, iodides, and triflates) utilizing the bench stable and atom economical bis-boronic acid (BBA) to access boronic acids and derivatives (Scheme 1, eqs 1 and 2).23, 24 Early in the development of these methods we also briefly demonstrated that the boronic acid obtained after borylation efficiently undergoes the two-step, one-pot borylation/cross-coupling reaction, providing biaryl products in good to excellent yield without the need to synthesize excess boronic acid. (Scheme 1, eq 3).23 Additionally, the reactions were run in environmentally benign EtOH at reduced temperatures of 80 °C. Because of the use of extremely reactive preformed palladium catalysts, low loads could be efficiently realized without the need to re-charge the system for the second step. Our method, therefore, in whole or in part, represents an improvement of current methods to access biaryl species utilizing a one-pot Miyaura borylation/Suzuki cross-coupling reaction sequence (Scheme 1, eqs 3 and 4).
Scheme 1.

Diboron Reagents. Comparison of Current and Newly Developed One-Pot Miyaura Borylation/Suzuki Cross-Coupling Reaction Methods
In this article we report the full scope and limitations of the palladium-catalyzed one-pot Miyaura borylation/Suzuki cross-coupling reaction utilizing BBA as the borylating source. The method described herein is an improvement from our previous communication in that it has been expanded to include aryl bromides and heteroaryl halides. In our first report of this method, we made use of the first generation Buchwald preformed catalyst, XPhos-Pd-G1 (1A) with an efficient catalyst load of 2 mol % (Figure 1). Through the use of Buchwald’s newly available second generation XPhos preformed catalyst, XPhos-Pd-G2 (2A), the load has been reduced by half to 1 mol % (Figure 1). That 2 instantaneously forms Pd(0) under the reaction conditions at rt is of great significance and utility to the method, as BBA decomposes rapidly when exposed to Pd(II).
Figure 1.

Buchwald’s First Generation XPhos Preformed Catalyst XPhos-Pd-G1 (1), Second Generation XPhos Preformed Catalyst XPhos-Pd-G2 (2), and Ligands XPhos (A) and CataCXium A (B).
As all reagents and catalysts are commercially available and bench stable, solids are simply weighed on a bench top balance and then placed under an inert atmosphere of argon before the addition of degassed, non-anhydrous EtOH. The reactions are then heated to 80 °C until the borylation is complete. As noted in our previous work with 2A, once all of the first halide has undergone borylation (and thus all of the starting material has been consumed), a readily observable color change occurs: aryl chlorides turn from colorless to yellow, and aryl bromides turn from colorless to orange (Figure 2). This signals that the second base (K2CO3) followed by the second halide can now be added to the reaction. This visual cue greatly facilitates the ease by which the method is executed. It should be noted that the use of strong base not only promotes the subsequent cross-coupling reaction, but also facilitates the decomposition of any remaining BBA as well. Consequently, no BBA is present at this stage to borylate the second halide that is added to the reaction mixture.
Figure 2.

Color Change Indicating Completion of Borylation Reaction. From Left to Right: Completion of Borylation on an Aryl Chloride, and Completion of Borylation of an Aryl Bromide.
We began our examination of the one-pot method by looking at simple aryl-aryl couplings to explore functional group compatibility (Table 1). In the first few substrates synthesized, we utilized 1.3 equivalents of the second aryl halide (Table 1, entries 1–4). As we sought to provide a general method that did not rely on an excess of either the boronic acid equivalent or the aryl halide component, we attempted the one-pot reaction with a 1:1 ratio of both halides (Table 1, entry 4) with no appreciable loss in yield. Going forward then, all reactions were carried out in this manner.
Table 1.
Two-Step, One-Pot Aryl-Aryl Cross-Coupling Demonstrating Functional Group Tolerability
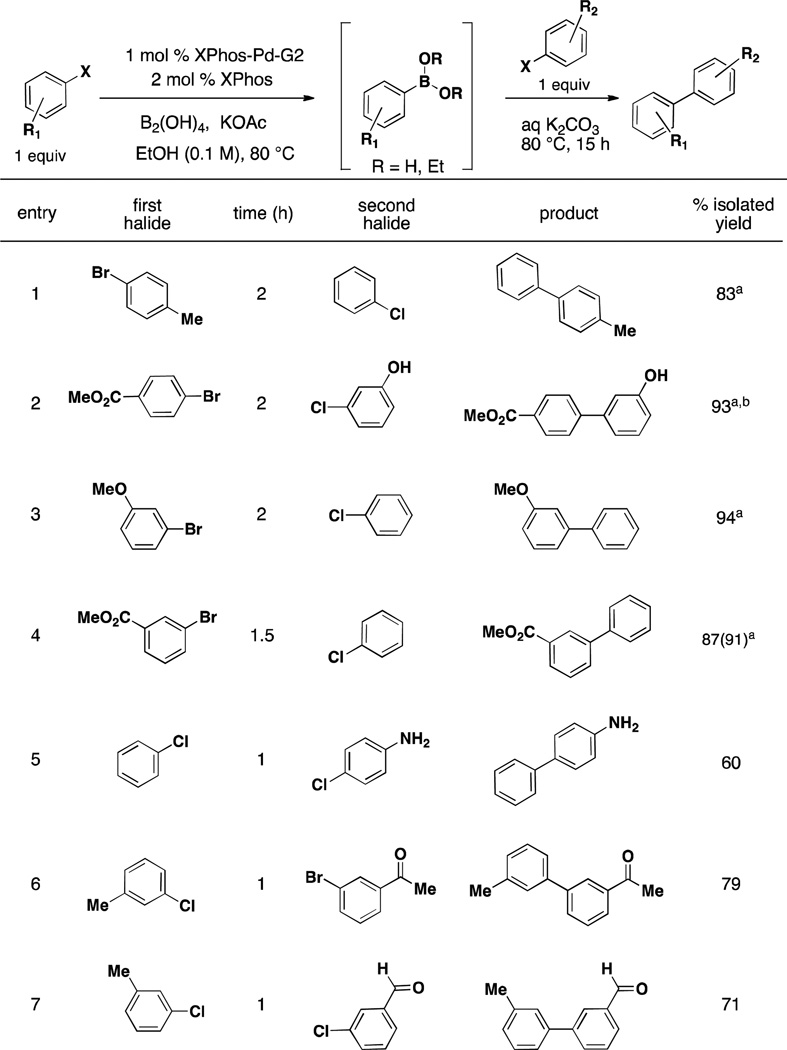 |
General conditions: a) 1 mol % XPhos-Pd-G2, 2 mol % XPhos, 3.0 equiv KOAc, 3.0 equiv B2(OH)4, EtOH (0.1 M), 80 °C for time indicated. b) 3 equiv 1.8 M K2CO3. 1 equiv second halide. 80 °C, 15 h.
1.3 equiv second halide added in second step.
MeOH used as solvent.
Methyl esters perform well in the reaction (Table 1, entries 2 and 4) with minimal amounts (<5%) of transesterification with EtOH observed. This can, however, be completely avoided through the use of MeOH as solvent. Unprotected amines and hydroxyl groups are well tolerated, leading to good to excellent yields after isolation of the cross-coupled product (Table 1, entries 2 and 5). We observe no α-arylation of ketones when used under these reaction conditions (Table 1, entry 6)21 and no reduction of either the ketone or aldehyde as previously observed in the haloaryl borylation of aldehydes and ketones.23, 24
We next turned our attention to comparing the reactivities of aryl bromides and chlorides. As seen in Table 2, both aryl bromides and chlorides perform well, but chlorides consistently provide superior results when utilized in the second step. We believe this result is due to the character and therefore inherent reactivity of the Pd-X bond. Similar to the mechanism proposed for borylation, the transmetalation in this system is the rate-determining step. Therefore, the more electronegative halide (Cl > Br) increases the electrophilicity of the organopalladium halide intermediate and escalates the rate at which this oxidative addition adduct undergoes transmetalation with the newly formed boronic acid (or BBA in the case of the borylation mechanism, Scheme 2).6, 24, 25 It should further be noted that this finding is in agreement with that proposed by Buchwald et al. in Suzuki/Miyaura cross-couplings with XPhos-Pd-G2, where they demonstrated that the rate order of transmetalation was ArCl > ArBr > ArI.6 As aryl chlorides are currently less expensive, more abundant, and more diversely substituted than aryl bromides, this reactivity feature is especially useful.
Table 2.
Comparison of Borylation with Aryl Chlorides and Bromides Utilizing BBA and XPhos-Pd-G2
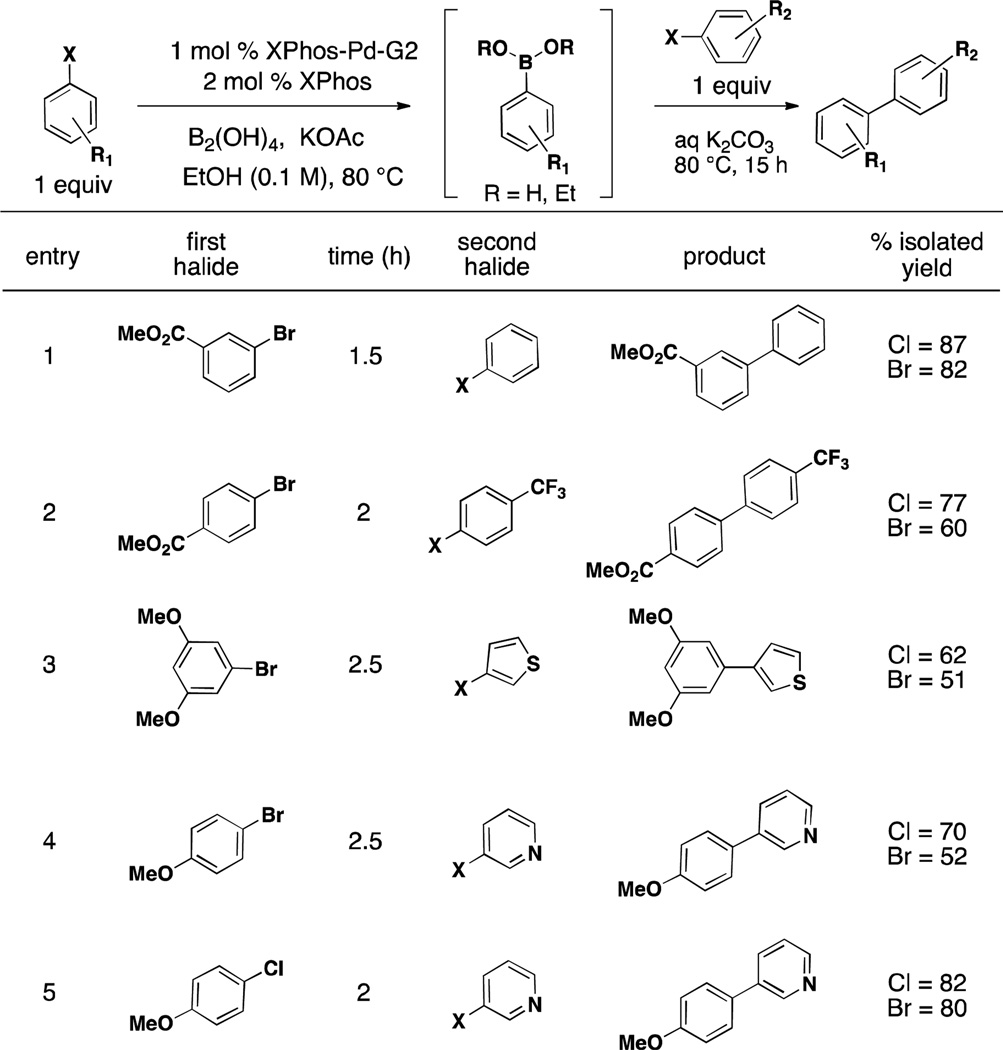 |
General conditions: a) 1 mol % XPhos-Pd-G2, 2 mol % XPhos, 3.0 equiv KOAc, 3.0 equiv B2(OH)4, EtOH (0.1 M), 80 °C for time indicated, b) 3 equiv 1.8 M K2CO3. 1 equiv second halide. 80°C, 15 h.
Scheme 2.

Proposed Mechanisms of the Two-Step, One-Pot Borylation/Suzuki Reaction with BBA and XPhos-Pd-G2
Encouraged by these early results, we next turned our attention to those substrates that provided low yields after borylation and subsequent conversion to the corresponding trifluoroborate in our previous studies (eq 5, Table 3).24 As these functional groups are desired in cross-coupled products, it was important to provide more efficient access to them. We surmised that even if they did not undergo the initial borylation in high yield, they should perform well as the second halide in the cross-coupling reaction. This was, in fact, observed. As outlined in Table 3, eq 5, with the exception of entry 7, all substrates that provided low yields of the trifluoroborate in our previously published borylation studies24 provided good to excellent yields of the cross-coupled product when used in the second step. Most notable are entries 5 and 6, which afforded no borylated product but provided excellent yields of cross-coupled product.
Table 3.
Aryl Chlorides Resulting in Low-Yielding Borylation used as the Second Partner in the One-Pot Miyaura Borylation/Suzuki Cross-Coupling Reaction
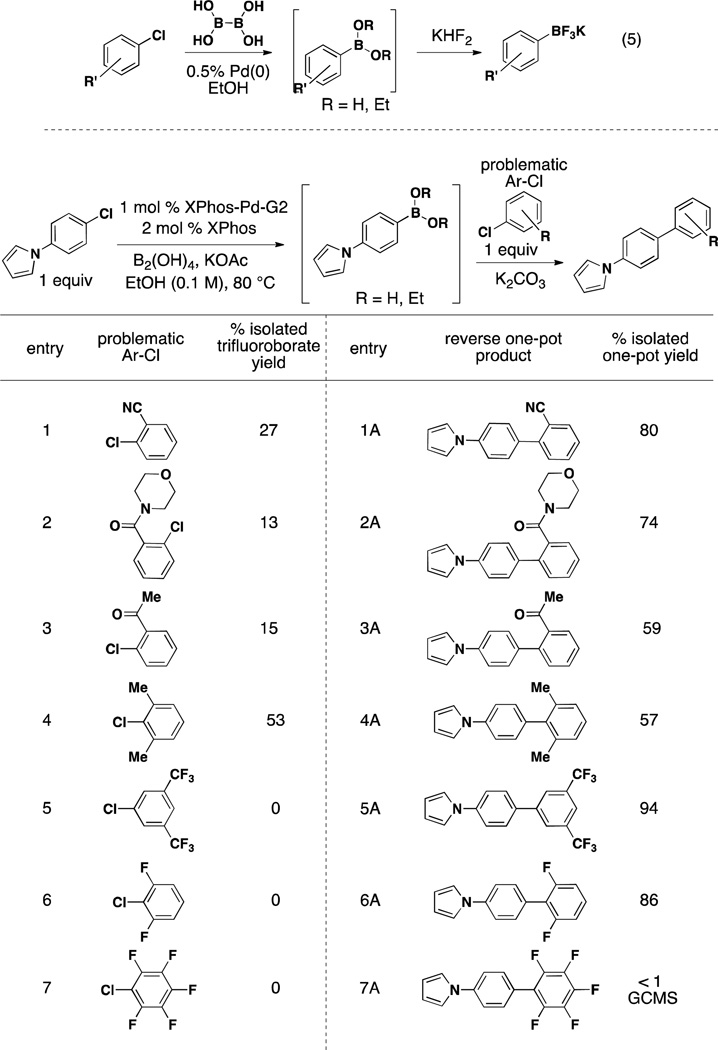 |
General conditions for the one-pot sequence: a) 1 mol % XPhos-Pd-G2, 2 mol % XPhos, 3.0 equiv KOAc, 3.0 equiv B2(OH)4, EtOH (0.1 M), 80 °C for 2 h. b) 3 equiv 1.8 M K2CO3. 1 equiv second aryl chloride, 80 °C, 15 h.
Because of the success encountered with the aforementioned strategy, its synthetic utility was explored still further. As mentioned above, one of the major advantages of the one-pot Miyaura borylation/Suzuki cross-coupling reaction is the use of two aryl halides. This distinguishing feature therefore allows a choice in the order of addition when carrying out the reaction: which halide is borylated and which is used as the electrophile in the second step can easily be reversed. As demonstrated in Table 4, the order in which the halides are used can have a significant effect on the overall result of the reaction.
Table 4.
Reverse-Order Strategy Comparisons in the Cross-Coupling of Two Aryl Halides
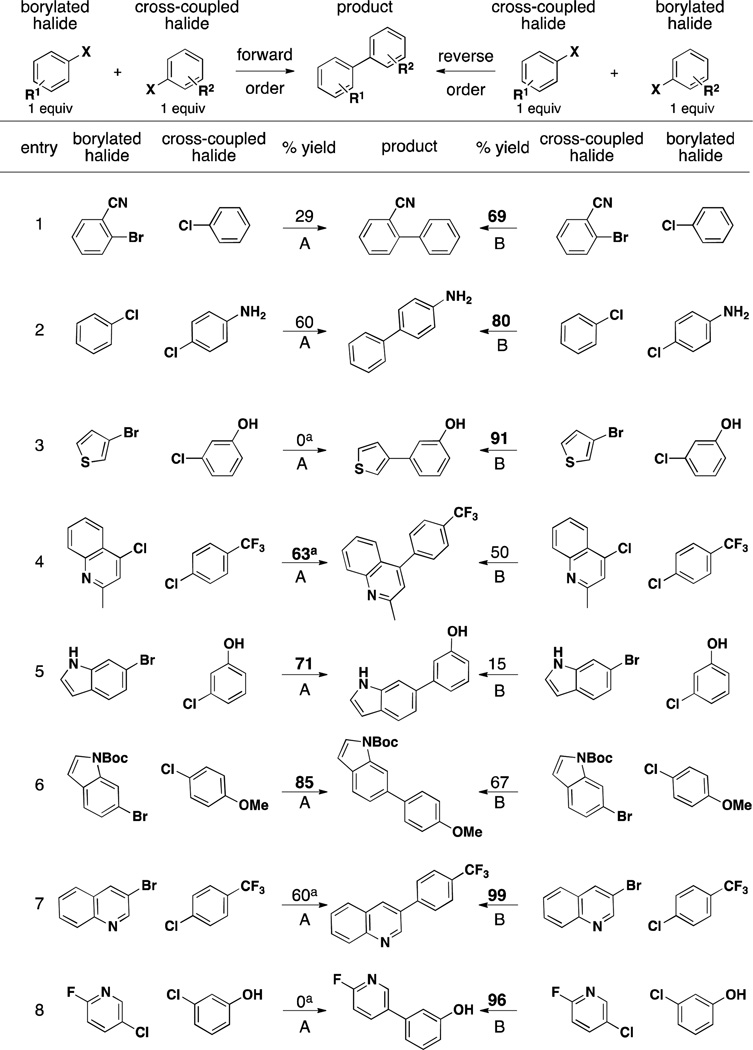 |
General conditions: a) 1 mol % XPhos-Pd-G2, 2 mol % XPhos, 3.0 equiv KOAc, 3.0 equiv B2(OH)4, EtOH (0.1 M), 80 °C. b) 3 equiv 1.8 M K2CO3. 1 equiv second aryl halide. 80 °C, 15 h.
5 mol % CataCXium A-Pd-G2, 3 equiv i-Pr2NEt, 3 equiv B2(OH)4, MeOH (0.2 M), 50 °C. b) 3 equiv 1 M K3PO4, 1 equiv second halide, 50 °C, 15 h.
For example, some heteroarylboronic acids, once synthesized using our optimized method with the CataCXium A preformed precatalyst (Figure 1, 2B),24 readily decompose under the reaction conditions as evinced by the fact that the reaction goes to full conversion, but little to no product formation of the corresponding trifluoroborate is observed. Therefore, yields for products containing heteroaryls are typically better if the heteroaryl is used in the second step (Table 4, entries 3, 7, and 8). Interestingly, this is not always the case, as 4-chloroquinaldine provides a superior yield when it is used to make the boronic acid (Table 4, entry 4). Both protected and unprotected indoles provide better results when they are used in the first step (Table 4, entries 5 and 6). Similarly, the unprotected amine performed better when used in the first step (Table 4, entry 2). If care is taken with respect to the order of addition, excellent yields can be obtained with most coupling partners. The results in Table 4 demonstrate the benefits of probing both combinations on small scale to find the highest yielding order before scaling up the reaction.
We next turned our attention to exploring the scope of heteroaryl halides. As few heteroaryls perform exceptionally well (specifically those with the heteroatom in the same ring as the halide) under the general borylating conditions, we focused on their use as coupling partners in the second step. As outlined in Table 5, substituted heteroaryl halides all undergo efficient cross-coupling. Pyridines substituted at the 3-position couple well with electron withdrawing or electron donating substituents or with no substitution at all (Table 5, entries 1–3). Similarly, 3-substituted pyrimidines and furans provide good to excellent yields (Table 5 entries, 4 and 5). 2-Chlorothiophenes couple well (Table 5, entries 6–8) and even 2-chlorobenzoxazole provided reasonable yield over two steps (Table 5, entry 11). The 2-chlorosubstituted pyrimidine and pyridine provided modest yields (Table 5, entries 9 and 10) while the 4-chloroquinaldine coupled well, providing product in excellent yield (Table 5, entry 12).
Table 5.
Use of Substituted Heteroaryls as Electrophiles in the One-Pot Miyaura Borylation/Suzuki Cross-Coupling Reaction Utilizing BBA and XPhos-Pd-G2
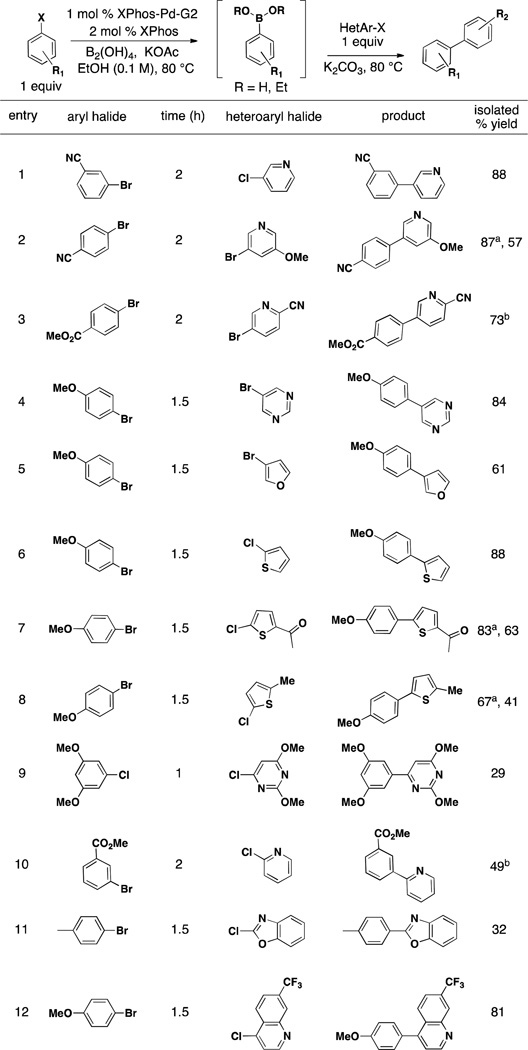 |
General conditions: a) 1 mol % XPhos-Pd-G2, 2 mol % XPhos, 3.0 equlv KOAc, 3.0 equiv B2(OH)4, EtOH (0.1 M), 80 °C for time indicated, b) 3 equiv 1.8 M K2CO3. 1 equiv second halide. 80 °C, 15 h.
1.3 equiv halide added in second step.
< 5% transesterification observed.
In a few cases where products were obtained in low yield using the general procedure, we found that the addition of 1.3 equivalents of the heteroaryl halide in the second step (as compared to 1.0 equivalents so that the boronic acid and heteroaryl halide are 1:1) significantly increased the yield of the isolated product (Table 5, entries 2, 7, and 8). This is most likely a result of the protodehalogentation of the heteroaryl halide under the reaction conditions. This commonly observed side product occurs to different extents with heteroaryl halides depending on their substitution pattern in terms of both of functional groups and the halide. Finally, although we attempted the coupling of two heteroaryl halides under these reaction conditions, products were only obtained in low yield. Therefore, the development of a method utilizing BBA to address this important area of research remains an active pursuit.
We sought to demonstrate the power of this one-pot method further within the context of parallel synthesis. Being careful to choose substrates with polarity differences, an efficient borylation/one-pot Suzuki reaction can be performed, providing three distinct products in excellent yields after chromatography. As outlined in Scheme 3, the first halide undergoes the borylation, with subsequent addition of base and three aryl/heteroaryl halides in equimolar amounts affording the desired cross-coupled products. To the best of our knowledge, this is the first time such a reaction has been demonstrated. It should also be noted that the one-pot sequence can effectively be scaled, as the one-pot reaction demonstrated in Scheme 3 was run on 4.5 mmol scale, with all products isolated in excellent yield.
Scheme 3.

One Pot Borylation/Suzuki Reaction Utilizing BBA and XPhos-Pd-G2 Yielding Three Distinct Products
Another application makes use of the fact that triflates borylate in high yield under the general reaction conditions. Therefore, triflates (and the alcohols from which they are derived) can essentially be used as a masked boronic acid or halide in a synthetic sequence in which an aryl halide exists in the same molecule. Using the strategy outlined in Scheme 4, the first cross-coupled product containing a hydroxyl group is isolated in a yield of 94% after undergoing the two-step, one-pot Miyaura borylation/Suzuki cross-coupling reaction with BBA and XPhos-Pd-G2. The alcohol moiety of this product is then converted to the triflate. This triflate product is then subjected to a second one-pot, two-step sequence. The final teraryl product was thus obtained in a combined yield of 62% over the 5-step sequence from simple aryl halides without ever purchasing or isolating a boronic acid.
Scheme 4.

Efficient Teraryl Synthesis Utilizing Alcohols as Masked Halides and Boronic Acids
CONCLUSION
The Suzuki-Miyaura reaction has emerged as one of the key transformations in modern synthetic organic chemistry. However, some undesirable aspects still remain, largely the requisite use of excess boronic acid to ensure efficient coupling. The advent of the two-step, one-pot borylation/Suzuki coupling reaction first demonstrated by Miyaura in 1997 and improved upon by other groups sought to help reduce this burden. Although advances were made, other reaction conditions, such as the use of B2Pin2 in the borylating step and refluxing ethereal solvents, still made these methods undesirable. Through the use of BBA and the first and second generation Buchwald preformed catalysts, we have developed a method that allows the efficient coupling of two aryl halides in one-pot without the need to synthesize the boronic acid in excess. The user-friendly process uses environmentally benign ethanol, and the readily observable color change indicates when the strong base and second halide can be added.
Additional advances include a parallel-synthesis application wherein three distinct cross-coupled products can be obtained after the two-step sequence in high yield. Further, the method allows quick access to teraryl species without the need to purchase or isolate a boronic acid. The only current limitation to the method is the efficient coupling of two heteroaryl halides. Efforts remain ongoing in this area.
EXPERIMENTAL SECTION
General Considerations
All reactions were carried out under an atmosphere of argon. Ethanol (non-anhydrous, 200 proof) was thoroughly degassed with argon directly before use. All aryl chlorides, XPhos-Pd-G1, XPhos-Pd-G2, and XPhos were purchased from commercial sources and used as received. Diisopropylethylamine (i-Pr2NEt) was distilled and stored under argon. KOAc and K2CO3 were dried in an oven overnight before use. All reagents (with the exception of the aryl chlorides), were stored in a bench-top desiccator. BBA was provided by BASF and used as received.
Analytical Methods
All new compounds were characterized by 1H NMR, 13C NMR, IR spectroscopy, high-resolution mass spectrometry (TOF), and melting point determination (for solids). All known compounds were characterized by 1H NMR and 13C NMR, and compared to literature values. 1H and 13C were recorded at 500 MHz and 125.8 MHz, respectively. Melting points are uncorrected.
General Procedure A: Palladium-Catalyzed Borylation of Aryl Halides and Their Suzuki Cross-Coupling with Aryl or Heteroaryl Halides
To an oven dried glass vessel capable of being sealed with a Teflon cap (for microwave vials) was added XPhos-Pd-G2 (11.78 mg, 15 µmol), XPhos (14.28 mg, 30 µmol), tetrahydroxydiboron (405 mg, 4.5 mmol), and KOAc (441 mg, 4.5 mmol). The vessel was sealed and then evacuated and backfilled with Ar (process was repeated four times). EtOH (15 mL degassed) was added via syringe followed by the addition of the first halide (1.5 mmol) in a similar manner (solid halides were added with the other solid reagents before sealing). The reaction was then heated to 80 °C for the time indicated, then a needle attached to a manifold under argon was inserted into the septum, and 3 equivalents (2.5 mL, 4.5 mmol) of 1.8 M degassed aqueous K2CO3 was added via syringe followed by the addition of the second halide (1.5 mmol) in a similar manner (in a solution of 500 µL degassed EtOH or THF if solid). The manifold needle was removed, and the reaction was further heated to 80 °C for 15 h. The reaction was cooled to rt, filtered through a thin pad of Celite (eluting with 50 mL EtOAc) and concentrated. The crude solid was extracted with EtOAc (3 × 5 mL), the combined organics were dried (Na2SO4) and then concentrated under reduced pressure. The desired compound was purified by column chromatography (40–63 µm silica gel stationary phase), eluting with EtOAc/hexane unless otherwise indicated.
General Procedure B: Palladium-Catalyzed Borylation of Heteroaryl Halides and their Suzuki Coupling with Aryl Halides
To an oven dried glass vessel capable of being sealed with a Teflon cap (for microwave vials) was added CataCXium A preformed catalyst (50 mg, 75 µmol) and B2(OH)4 (405 mg, 4.5 mmol). The vessel was sealed and then evacuated and backfilled with Ar (process was repeated four times). MeOH (7.5 mL, degassed) was added via syringe followed by the addition of the halide (1.5 mmol) and i-Pr2NEt (784 µL, 4.5 mmol) in a similar manner (solid halides were added with the other solid reagents before sealing). The reaction was then heated to 50 °C until the starting material was consumed (as monitored by GC). Subsequently, a needle attached to a manifold under argon was inserted into the septum and 3 equiv (4.5 mL, 4.5 mmol) of 1.0 M degassed aqueous K3PO4 was added via syringe followed by the second halide (1.5 mmol) in a similar manner (in a solution of 500 µL degassed EtOH or THF if solid). The manifold needle was removed, and the reaction was further heated to 50 °C for 15 h. The reaction was cooled to rt, filtered through a thin pad of Celite (eluting with 5 × 10 mL EtOAc) and concentrated. The crude solid was extracted with EtOAc (3× 5 mL), the combined organics were dried (Na2SO4) and then concentrated under reduced pressure. The desired compound was purified by column chromatography (40–63 µm silica gel stationary phase), eluting with EtOAc/hexane unless otherwise indicated.
4-Methyl-1,1'-biphenyl (Table 1, entry 1).26
General procedure A was employed with the only modification being that 1.3 equivalents of the second halide were employed instead of 1.0 equivalent. Column chromatography (1:9 → 1:3 EtOAc/hexane) provided the title compound as white crystals in 83% yield (209 mg). Spectral data were in accordance with those of published results. mp 42 – 43 °C; 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 7.9 Hz, 2H), 7.45 (t, J = 7.6 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H), 7.28 (d, J = 7.7 Hz, 2H), 2.43 (s, 3H); 13C NMR (125.8 MHz, CDCl3) δ141.3, 138.5, 137.2, 129.6, 128.9, 127.2, 127.1 (2), 21.3.
Methyl 3'-Hydroxy-[1,1'-biphenyl]-4-carboxylate (Table 1, entry 2).27
General procedure A was employed with the only modification being that 1.3 equivalents of the second halide were employed instead of 1.0 equivalent. Column chromatography (1:4 EtOAc/hexane) provided the title compound as off-white crystals in 93% yield (330 mg). Spectral data were in accordance with those of published results. mp = 145 °C decomposition. 1H NMR (500 MHz, CDCl3) δ 8.13 – 8.06 (m, 2H), 7.65 – 7.60 (m, 2H), 7.33 (t, J = 7.9 Hz, 1H), 7.19 (ddd, J = 7.7, 1.6, 0.9 Hz, 1H), 7.13 – 7.08 (m, 1H), 6.88 (ddd, J = 8.1, 2.5, 0.9 Hz, 1H), 5.33 (s, 1H), 3.95 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 167.4, 156.3, 145.4, 141.8, 130.3, 130.3, 129.1, 127.2, 119.9, 115.3, 114.4, 52.4.
3-Methoxy-1,1'-Biphenyl (Table 1, entry 3).28
General procedure A was employed with the only modification being that 1.3 equivalents of the second halide were employed instead of 1.0 equivalent. Column chromatography (1:49 EtOAc/hexane) provided the title compound as clear, pale yellow oil in 94% yield (261 mg). Spectral data were in accordance with those of published results. 1H NMR (500 MHz, CDCl3) δ 7.65 (dd, J = 8.3, 1.2 Hz, 2H), 7.52 – 7.47 (m, 2H), 7.44 – 7.38 (m, 2H), 7.27 – 7.23 (m, 1H), 7.21 – 7.19 (m, 1H), 6.96 (ddd, J = 8.2, 2.6, 0.8 Hz, 1H), 3.91 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 160.1, 142.9, 141.2, 129.9, 128.9, 127.6, 127.3, 119.8, 113.0, 112.8, 55.4.
Methyl [1,1'-Biphenyl]-3-carboxylate (Table 1, entry 4).29
General procedure A was employed with the only modification being that 1.3 equivalents of the second halide were employed instead of 1.0 equivalent. Column chromatography (1:49 EtOAc/hexane) provided the title compound as a clear, colorless oil in 91% yield (290 mg). Under general procedure A utilizing 1.0 equivalents of the second halide, the title compound was isolated in 87% yield (271 mg). Spectral data were in accordance with those of published results. 1H NMR (500 MHz, CDCl3) δ 8.29 (s, 1H), 8.03 (d, J = 7.7 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.63 (d, J = 8.3 Hz, 2H), 7.52 (t, J = 7.7 Hz, 1H), 7.47 (t, J = 7.8 Hz, 2H), 7.39 (t, J = 7.4 Hz, 1H), 3.95 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 167.2, 141.6, 140.3, 131.7, 130.8, 129.0, 129.0, 128.5, 128.4, 127.9, 127.3, 52.3.
[1,1'-Biphenyl]-4-amine (Table 1, entry 5).30, 31
General procedure A was employed. Column chromatography (1:2 → 1:1 EtOAc/hexane) provided the title compound as pale yellow crystals in 60% yield (150 mg). Spectral data were in accordance with those of published results. mp = 49 – 45 °C. 1H NMR (500 MHz, CDCl3) δ 7.55 (d, J = 7.2 Hz, 2H), 7.47 – 7.38 (m, 4H), 7.28 (t, J = 7.4 Hz, 1H), 6.77 (d, J = 8.3 Hz, 2H), 3.69 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 145.9, 141.2, 131.5, 128.7, 128.0, 126.4, 126.3, 115.5.
1-(3'-Methyl-[1,1'-biphenyl]-3-yl)ethanone (Table 1, entry 6)
General procedure A was employed. Column chromatography (1:20 EtOAc/hexane) provided the title compound as pale yellow oil in 79% yield (250 mg). 1H NMR (500 MHz, CDCl3) δ 8.18 (t, J = 1.6 Hz, 1H), 7.93 (dd, J = 8.3, 2.0 Hz, 1H), 7.79 (dt, J = 7.7, 1.4 Hz, 1H), 7.53 (t, J = 7.7 Hz, 1H), 7.43 (d, J = 12.4 Hz, 2H), 7.36 (t, J = 7.5 Hz, 1H), 7.21 (d, J = 7.4 Hz, 1H), 2.67 (s, 3H), 2.44 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 198.2, 141.9, 140.3, 138.7, 137.7, 131.9, 129.1, 128.9, 128.7, 128.1, 127.2, 127.0, 124.4, 26.9, 21.6. IR (dry film): 3030, 2359, 2340, 1684, 1500 cm−1. HRMS (ES+) calcd. for C15H15O (M+1) 211.1123, found 211.1121.
3'-Methyl-[1,1'-biphenyl]-3-carbaldehyde (Table 1, entry 7)
General procedure A was employed. Column chromatography (0–10% EtOAc/hexane) provided the title compound as offwhite crystals in 71% yield (210 mg). Prolonged storage on the bench led to decomposition and therefore a reasonable 13C NMR spectra could not be obtained. Low melting solid. 1H NMR (500 MHz, CDCl3) δ 10.11 (s, 1H), 8.11 (t, J = 1.5 Hz, 1H), 7.91 – 7.82 (m, 2H), 7.62 (t, J = 7.6 Hz, 1H), 7.49 – 7.42 (m, 2H), 7.38 (t, J = 7.6 Hz, 1H), 7.23 (d, J = 7.5 Hz, 1H), 2.46 (s, 3H). IR (dry film): 3022, 1690, 1583, 753 cm−1. HRMS (CI+) calcd. for C14H12O (M+H) 197.0966, found 197.0964.
Methyl [1,1'-Biphenyl]-3-carboxylate (Table 2, entry 1, aryl bromide second step).29
General procedure A was employed. Column chromatography (1:49 EtOAc/hexane) provided the title compound as a clear colorless oil in 82% yield (270 mg). Spectral data were in accordance with those of published results. 1H NMR (500 MHz, CDCl3) δ 8.29 (t, J = 1.6 Hz, 1H), 8.03 (dt, J = 7.8, 1.4 Hz, 1H), 7.81 – 7.77 (m, 1H), 7.63 (dd, J = 8.3, 1.2 Hz, 2H), 7.52 (t, J = 7.7 Hz, 1H), 7.47 (t, J = 7.6 Hz, 2H), 7.41 – 7.35 (m, 1H), 3.95 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 167.2, 141.6, 140.3, 131.7, 130.8, 129.0, 129.0, 128.5, 128.4, 127.9, 127.3, 52.3.
Methyl 4'-(Trifluoromethyl)-[1,1'-biphenyl]-4-carboxylate (Table 2, entry 2, aryl chloride in second step).32
General procedure A was employed. Column chromatography (0–5% EtOAc/hexane) provided the title compound as a white solid 77% yield (323 mg). Spectral data were in accordance with those of published results. mp 118–120 °C. 1H NMR (500 MHz, CDCl3) δ 8.14 (d, J = 8.3 Hz, 2H), 7.73 (s, 4H), 7.67 (d, J = 8.3 Hz, 2H), 3.96 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 166.9, 144.2, 143.7, 130.4, 130.4, 130.2, 129.9, 127.8, 127.4, 126.0 (q, J = 3.7 Hz), 52.4.
Methyl 4'-(Trifluoromethyl)-[1,1'-biphenyl]-4-carboxylate (Table 2, entry 2, aryl bromide in second step).32
General procedure A was employed. Column chromatography (0–5% EtOAc/hexane) provided the title compound as a white solid in 60% yield (249 mg). Spectral data were in accordance with those of published results. mp 119–121 °C. 1H NMR (500 MHz, CDCl3) δ 8.14 (d, J = 8.2 Hz, 2H), 7.73 (s, 4H), 7.67 (d, J = 8.3 Hz, 2H), 3.96 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 166.9, 144.2, 143.7, 143.7, 130.4, 129.9, 127.8, 127.4, 126.1, 126.0 (q, J = 3.8 Hz), 52.4.
3-(3,5-Dimethoxyphenyl)thiophene (Table 2, entry 3, aryl chloride in second step)
Column chromatography (0–5% EtOAc/hexane) provided the title compound as yellow oil in 62% yield (206 mg). 1H NMR (500 MHz, CDCl3) δ 7.45 (s, 1H), 7.40 – 7.35 (m, 2H), 6.75 (s, 2H), 6.43 (s, 1H), 3.85 (s, 6H). 13C NMR (125.8 MHz, CDCl3) δ 161.3, 142.5, 138.0, 126.7, 126.3, 120.9, 105.0, 99.3, 55.56. IR (dry film): 3000, 1203, 838 cm−1. HRMS (CI+) calcd. for C12H13O2S (M+H) 221.0636, found 221.0638.
3-(3,5-Dimethoxyphenyl)thiophene (Table 2, entry 3, aryl bromide in second step)
General procedure A was employed. Column chromatography (0–5% EtOAc/hexane) provided the title compound as a yellow oil in 51% yield (170 mg). 1H NMR (500 MHz, CDCl3) δ 7.45 (s, 1H), 7.39 – 7.35 (m, 2H), 6.75 (s, 2H), 6.43 (s, 1H), 3.84 (s, 6H). 13C NMR (125.8 MHz, CDCl3) δ 161.2, 142.5, 138.0, 126.6, 126.3, 120.9, 105.0, 99.3, 55.6. IR (dry film): 3000, 1201, 838 cm−1. HRMS (ES−) calcd. for C12H11O2S (M−H) 219.0480, found 219.0471.
3-(4-Methoxyphenyl)pyridine (Table 2, entry 4, aryl chloride in second step).33
General procedure A was employed. Column chromatography (0–50% EtOAc/hexane) provided the title compound as a pale yellow solid in 70% yield (194 mg). Spectral data were in accordance with those of published results. mp 64–66 °C. 1H NMR (500 MHz, CDCl3) δ 8.82 (d, J = 1.7 Hz, 1H), 8.55 (d, J = 3.9 Hz, 1H), 7.83 (d, J = 7.9 Hz, 1H), 7.52 (d, J = 8.7 Hz, 2H), 7.33 (dd, J = 7.8, 4.8 Hz, 1H), 7.02 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 159.9, 148.1, 148.0, 136.4, 134.0, 130.4, 128.4, 123.7, 114.7, 55.5.
3-(4-Methoxyphenyl)pyridine (Table 2, entry 4, aryl bromide in second step).33
General procedure A was employed. Column chromatography (0–50% EtOAc/hexane) provided the title compound as a pale yellow solid in 52% yield (144 mg). Spectral data were in accordance with those of published results. mp 64–66 °C. 1H NMR (500 MHz, CDCl3) δ 8.82 (s, 1H), 8.55 (d, J = 4.6 Hz, 1H), 7.83 (d, J = 7.9 Hz, 1H), 7.52 (d, J = 8.6 Hz, 2H), 7.33 (dd, J = 7.8, 4.8 Hz, 1H), 7.01 (d, J = 8.6 Hz, 2H), 3.86 (s, 4H). 13C NMR (125.8 MHz, CDCl3) δ 159.9, 148.2, 148.0, 136.4, 133.9, 130.4, 128.4, 123.6, 114.7, 55.5.
3-(4-Methoxyphenyl)pyridine (Table 2, entry 5, aryl chloride in second step).33
General procedure A was employed. Column chromatography (0–50% EtOAc/hexane) provided the title compound as a pale yellow solid in 82% yield (229 mg). Spectral data were in accordance with those of published results. mp 63–65 °C. 1H NMR (500 MHz, CDCl3) δ 8.82 (d, J = 1.9 Hz, 1H), 8.55 (d, J = 3.7 Hz, 1H), 7.83 (dd, J = 7.9, 1.7 Hz, 1H), 7.52 (d, J = 8.7 Hz, 2H), 7.34 (dd, J = 7.8, 4.8 Hz, 1H), 7.02 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H).13C NMR (125.8 MHz, CDCl3) δ 159.9, 148.2, 148.0, 136.4, 134.0, 130.4, 128.4, 123.7, 114.7, 55.5.
3-(4-Methoxyphenyl)pyridine (Table 2, entry 5, aryl bromide in second step).33
General procedure A was employed. Column chromatography (0–50% EtOAc/hexane) provided the title compound as a pale yellow solid in 80% yield (223 mg). Spectral data were in accordance with those of published results. mp 63–65 °C. 1H NMR (500 MHz, CDCl3) δ 8.82 (d, J = 1.7 Hz, 1H), 8.55 (dd, J = 4.8, 1.6 Hz, 1H), 7.85-7.82 (m, 1 H), 7.54 – 7.48 (m, 2H), 7.35-7.33 (m, 1 H), 7.03 – 6.97 (m, 2H), 3.85 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 159.9, 148.1, 148.0, 136.4, 133.9, 130.4, 128.4, 123.6, 114.7, 55.5.
4'-(1H-Pyrrol-1-yl)-[1,1'-biphenyl]-2-carbonitrile (Table 3, entry 1A)
General procedure A was employed. Column chromatography (1:9 EtOAc/hexane) provided the title compound as off-white crystals in 80% yield (294 mg). mp = 100 – 101 °C. 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 7.7 Hz, 1H), 7.66 (dd, J = 16.1, 8.1 Hz, 3H), 7.53 (t, J = 9.1 Hz, 3H), 7.46 (t, J = 7.6 Hz, 1H), 7.21 – 7.11 (m, 2H), 6.49 – 6.34 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 144.5, 141.0, 135.3, 133.9, 133.0, 130.1, 130.0, 127.8, 120.5, 119.3, 118.8, 111.2, 111.0. IR (dry film): 3144, 3058, 2882, 2359, 2225, 1608, 1524 cm−1. HRMS (ES+) calcd. for C17H13N2 (M+1) 245.1079, found 245.1074.
4'-(1H-Pyrrol-1-yl)-[1,1'-biphenyl]-2-yl)(morpholino)methanone (Table 3, entry 2A)
General procedure A was employed. Column chromatography (1:9 → 1:1 EtOAc/hexane) provided the title compound as off-white crystals in 74% yield (368 mg). 1H NMR (500 MHz, CDCl3) δ 7.58 – 7.35 (m, 8H), 7.14 (s, 2H), 6.38 (s, 2H), 3.59 (dt, J = 35.5, 13.1 Hz, 3H), 3.36 – 3.25 (m, 2H), 3.05 – 2.95 (m, 1H), 2.81 – 2.62 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 170.4, 169.9, 140.3, 137.6, 136.9, 135.5, 134.9, 130.1, 130.0, 129.8, 129.3, 128.7, 128.1, 127.9, 127.2, 120.1, 119.2, 111.1, 66.9, 66.2, 46.9, 41.9. IR (dry film): 3101, 3041, 2986, 2851, 2320, 1618 cm−1. HRMS (ES+) calcd. for C21H21N2O2 (M+1) 333.1603, found 333.1594.
1-(4'-(1H-Pyrrol-1-yl)-[1,1'-biphenyl]-2-yl)ethanone (Table 3, entry 3A)
General procedure A was employed. Column chromatography (0–10% EtOAc/hexane) provided the title compound as a yellow solid in 59% yield (230 mg). mp 119–121 °C. 1H NMR (500 MHz, CDCl3) δ 7.61 – 7.52 (m, 2H), 7.49 – 7.44 (m, 3H), 7.41 (m, 3H), 7.16 (t, J = 2.2 Hz, 2H), 6.39 (t, J = 2.2 Hz, 2H), 2.12 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 204.7, 140.9, 140.5, 139.7, 138.1, 131.0, 130.4, 130.2, 128.2, 127.8, 120.5, 119.3, 110.9, 30.7. IR (dry film): 2977, 1673, 1613, 833, 761 cm−1. HRMS (CI+) calcd. for C18H16NO (M+H) 262.1232, found 262.1220.
1-(2',6'-Dimethyl-[1,1'-biphenyl]-4-yl)-1H-pyrrole (Table 3, entry 4A)
General procedure A was employed. Column chromatography (1:19 EtOAc/hexane) provided the title compound as pale yellow crystals in 57% yield (211 mg). mp = 88 – 89 °C. 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J = 8.4 Hz, 2H), 7.22 – 7.11 (m, 7H), 6.38 (t, J = 2.1 Hz, 2H), 2.07 (s, 6H). 13C NMR (125.8 MHz, CDCl3) δ 141.0, 139.4, 138.5, 136.3, 130.3, 127.5, 127.4, 120.5, 119.3, 110.5, 21.0. IR (dry film): 3065, 2960, 2396, 2342, 1610, 1518 cm−1. HRMS (ES+) calcd. for C18H17N (M+1) 248.1439, found 248.1432.
1-(3',5'-Bis(trifluoromethyl)-[1,1'-biphenyl]-4-yl)-1H-pyrrole (Table 3, entry 5A).34
General procedure A was employed. Column chromatography (1:19 EtOAc/hexane) provided the title compound as pale yellow crystals in 94% yield (499 mg). mp = 156 – 157 °C. 1H NMR (500 MHz, CDCl3) δ 8.03 (s, 2H), 7.88 (s, 1H), 7.68 (d, J = 8.1 Hz, 2H), 7.54 (d, J = 8.1 Hz, 2H), 7.17 (s, 2H), 6.41 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 142.4, 141.3, 135.4, 132.8, 132.5, 132.3, 132.0, 128.6, 127.1, 126.8, 124.6, 122.4, 121.1, 121.1, 121.1, 121.0, 120.9, 120.3, 119.2, 111.2. IR (dry film): 3142, 3104, 3021, 1901, 1817, 1613 cm−1. HRMS (ES+) calcd. for C18H12F6N (M+1) 356.0874, found 356.0866.
1-(2',6'-Difluoro-[1,1'-biphenyl]-4-yl)-1H-pyrrole (Table 3, entry 6A)
General procedure A was employed. Column chromatography (1:19 EtOAc/hexane) provided the title compound as off-white crystals in 86% yield (328 mg). mp = 143 – 144 °C. 1H NMR (500 MHz, CDCl3) δ 7.55 (d, J = 8.1 Hz, 2H), 7.49 (d, J = 8.5 Hz, 2H), 7.30 (ddd, J = 14.6, 8.3, 6.5 Hz, 1H), 7.18 – 7.13 (m, 2H), 7.01 (t, J = 7.8 Hz, 2H), 6.41 – 6.35 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 156.3, 156.2, 154.3, 154.2, 135.6, 126.7, 124.2, 124.1, 124.0, 121.5, 115.3, 114.4, 112.7, 107.0, 106.9, 106.8, 106.8, 105.8. IR (dry film): 3144, 3043, 2926, 1901, 1613, 1531 cm−1. HRMS (ES+) calcd. for C16H12F2N (M+1) 256.0938, found 256.0949.
[1,1'-Biphenyl]-2-carbonitrile (Table 4, entry 1, route A).22
General procedure A was employed. Column chromatography (1:49 EtOAc/hexane) provided the title compound as a clear colorless oil in 25% yield (81 mg). Spectral data were in accordance with those of published results. 1H NMR (500 MHz, CDCl3) δ 7.77 (dd, J = 7.5, 1.6 Hz, 1H), 7.65 (td, J = 7.7, 1.4 Hz, 1H), 7.59 – 7.55 (m, 2H), 7.52 (dd, J = 8.7, 1.0 Hz, 1H), 7.51 – 7.48 (m, 2H), 7.48 – 7.42 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 145.6, 138.3, 133.9, 132.9, 130.2, 129.6, 128.9, 128.9, 127.7, 118.8, 111.4.
[1,1'-Biphenyl]-2-carbonitrile (Table 4, entry 1, route B).22
General procedure A was employed. Column chromatography (1:49 EtOAc/hexane) (1:49 → 1:19 EtOAc/hexane) provided the title compound as clear colorless oil in 69% yield (185 mg). Spectral data were in accordance with those of published results. 1H NMR (500 MHz, CDCl3) δ 7.79 – 7.76 (m, 1H), 7.65 (td, J = 7.7, 1.3 Hz, 1H), 7.59 – 7.55 (m, 2H), 7.54 – 7.48 (m, 3H), 7.45 (tdd, J = 7.8, 3.4, 1.2 Hz, 2H). 13C NMR (125.8 MHz, CDCl3) δ 145.6, 138.3, 133.9, 132.9, 130.2, 128.9, 128.8 (2), 127.7, 118.8, 111.4.
[1,1'-Biphenyl]-4-amine (Table 4, entry 2, route A).31
General procedure A was employed. Column chromatography (1:2 → 1:1 EtOAc/hexane) provided the title compound as pale yellow crystals in 60% yield (150 mg). Spectral data were in accordance with those of published results. mp = 49 – 45 °C. 1H NMR (500 MHz, CDCl3) δ 7.55 (d, J = 7.2 Hz, 2H), 7.47 – 7.38 (m, 4H), 7.28 (t, J = 7.4 Hz, 1H), 6.77 (d, J = 8.3 Hz, 2H), 3.69 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 145.9, 141.2, 131.5, 128.7, 128.0, 126.4, 126.3, 115.5.
[1,1'-Biphenyl]-4-amine (Table 4, entry 2, route B).31
General procedure A was employed. Column chromatography (1:4 → 1:2 EtOAc/hexane) provided the title compound as pale brown crystals in 80% yield (200 mg). Spectral data were in accordance with those of published results. mp = 50 – 51 °C. 1H NMR (500 MHz, CDCl3) δ 7.62 (d, J = 7.7 Hz, 2H), 7.53 – 7.41 (m, 4H), 7.34 (t, J = 7.2 Hz, 1H), 6.79 (d, J = 7.7 Hz, 2H), 3.74 (brs, 2H). 13C NMR (125.8 MHz, CDCl3) δ 146.0, 141.2, 131.5, 128.7, 128.1, 126.5, 126.3, 115.4.
3-(Thiophen-3-yl)phenol (Table 4, entry 3, route B)
General procedure A was employed. Column chromatography (0–10% EtOAc/hexane) provided the title compound as offwhite crystals in 91% yield (240 mg). mp 95–97 °C. 1H NMR (500 MHz, CDCl3) δ 7.45 (dd, J = 2.8, 1.4 Hz, 1H), 7.38-7.28 (m, 2H), 7.20-7.19 (m, 1H), 7.10 – 7.08 (m, 1H), 6.80 – 6.75 (m, 1H), 4.75 (s, 1H). 13C NMR (125.8 MHz, CDCl3) δ155.95, 142.0, 137.7, 130.2, 126.5, 126.4, 120.8, 119.3, 114.2, 113.5. IR (dry film): 2365, 1581, 1219, 769 cm−1. HRMS (CI+) calcd. for C10H9OS (M+H) 177.0374, found 177.0370.
2-Methyl-4-(4-(trifluoromethyl)phenyl)quinolone (Table 4, entry 4, route A).24
General procedure B was employed. Column chromatography (0–5% MeOH/CH2Cl2) provided the title compound as off-white crystals in 63% yield (275 mg). Spectral data were in accordance with those of published results. mp 104–106 °C. 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 8.0 Hz, 2H), 7.78 – 7.70 (m, 2H), 7.62 (d, J = 8.0 Hz, 2H), 7.46 (t, J = 7.5 Hz, 1H), 7.23 (s, 1H), 2.80 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 158.7, 148.5, 147.1, 141.9, 131.1, 130.8, 130.6, 130.3, 130.0, 129.7, 129.4, 126.3, 125.65 (q, J = 3.7 Hz), 122.3, 25.5.
2-Methyl-4-(4-(trifluoromethyl)phenyl)quinolone (Table 4, entry 4, route B).24
General procedure A was employed. Column chromatography (0–5% MeOH/CH2Cl2) provided the title compound as a white solid in 50% yield (215 mg). Spectral data were in accordance with those of published results. mp 105–107 °C. 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 7.7 Hz, 2H), 7.77 – 7.69 (m, 2H), 7.62 (d, J = 7.7 Hz, 2H), 7.46 (t, J = 7.6 Hz, 1H), 7.23 (s, 1H), 2.80 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 158.7, 148.5, 147.1, 141.9, 130.8, 130.6, 130.0, 129.8, 129.4, 126.3, 125.6 (q, J = 3.8), 125.2, 124.8, 122.3, 25.5.
3-(1H-Indol-6-yl)phenol (Table 4, entry 5, route A)
General procedure A was employed. Column chromatography (1:4 → 1:1 EtOAc/hexane) provided the title compound as light brown crystals in 71% yield (223 mg). mp = 46 – 47 °C. 1H NMR (500 MHz, CDCl3) 8.06 (s, 1H), 7.85 (s, 1H), 7.45 – 7.37 (m, 1H), 7.36 – 7.23 (m, 3H), 7.16 – 7.06 (m, 2H), 6.83 – 6.76 (m, 2H), 6.59 (s, 1H). 13C NMR (125.8 MHz, CDCl3) δ 155.8, 144.4, 135.5, 132.9, 130.0, 128.4, 125.2, 121.9, 120.1, 119.3, 114.4, 113.4, 111.4, 103.0. IR (dry film): 3400, 3258, 3100, 3051 2364, 2343, 1932, 1616 cm−1. HRMS (ES+) calcd. for C14H12NO (M+1) 210.0919, found 210.0910.
3-(1H-Indol-6-yl)phenol (Table 4, entry 5, route B)
General procedure A was employed. Column chromatography (1:4 → 1:1 EtOAc/hexane) provided the title compound as light brown crystals in 15% yield (51 mg). mp = 46 – 47 °C. 1H NMR (500 MHz, CDCl3) δ 8.06 (s, 1H), 7.85 (s, 1H), 7.45 – 7.37 (m, 1H), 7.36 – 7.23 (m, 3H), 7.16 – 7.06 (m, 2H), 6.83 – 6.76 (m, 2H), 6.59 (s, 1H). 13C NMR (125.8 MHz, CDCl3) δ 155.8, 144.4, 135.5, 132.9, 130.0, 128.4, 125.2, 121.9, 120.1, 119.3, 114.4, 113.4, 111.4, 103.0. IR (dry film): 3400, 3258, 3100, 3051 2364, 2343, 1932, 1616 cm−1. HRMS (ES+) calcd. for C14H12NO (M+1) 210.0919, found 210.0910.
tert-Butyl 5-(4-Methoxyphenyl)-1H-indole-1-carboxylate (Table 4, entry 6, route A).35
General procedure A was employed. Column chromatography (1:9 → 1:1 EtOAc/hexane) provided the title compound as a clear colorless oil in 67% yield (325 mg) plus 18% yield (60 mg) of 5-(4-methoxyphenyl)-1H-indole. Spectral data were in accordance with those of published results. 1H NMR (500 MHz, CDCl3) δ 8.23 (s, 1H), 7.76 (s, 1H), 7.67 (s, 1H), 7.61 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.5 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 3.0 Hz, 1H), 3.87 (s, 3H), 1.73 (s, 9H). 13C NMR (125.8 MHz, CDCl3) δ 159.1, 149.9, 135.9, 134.4, 131.3, 128.5, 127.9, 126.6, 123.7, 119.1, 115.5, 114.4, 107.7, 83.9, 55.5, 28.4. 5-(4-Methoxyphenyl)-1H-indole: 1H NMR (500 MHz, CDCl3) δ 8.16 (s, 1H), 7.81 (s, 1H), 7.61 – 7.55 (m, 2H), 7.48 – 7.39 (m, 2H), 7.25 – 7.22 (m, 1H), 7.03 – 6.96 (m, 2H), 6.61 – 6.56 (m, 1H), 3.86 (s, 3H).
tert-Butyl 5-(4-Methoxyphenyl)-1H-indole-1-carboxylate (Table 4, entry 6, route B).35
General procedure A was employed. Column chromatography (1:20 → 1:4 EtOAc/hexane) provided the title compound as a pale yellow oil in 67% yield (327 mg). Spectral data were in accordance with those of published results. 1H NMR (500 MHz, CDCl3) δ 8.15 (s, 1H), 7.72 (s, 1H), 7.64 – 7.55 (m, 2H), 7.52 (dd, J = 8.6, 1.6 Hz, 1H), 7.26 (s, 1H), 6.99 (d, J = 8.7 Hz, 2H), 6.61 (d, J = 3.5 Hz, 1H), 3.86 (s, 3H), 1.69 (s, 9H). 13C NMR (125.8 MHz, CDCl3) δ 159.0, 149.9, 135.8, 134.4, 131.3, 128.4, 127.9, 126.6, 123.6, 119.0, 115.4, 114.3, 107.6, 83.8, 55.5, 28.4.
3-(4-(Trifluoromethyl)phenyl)quinolone (Table 4, entry 7, route A).24
General procedure B was employed. Column chromatography (0–10% EtOAc/hexane) provided the title compound as a pale yellow solid in 60% yield (245 mg). mp = 135–137 °C. 1H NMR (500 MHz, CDCl3) δ 9.18 (d, J = 2.2 Hz, 1H), 8.34 (d, J = 1.7 Hz, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.1 Hz, 1H), 7.85 – 7.81 (m, 2H), 7.81 – 7.75 (m, 3H), 7.62 (t, J = 7.5 Hz, 1H). 13C NMR (125.8 MHz, CDCl3) δ 149.6, 147.9, 141.6, 133.9, 132.6, 130.5, 130.1, 129.5, 128.3, 127.9, 127.8, 127.5, 126.3 (q, J = 3.8 Hz), 125.4, 123.2, 100.1. IR (dry film): 2912, 2365. HRMS (ES+) calcd. for C16H11F3N: 274.0844 (M+H), found 274.0857.
3-(4-(Trifluoromethyl)phenyl)quinolone (Table 4, entry 7, route B).24
General procedure B was employed. Column chromatography (1:9 → 1:2 EtOAc/hexane) provided the title compound as white crystals in 99% yield (420 mg). Spectral data were in accordance with those of published results. mp = 139 – 140 °C. 1H NMR (500 MHz, CDCl3) δ 9.18 (d, J = 2.3 Hz, 1H), 8.34 (d, J = 1.8 Hz, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.87 – 7.73 (m, 5H), 7.62 (t, J = 8.0 Hz, 1H). 13C NMR (125.8 MHz, CDCl3) δ 149.6, 147.9, 141.6, 133.9, 132.5, 130.5, 130.2, 130.1, 129.5, 128.2, 127.9, 127.9, 127.5, 126.3, 126.3, 126.2, 126.2, 125.3, 123.2.
3-(6-Fluoropyridin-3-yl)phenol (Table 4, entry 8, route B)
General procedure B was employed. Column chromatography (1:2 → 1:1 EtOAc/hexane) provided the title compound as white crystals in 96% yield (273 mg). mp = 112 – 113 °C. 1H NMR (500 MHz, CDCl3) δ 8.43 (d, J = 2.5 Hz, 1H), 7.98 (td, J = 8.3, 2.6 Hz, 1H), 7.35 (t, J = 7.9 Hz, 1H), 7.11 – 7.01 (m, 3H), 6.93 (dd, J = 8.1, 2.0 Hz, 1H), 6.29 (s, 1H). 13C NMR (125.8 MHz, CDCl3) δ 164.2, 162.3, 156.9, 145.5, 145.4, 140.3, 140.3, 138.1, 135.0, 135.0, 130.6, 119.3, 115.6, 114.2, 110.0, 109.7. IR (dry film): 3241 br, 3040, 2359, 2339, 1595, 1579 cm−1. HRMS (ES+) calcd. for C11H9FNO (M+1) 190.0668, found 190.0663.
3-(Pyridin-3-yl)benzonitrile (Table 5, entry 1)
General procedure A was employed. Column chromatography (1:20 EtOAc/hexane) provided the title compound as off-white crystals in 88% yield (232 mg). mp = 87 – 88 °C. 1H NMR (500 MHz, CDCl3) δ 8.75 (d, J = 82.0 Hz, 2H), 7.86 – 7.42 (m, 6H). 13C NMR (125.8 MHz, CDCl3) δ 149.7, 148.3, 139.3, 134.6, 134.5, 131.7, 131.6, 130.8, 130.1, 123.9, 118.6, 113.6. IR (dry film): 3067, 3007, 2354, 2232, 1582, 1466 cm−1. HRMS (ES+) calcd. for C12H9N2 (M+1) 181.0766, found 181.0759.
4-(5-Methoxypyridin-3-yl)benzonitrile (Table 5, entry 2)
General procedure A was employed with the only modification being that 1.3 equivalents of the second halide were employed instead of 1.0 equivalent. Column chromatography (1:9 → 1:3 EtOAc/hexane) provided the title compound as a white powder in 87% yield (275 mg). mp = 122 – 123 °C. 1H NMR (500 MHz, CDCl3) δ 8.46 (s, 1H), 8.37 (s, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 4.6 Hz, 1H), 3.94 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 156.0, 142.3, 140.5, 137.3, 135.7, 133.0, 128.1, 119.4, 118.7, 112.2, 55.9. HRMS (ES+) calcd. for C13H11N2O (M+1) 211.0871, found 211.0864.
Methyl 4-(6-Cyanopyridin-3-yl)benzoate (Table 5, entry 3).36
General procedure A was employed. Column chromatography (1:4 → 1:3 EtOAc/hexane) provided the title compound as white crystals in 73% yield (206 mg). Spectral data were in accordance with those of published results. mp = 157 – 158 °C. 1H NMR (500 MHz, CDCl3) δ 8.97 (d, J = 2.3 Hz, 1H), 8.19 (d, J = 8.2 Hz, 2H), 8.05 (dd, J = 8.1, 2.3 Hz, 1H), 7.81 (d, J = 8.1 Hz, 1H), 7.75 – 7.60 (m, 2H), 3.96 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 166.5, 149.8, 140.2, 138.9, 135.3, 133.2, 131.1, 130.8, 128.6, 127.5, 117.3, 52.6. HRMS (ES+) calcd. for C14H11N2O2 (M+1) 239.0821, found 239.0814.
Methyl 4-(Pyrimidin-5-yl)benzoate (Table 5, entry 4).37
General procedure A was employed. Column chromatography (1:2 EtOAc/hexane) provided the title compound as white crystals in 87% yield (280 mg). Spectral data were in accordance with those of published results. mp = 142 – 143 °C. 1H NMR (500 MHz, CDCl3) δ 9.25 (s, 1H), 9.00 (s, 2H), 8.19 (d, J = 8.6 Hz, 2H), 7.67 (d, J = 8.6 Hz, 2H), 3.96 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 166.6, 158.3, 155.2, 138.7, 130.8, 127.1 (2), 100.1, 52.5. HRMS (ES+) calcd. for C12H11N2O2 (M+1) 215.0821, found 215.0820.
3-(4-Methoxyphenyl)furan (Table 5, entry 5).38
General procedure A was employed. Column chromatography (1:49 EtOAc/hexane) provided the title compound as white crystals in 67% yield (176 mg). Spectral data were in accordance with those of published results. mp = 55 – 56 °C. 1H NMR (500 MHz, CDCl3) δ 7.55 – 7.51 (m, 2H), 7.39 – 7.32 (m, 3H), 6.96 – 6.92 (m, 2H), 3.84 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 159.0, 142.2, 128.9, 127.7, 126.4, 126.2, 119.1, 114.4, 55.5.
2-(4-Methoxyphenyl)thiophene (Table 5, entry 6).38
General procedure A was employed. Column chromatography (1:19 EtOAc/hexane) provided the title compound as white crystals in 96% yield (275 mg). Spectral data were in accordance with those of published results. mp = 105 – 106 °C. 1H NMR (500 MHz, CDCl3) δ 7.61 – 7.48 (m, 2H), 7.24 – 7.19 (m, 2H), 7.06 (dd, J = 5.1, 3.6 Hz, 1H), 6.97 – 6.86 (m, 2H), 3.84 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 159.3, 144.5, 128.1, 127.4 (2), 124.0, 122.2, 114.4, 55.5. HRMS (ES+) calcd. for C11H11OS (M+1) 191.0531, found 191.0540.
1-(5-(4-Methoxyphenyl)thiophen-2-yl)ethanone (Table 5, entry 7).39
General procedure A was employed with the only modification being that 1.3 equivalents of the second halide were employed instead of 1.0 equivalent. Column chromatography (1:19 → 1:5 EtOAc/hexane) provided the title compound as pale yellow crystals in 83% yield (288 mg). Spectral data were in accordance with those of published results. mp = 149 – 150 °C. 1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 4.0 Hz, 1H), 7.61 – 7.56 (m, 2H), 7.21 (d, J = 4.0 Hz, 1H), 6.96 – 6.92 (m, 2H), 3.85 (s, 3H), 2.55 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 190.8, 160.8, 153.3, 142.5, 134.0, 128.0, 126.5, 123.2, 114.9, 55.8, 26.8.
2-(4-Methoxyphenyl)-5-methylthiophene (Table 5, entry 8).40
General procedure A was employed with the only modification being that 1.3 equivalents of the second halide were employed instead of 1.0 equivalent. Column chromatography (1:4 → 1:3 EtOAc/hexane) provided the title compound as white crystals in 67% yield (204 mg). Spectral data were in accordance with those of published results. mp = 94 – 95 °C. 1H NMR (500 MHz, CDCl3) δ 7.49 – 7.45 (m, 2H), 6.98 (d, J = 3.5 Hz, 1H), 6.91 – 6.87 (m, 2H), 6.69 (dd, J = 3.5, 1.1 Hz, 1H), 3.83 (s, 3H), 2.49 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 159.0, 142.1, 138.6, 127.8, 126.9, 126.2, 122.0, 114.4, 55.5, 15.6.
4-(3,5-Dimethoxyphenyl)-2,6-dimethoxypyrimidine (Table 5, entry 9)
General procedure A was employed. Column chromatography (1:4 → 1:1 EtOAc/hexane) provided the title compound as off-white crystals in 29% yield (120 mg). 1H NMR (500 MHz, CDCl3) δ 7.18 (d, J = 2.1 Hz, 2H), 6.72 (s, 1H), 6.55 (s, 1H), 4.06 (s, 3H), 3.99 (s, 3H), 3.83 (s, 6H). 13C NMR (125.8 MHz, CDCl3) δ 172.9, 165.7, 161.4, 138.9, 105.1, 102.6, 97.0, 55.0, 54.1, 53.4. HRMS (ES+) calcd. for C14H17N2O4 (M+H) 277.1188, found 277.1184.
Methyl 3-(Pyridin-2-yl)benzoate (Table 5, entry 10).41
General procedure A was employed. Column chromatography (1:9 → 1:4 EtOAc/hexane) provided the title compound as white crystals in 49% yield (157 mg). Spectral data were in accordance with those of published results. mp = 49 – 50 °C. 1H NMR (500 MHz, CDCl3) δ 8.73 – 8.69 (m, 1H), 8.66 – 8.61 (m, 1H), 8.25 – 8.22(m, 1H), 8.11 – 8.07 (m, 1H), 7.81 – 7.74 (m, 2H), 7.58 – 7.53 (m, 1H), 7.28 – 7.23 (m, 1H), 3.95 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 167.1, 156.5, 149.9, 139.8, 137.0, 131.5, 130.8, 130.1, 129.0, 128.1, 122.7, 120.7, 52.3.
2-(p-Tolyl)benzoxazole (Table 5, entry 11).42
General procedure A was employed. Column chromatography (1:19 EtOAc/hexane) provided the title compound as pale yellow crystals in 32% yield (100 mg). Spectral data were in accordance with those of published results. mp = 105 – 106 °C. 1H NMR (500 MHz, CDCl3) δ 8.15 (d, J = 8.2 Hz, 2H), 7.86 – 7.67 (m, 1H), 7.69 – 7.48 (m, 1H), 7.37 – 7.33 (m, 4H), 2.45 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 163.5, 150.8, 142.2, 129.8 (2), 127.8 (2), 125.0, 124.6, 120.0, 110.6, 21.8.
4-(4-Methoxyphenyl)-7-(trifluoromethyl)quinoline (Table 5, entry 12)
General procedure A was employed. Column chromatography (1:20 EtOAc/hexane) provided the title compound as white crystals in 87% yield (397 mg). mp = 125 – 126 °C. 1H NMR (500 MHz, CDCl3) δ 9.01 (d, J = 4.4 Hz, 1H), 8.47 (s, 1H), 8.10 (d, J = 8.8 Hz, 1H), 7.67 (dd, J = 8.8, 1.8 Hz, 1H), 7.48 – 7.40 (m, 3H), 7.13 – 7.04 (m, 2H), 3.92 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 160.4, 151.9, 151.5, 148.5, 148.0, 136.0, 131.5, 131.3, 131.0, 130.9, 129.6, 129.2, 128.7, 127.9, 127.9, 127.8, 127.5, 125.2, 123.0, 122.2, 114.4, 55.6. IR (dry film): 3065, 2980, 2360, 1609, 1505 cm−1. HRMS (ES+) calcd. for C17H13F3NO (M+1) 304.0949, found 304.0949.
One-Pot (Scheme 3)
General procedure A was employed with the following modifications: 1) the first step was performed on 4.5 mmol scale and 2) all aryl halides (1.5 mmol) were subsequently added one after the other to the same pot 3) one column was performed on the crude mixture of all three compounds (1:20 → 1:3 → 1:1 EtOAc/hexane) to provide the title compounds.
3-(Pyrimidin-5-yl)phenol (Scheme 3, product 1)
was obtained as white crystals in 80% yield (203 mg). 1H NMR (500 MHz, acetone-d6) δ 9.13 (s, 1H), 9.01 (s, 2H), 8.79 (s, 1H), 7.37 (t, J = 7.8 Hz, 1H), 7.24 – 7.17 (m, 2H), 6.97 (d, J = 8.1 Hz, 1H). 13C NMR (125.8 MHz, acetone-d6) δ 159.1, 158.2, 155.5, 136.7, 134.9, 131.3, 119.0, 116.7, 114.6. IR (dry film): 3338, 3056, 2808, 2678, 2363, 2337, 1584, cm−1. HRMS (ES+) calcd. for C10H9N2O (M+1) 173.0715, found 173.0708.
3-(Quinolin-3-yl)phenol (Scheme 3, product 2)43
was obtained as pale yellow crystals in 93% yield (310 mg). Spectral data were in accordance with those of published results. 1H NMR (500 MHz, acetone-d6 + two drops DMSO-d6) δ 9.33 (s, 1H), 9.20 (d, J = 2.1 Hz, 1H), 8.52 – 8.46 (m, 1H), 8.09 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 8.1 Hz, 1H), 7.79 – 7.71 (m, 1H), 7.62 (t, J = 7.4 Hz, 1H), 7.36 (t, J = 7.8 Hz, 1H), 7.29 (dd, J = 9.9, 4.7 Hz, 2H), 6.98 – 6.90 (m, 1H). 13C NMR (125.8 MHz, acetone-d6 + two drops DMSO-d6) δ 158.8, 149.8, 147.6, 139.2, 133.9, 132.9, 130.4, 129.4, 129.2, 128.5, 128.3, 127.1, 118.3, 115.5, 114.4.
4'-(Trifluoromethyl)-[1,1'-biphenyl]-3-ol (Scheme 3, product 3)44
was obtained as off-white crystals in 93% yield (331 mg). Spectral data were in accordance with those of published results. 1H NMR (500 MHz, CDCl3) δ 7.65 (q, J = 7.9 Hz, 4H), 7.36 (t, J = 7.8 Hz, 1H), 7.19 (d, J = 7.4 Hz, 1H), 7.10 (s, 1H), 6.92 (d, J = 7.6 Hz, 1H), 5.71 (s, 1H). 13C NMR (125.8 MHz, CDCl3) δ 156.0, 144.2, 141.7, 130.5, 129.7 (q, J = 32.5 Hz), 127.5, 125.9 (q, J = 3.6 Hz), 124.5 (q, J = 271.9 Hz), 120.2, 115.4, 114.5.
Teraryl Synthesis (Scheme 4)
For the synthesis of Methyl 3'-hydroxy-[1,1'-biphenyl]-4-carboxylate (Scheme 4, product 4, see Table 1, entry 2).
Methyl 3'-(((Trifluoromethyl)sulfonyl)oxy)-[1,1'-biphenyl]-4-carboxylate (Scheme 4, product 5)
To methyl 3'-hydroxy-[1,1'-biphenyl]-4-carboxylate (114 mg, 0.5 mmol) in 1.5 mL CH2Cl2 at −5 °C was added triethylamine (290 mg, 210 µL, 1.5 mmol) prior to the dropwise addition of triflic anhydride (80 mg, 133 µL, 0.75 mmol). The reaction mixture was allowed to stir at −5 °C for 1 h before quenching the reaction with the slow addition of water (5 mL, cooled to −5 °C prior to addition). The organic layer was separated, and the aqueous layer was further extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered through a small pad of silica gel, concentrated, and dried in vacuo for 30 min. The crude product was used in the one-pot borylation/Suzuki-Miyaura reaction without further purification.
Methyl 3'-(Quinolin-3-yl)-[1,1'-biphenyl]-4-carboxylate (Scheme 4, product 6)
General procedure A was employed with the only modification being that the reaction was run on 0.36 mmol scale. Column chromatography (40–63 µm silica gel stationary phase, 1:9 EtOAc/hexane) provided the title compound as white crystals in 92% yield (113 mg). mp = 126 – 127 °C. 1H NMR (500 MHz, CDCl3) δ 9.22 (s, 1H), 8.35 (s, 1H), 8.20 – 8.09 (m, 3H), 7.97 – 7.83 (m, 2H), 7.79 – 7.53 (m, 8H), 3.95 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 167.1, 150.0, 147.7, 145.3, 141.2, 138.8, 133.7, 133.6, 130.4, 129.9, 129.7, 129.5, 129.4, 128.2, 128.1, 127.3, 127.3, 127.2, 126.6, 52.3. IR (dry film): 3043, 2955, 2369, 2336, 1719, 1610 cm−1. HRMS (ES+) calcd. for C23H18NO2 (M+1) 340.1338, found 340.1331.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by the NIGMS (R01 GM-081376) and an NSF GOALI Grant. SLJT is a Merck Doctoral Fellow, funded through the MRL Doctoral Program. We thank BASF for their generous donation of BBA. Rakesh Kohli (University of Pennsylvania) in acknowledged for his help in obtaining HRMS data. The authors also wish to thank Dr. Matthew T. Tudge (Merck Research Laboratories) for supplying the CataCXium A preformed catalyst.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
SUPPORTING INFORMATION
1H and 13C NMR spectra for all compounds synthesized. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- 1.Bellina F, Carpita A, Rossi R. Synthesis. 2004:2419–2440. [Google Scholar]
- 2.Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002;58:9633–9695. [Google Scholar]
- 3.Miyaura N, Yanagi T, Suzuki A. Synth. Commun. 1981:513–519. [Google Scholar]
- 4.Suzuki A. Acc. Chem. Res. 1982;15:178–184. [Google Scholar]
- 5.Biscoe MR, Fors BP, Buchwald SL. J. Am. Chem. Soc. 2008;130:6686–6687. doi: 10.1021/ja801137k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kinzel T, Zhang Y, Buchwald SL. J. Am. Chem. Soc. 2010;132:14073–14075. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knapp DM, Gillis EP, Burke MD. J. Am. Chem. Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molander GA, Canturk B. Angew. Chem. Int. Ed. 2009;48:9240–9261. doi: 10.1002/anie.200904306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown HC, Cole TE. Organometallics. 1983;2:1316–1319. [Google Scholar]
- 10.Deng H, Jung JK, Liu T, Kuntz KW, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2003;125:9032–9034. doi: 10.1021/ja030249r. [DOI] [PubMed] [Google Scholar]
- 11.Ishiyama T, Isou H, Kikuchi T, Miyaura N. Chem. Commun. 2010:159–161. doi: 10.1039/b910298a. [DOI] [PubMed] [Google Scholar]
- 12.Ishiyama T, Itoh Y, Kitano T, Miyaura N. Tetrahedron Lett. 1997;38:3447–3450. [Google Scholar]
- 13.Ishiyama T, Murata M, Miyaura N. J. Org. Chem. 1995;60:7508–7510. [Google Scholar]
- 14.Jung ME, Lazarova TI. J. Org. Chem. 1999;64:2976–2977. doi: 10.1021/jo9902751. [DOI] [PubMed] [Google Scholar]
- 15.Leermann T, Leroux FR, Colobert F. Org. Lett. 2011;13:4479–4481. doi: 10.1021/ol2016252. [DOI] [PubMed] [Google Scholar]
- 16.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem. Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 17.Pennington T, Kardiman C, Hutton C. Tetrahedron Lett. 2004;45:6657–6660. [Google Scholar]
- 18.Yang W, He H, Drueckhammer DG. Angew. Chem. Int. Ed. 2001;40:1714–1718. [PubMed] [Google Scholar]
- 19.Baudoin O, Cesario M, Guenard D, Gueritte F. J. Org. Chem. 2002;67:1199–1207. doi: 10.1021/jo0160726. [DOI] [PubMed] [Google Scholar]
- 20.Baudoin O, Guenard D, Gueritte F. J. Org. Chem. 2000;65:9268–9271. doi: 10.1021/jo005663d. [DOI] [PubMed] [Google Scholar]
- 21.Billingsley K, Barder T, Buchwald S. Angew. Chem. Int. Ed. 2007;46:5359–5363. doi: 10.1002/anie.200701551. [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Cui X, Zhu Z, Wu Y, Li J, Wu Y. Eur. J. Org. Chem. 2012:595–603. [Google Scholar]
- 23.Molander GA, Trice SLJ, Dreher SD. J. Am. Chem. Soc. 2010;132:17701–17703. doi: 10.1021/ja1089759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molander GA, Trice SLJ, Kennedy SM, Dreher SD, Tudge MT. J. Am. Chem. Soc. 2012;134:11667–116673. doi: 10.1021/ja303181m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stille JK, Lau KSY. Acc. Chem. Res. 1977;10:434–442. [Google Scholar]
- 26.Sau SC, Santra S, Sen TK, Mandal SK, Koley D. Chem. Commun. 2012;48:555–557. doi: 10.1039/c1cc15732a. [DOI] [PubMed] [Google Scholar]
- 27.Han Z, Obermann R, Nolan W, Wildman SA, Hobbs D, Ellenberger T, Janetka JW, Pinkner JS, Cusumano CK, Hultgren SJ, Ford B. J. Med. Chem. 2010;53:4779–4792. doi: 10.1021/jm100438s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han J, Liu Y, Guo R. J. Am. Chem. Soc. 2009;131:2060–2061. doi: 10.1021/ja808935n. [DOI] [PubMed] [Google Scholar]
- 29.Zhou G, Wu D, Kaur H, Gochin M, Snyder B, Ptak RG. J. Med. Chem. 2011;54:7220–7231. doi: 10.1021/jm200791z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alacid E, Najera C. Org. Lett. 2008;10:5011–5014. doi: 10.1021/ol802024j. [DOI] [PubMed] [Google Scholar]
- 31.Yamada YMA, Sarkar SM, Uozumi Y. J. Am. Chem. Soc. 2012;134:3190–3198. doi: 10.1021/ja210772v. [DOI] [PubMed] [Google Scholar]
- 32.Amatore M, Gosmini C. Angew. Chem. Int. Ed. 2008;47:2089–2092. doi: 10.1002/anie.200704402. [DOI] [PubMed] [Google Scholar]
- 33.Quasdorf KW, Riener M, Petrova KV, Garg NK. J. Am. Chem. Soc. 2009;131:17748–17749. doi: 10.1021/ja906477r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nad S, Roller S, Haag R, Breinbauer R. Org Lett. 2006;8:403–406. doi: 10.1021/ol052096d. [DOI] [PubMed] [Google Scholar]
- 35.Prieto M, Zurita E, Rosa E, Munoz L, Lloyd-Williams P, Giralt E. J. Org. Chem. 2004;69:6812–6820. doi: 10.1021/jo0491612. [DOI] [PubMed] [Google Scholar]
- 36.Bruton G, Rana KK, Huxley A, Orlek BS. WO2005/40144. Patent. 2005:A1.
- 37.Rong D, Phillips VA, Wheelhouse RT, Rubio RS, Angeles Castro M. Tetrahedron Lett. 2008;49:6933–6935. [Google Scholar]
- 38.Tang YQ, Lv H, Lu JM, Shao LX. J. Organomet. Chem. 2011;696:2576–2579. [Google Scholar]
- 39.Roger J, Pozgan F, Doucet H. Green Chem. 2009;11:425–432. [Google Scholar]
- 40.Join B, Yamamoto T, Itami K. Angew. Chem. Int. Ed. 2009;48:3644–3647. doi: 10.1002/anie.200806358. [DOI] [PubMed] [Google Scholar]
- 41.Gosmini C, Bassene-Ernst C, Durandetti M. Tetrahedron. 2009;65:6141–6146. [Google Scholar]
- 42.Ranjit S, Liu X. Chem. Eur. J. 2011;17:1105–1108. doi: 10.1002/chem.201002787. [DOI] [PubMed] [Google Scholar]
- 43.Frotscher M, Ziegler E, Marchais-Oberwinkler S, Kruchten P, Neugebauer A, Fetzer L, Scherer C, Muller-Vieira U, Messinger J, Thole H, Hartmann RW. J. Med. Chem. 2008;51:2158–2169. doi: 10.1021/jm701447v. [DOI] [PubMed] [Google Scholar]
- 44.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. J. Med. Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.