

SUMMARY
Background and Aims
Crown-like structures (CLS) are characteristic histopathology features of inflamed adipose tissues in obese mice and humans. In previous work, we suggested that these cells derived from macrophages primarily involved in the reabsorption of dead adipocytes. Here, we used a well-characterized transgenic mouse model in which the death of adipocytes in adult mice is inducible and highly synchronized. In this “FAT-ATTAC” model, apoptosis is induced through forced dimerization of a caspase-8 fusion protein.
Methods and Results
0, 0.5, 1, 2, 3 and 10 days post induction of adipocyte cell death, we analyzed mesenteric and epididymal adipose depots by histology, immunohistochemistry and electron microscopy. Upon induction of caspase-8 dimerization, numerous adipocytes lost immunoreactivity for perilipin, a marker for live adipocytes. In the same areas, we found adipocytes with hypertrophic mitochondria and signs of organelle degeneration. Neutrophils and lymphocytes were the main inflammatory cells present in the tissue, and the macrophages were predominantly Mac-2 negative. Over the course of ablation, Mac-2 positive macrophages substituted for Mac-2 negative macrophages, followed by CLS formation. All perilipin-negative, dead adipocytes were surrounded by CLS structures. The time course of histopathology was similar in both fat pads studied, but occurred at earlier stages and was more gradual in mesenteric fat.
Conclusion
Our data demonstrate that CLS formation results as a direct consequence of adipocyte death, and that infiltrating macrophages actively uptake remnant lipids of dead adipocytes. Upon induction of adipocyte apoptosis, inflammatory cells infiltrate adipose tissue initially consisting of neutrophils followed by macrophages that are involved in CLS formation.
Keywords: CLS, inflammation, fat, apoptosis, FAT-ATTAC, adipocytes
INTRODUCTION
The chronic subclinical inflammation of adipose tissue in obese mice and humans has been widely described in the literature [1, 2]. Macrophages have been identified as a strong mediator of adipose tissue inflammation and dysfunction. Macrophage infiltration into adipose tissue is of clinical importance due to the tight correlation with the onset of insulin resistance [3–7], a major component of the metabolic syndrome [8, 9]. While the presence of macrophages may dictate the tissue inflammatory environment subsequent to their infiltration, the underlying causes for the inflammatory phenotype of dysfunctional adipose tissue are not known. We hypothesized that inflammation is a direct consequence of adipocyte death [10]. Visceral adipose tissue is more inflamed during weight gain, suggesting that a higher rate of cell death occurs in the visceral pads compared to subcutaneous depot cells, possibly due to the fact that they are more prone to death [11]. Visceral fat accumulation is therefore more tightly linked to the metabolic syndrome [11–13]. Here we present new experimental data that strongly supports the hypothesis that macrophage infiltration is a primary and direct consequence of adipocyte death.
FAT-ATTAC mice are transgenic mice that undergo adipocyte specific apoptosis after dimerizer administration that forces dimerization and activation of caspase-8 [14, 15]. This tightly controlled, induced adipocyte apoptosis allows for high temporal resolution of the tissue events resulting from adipocyte death. We compared the morphology, immunohistochemistry and electron microscopy (EM) of two visceral fat depots upon dimerization and collected tissues at 0, 12, 24, 48, 72 hours and 10 days after dimerizer to resolve the initiation of immune cell infiltration and CLS formation. We demonstrate that CLS formation occurs specifically at sites of dead adipocytes whose remnants need to be resorbed, a process that occurs primarily by activated macrophages.
MATERIAL AND METHODS
Animals
The FAT ATTAC mice [15](“Fat Apotosis Through Targeted Activation of Caspase 8”) are a transgenic mouse model of inducible fat loss generated through the transgenic expression of a myristoylated caspase 8-FKBP fusion protein in adipocytes. Transgene expression is under the control of the aP2 promoter and is in this particular case expressed only in mature adipocytes [15]. The FKBP moiety was developed by Clackson and colleagues [16] that takes advantage of a mutated version of a FKBP-domain that selectively binds a chemical dimerizer (AP20187, a FK506 analogue) that force-dimerizes two mutant FKBP domains (but does not bind endogenous FKBP). In the FAT-ATTAC mice, treatment of adipocytes with AP20187 (Ariad Pharmaceuticals), forcedimerizes a membrane-associated caspase-8 that activates downstream signaling cascades resulting in selective apoptosis of adipocytes. This is a very specific way to induce apoptosis, since this is the only pathway known to be triggered by caspase-8 [15].
Six- to 8-wk old male FAT ATTAC mice (N = 11) in the genetic background of FVB mice and wild type (N = 8) were used. Mice were provided ad libitum access to standard chow diet and water and maintained on a 12 hour light/dark cycle. AP21087 was delivered on 4 consecutive days at 0.2μg/g body weight. Mice were sacrificed at the respective time points (7am or 7pm) after 0 (basal), 12, 24, 48, 72 hours and 10 days after dimerizer by exsanguination under isofluorane anesthesia. Cervical dislocation was used to verify death before tissues were collected.
Serum adiponectin analysis
Reduced circulating adiponectin was used to verify induction of adipocyte apoptosis. Approximately 10μL of blood was collected from the tail vein of mice at 2 and 8 days post-dimerization, the plasma was reduced, separated on a Criterion TGX gel (BioRad, Hercules, CA), and immunoblotted with rabbit anti-serum against adiponectin as previously described [17].
Light microscopy
Mesenteric and epididymal fat depots were dissected and fixed by immersion in 4% paraformaldehyde in 0.1 M sodium phosphate buffer, pH 7.4 overnight at 4°C, then dehydrated, cleared and paraffin-embedded. Three different biopsies of each depot were analysed. For each sample, 3 μm-thick serial sections were obtained: the first was stained by haematoxylin & eosin to assess morphology, the others were processed for immunohistochemistry. Tissue sections were observed with a Nikon Eclipse E800 light microscope using a x20 objective at x200 final magnification, and digital images were captured with a Nikon DXM 1200 camera.
Immunohistochemistry
For immunohistochemistry 3-μm dewaxed serial sections were incubated with anti-Mac-2/galectin-3 (Cedarlane Laboratories, Canada) and anti-perilipin (kindly provided by Andy Greenberg, Tufts University, Boston, USA) primary antibodies according to the ABC method [18]. We used 3% hydrogen peroxide to inactivate endogenous peroxidase followed by normal goat or horse serum to reduce non-specific staining. Consecutive serial sections were incubated overnight (4°C) with anti-Mac-2/galectin-3 (1:1000) and anti-perilipin (1:50) primary antibodies. Biotinylated HRP-conjugated secondary antibodies were goat anti-rabbit IgG (perilipin) and horse anti-mouse IgG (Mac-2/galectin-3; Vector Laboratories; Burlingame, CA, USA). Histochemical reactions were performed using Vector’s Vectastain ABC Kit and Sigma Fast 3,3′-diaminobenzidine as substrate (Sigma, St Louis, MO, USA). Sections were counterstained with haematoxylin.
Morphometry
CLS density was quantified by counting the total number of CLS in each section compared with the total number of adipocytes and was expressed as CLS number/10 000 adipocytes.
Adipocyte perilipin-negative density was obtained by counting the total number of perilipin negative adipocytes in each section compared with the total number of living adipocytes. Values are expressed as perilipin negative adipocyte number/10 000 adipocytes.
Electron Microscopy
Small tissue fragments were fixed in 2% glutaraldehyde-2% paraformaldehyde in 0.1 M PB, pH 7.4, for 4 h, postfixed in 1% osmium tetroxide and embedded in an Epon-Araldite mixture. Semithin sections (2 μm) were stained with toluidine blue; thin sections were obtained with an MT-X ultratome (RMC, Tucson, AZ, USA), stained with lead citrate and examined with a CM10 transmission electron microscope (Philips, Eindhoven, The Netherlands).
Inflammatory Cells Composition
Light and electron microscopy allowed us to distinguish the different inflammatory cells (lymphocytes, neutrophils and macrophages) infiltrating the fat depots. Mac-2 immunoreactivity was used to distinguish between populations of macrophages (Mac-2 positive, phagocytic active and Mac-2 negative, phagocytic inactive macrophages) [19]. Throughout this paper, we used the “classic” classification of the inflammatory cells [20]. Macrophages: The nucleus of macrophages is oval with several indentations. It contains several nucleoli and abundant, dense peripheral heterochromatin. The cell surface varies, depending on the location of the cell, but microvilli and pseudopods are numerous in the active phagocytic cells. The cytoplasm is abundant and contains a moderate amount of free ribosomes and a large amount of granular endoplasmic reticulum. The Golgi zone is prominent. Mitochondria are oval and few in number. The surface membrane has numerous pinocytotic invaginations, most of which are smooth but some are typically coated. There is no external lamina (basal lamina) associated with macrophages. The macrophage is filled with a varied number of vesicles, vacuoles, lysosomes, and residual bodies. Lymphocytes: Lymphocytes have a dense nucleus surrounded by a thin rim of cytoplasm. The nucleus is central, round, or slightly indented, very rich in randomly dispersed, heterochromatic masses; the nucleous is small. Electron microscopy shows a small Golgi apparatus and a few mitochondria. Free ribosomes in moderate numbers are scattered as single units throughout the cytoplasm; cisternae of the granular endoplasmic reticulum are found only exceptionally. Small numbers of lysosomes are also present. Neutrophils: The nucleus of neutrophils is multilobed in older cells. The lobes are connected by a narrow strand of nucleoplasm. Heavy masses of dense heterochromatin are distributed against the nuclear membrane, and the loosely arranged euchromatin is found mostly in the center. There is usually no well-delineated nucleolus. The cytoplasm contains a large number of membrane-bound granules. The cytoplasm contains a large number of glycogen particles, however, other organelles are scarce. The Golgi apparatus is rudimentary, and mitochondria are few in number and rod-shaped [20]. Inflammatory cells were counted in each section and compared with the total number of adipocytes. Results are represented as inflammatory cell density (inflammatory cells number/10 000 adipocytes), and as percentage of macrophage Mac-2 positive, macrophage Mac-2 negative, lymphocytes and neutrophils.
Statistical analysis
Results are given as mean ± standard error (SEM). Differences Statistical analysis were performerd by one-way ANOVA (InStat, GraphPad, San Diego, CA, USA). Significance was accepted at P≤0.05.
RESULTS
Adipose tissue morphology in both visceral tissues studied (mesenteric and epididymal) has a similar appearance in wild type and in FAT-ATTAC transgenic mice without dimerizer injection. Treatment with dimerizer did not cause weight loss in FAT-ATTAC mice (Fig. 1A) through our experimental time course, but did result in the selective apoptosis of adipocytes as evidenced by diminishing levels of adiponectin in circulation (Fig. 1B). The metabolic consequences of this system have been previously described [15].
Figure 1.
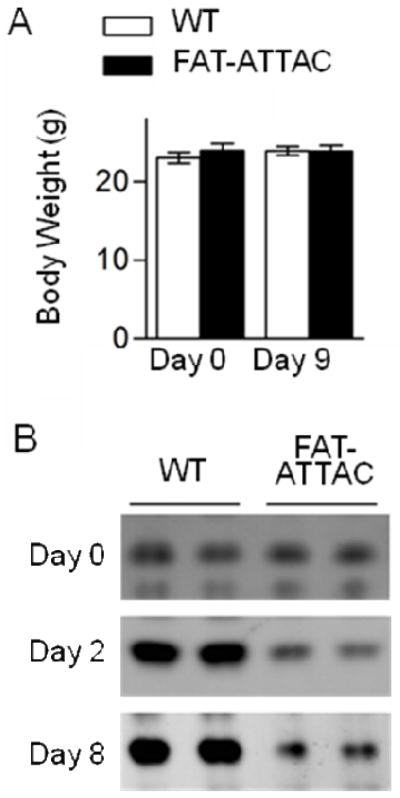
Average mouse body weights before and 9 days after initiation of dimerizer treatment in wild type (WT) and FAT-ATTAC mice demonstrate no change (N=12 samples) (A). Circulating adiponectin levels are greatly reduced following induced adipocyte apoptosis in FAT-ATTAC mice (B).
Induction of caspase-8 activation caused a progressive adipose tissue infiltration by inflammatory cells (Fig. 2A–B), and this infiltration was associated with a progressive increase in number of dead adipocytes (Fig. 2C–D), characterized by the loss for perilipin immunoreactivity [10, 15, 21, 22]. Of note, perilipin negative “adipocytes” (corresponding to remnants of adipocytes), were present at 12 and 24 hours in mesenteric fat and at 48 hours in epididymal fat of treated mice. Thereafter, CLS formation began suggesting that macrophages were recruited for the formation of CLS upon adipocyte death (Fig. 2C–D).
Figure 2.

Quantitative evaluation of all inflammatory cells in mesenteric (A) and epididymal (B) fat depots of wild type (N = 18 samples) and FAT ATTAC mice (N = 18 samples) at 0 (basal), 12, 24, 48, 72 hours and 10 days after dimerizer administration. Density of inflammatory cells was expressed as inflammatory cell number/10,000 adipocytes. Quantitative evaluation of CLS and perilipin negative adipocytes in mesenteric (C) and epididymal (D) fat depots of wild type (N = 18 samples) and FAT ATTAC mice (N = 18 samples) at 0 (basal), 12, 24, 48, 72 hours and 10 days after dimerizer administration. Density of CLS was expressed as CLS number/10 000 adipocytes, and density of perilipin negative adipocytes was expressed as perilipin negative adipocytes number/10 000 adipocytes. Wild-type values did not change during the time course (basal, 12, 24, 48, 72 hours and 10 days after dimerizer administration) so only the average values are presented. Data are shown as mean ± SEM. # P < 0.05 ## P < 0.01; ### P < 0.001 for comparison between wild type and FAT ATTAC mice at the indicated time points. * P < 0.05 ** P < 0.01; *** P < 0.001 refer to comparison between FAT ATTAC mice at the indicated time points and preceding FAT ATTAC mice.
Under the conditions chosen, 10 days following the start of dimerizer treatment, both fat pads reached the maximum number of dead adipocytes, along with high levels of immune cells present as judged mostly by Mac-2 immunoreactive macrophages when the largest number of CLS formation is observed (Fig. 2C–D). Both adipocyte cell death and immune cell infiltration were delayed in epididymal fat.
Adipose tissue from non-dimerized mice or regions of the adipose depots that exhibited limited apoptosis lacked CLS. Characteristic Mac-2 positive CLS were only found surrounding perilipin negative adipocytes (Fig. 3). These CLS presented the typical features previously described by electron microscopy [10] and shown in Figure 3: lipid laden macrophages surrounding the residual lipid droplet of dead adipocytes. The vast majority of dead adipocytes were part of a CLS (Fig. 3).
Figure 3.
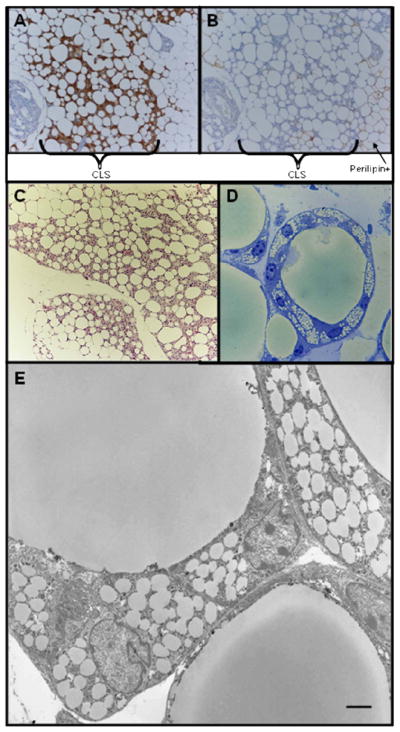
Consecutive serial sections of mesenteric fat depot after 10 days of dimerizer treatment showing that Mac-2 positive CLS (A) were present only in correspondence of perilipin negative (dead) adipocytes (B)
Hematoxylin-Eosin staining (C), toluidine blue staining of resin embedded tissue (D) and electron micrograph (E) showing that all dead adipocytes form classic CLS with lipid laden macrophages. Scale Bar: 130 μm for A and B; 150 μm for C; 15 μm for D; 2 μm for E.
Total numbers of immune cells increased in adipose tissue following dimerizer administration. The type of immune cells, however, may be more indicative of the tissue’s inflammatory state. Light and electron microscopy allowed us to distinguish the main immune populations (lymphocytes, neutrophils and macrophages) infiltrating the fat depots. Mac-2 immunoreactivity allowed us to distinguish two populations of macrophages (Mac-2 positive and Mac-2 negative), corresponding respectively to phagocytic active and inactive macrophages [19]. A quantitative evaluation of the percentage of the different type of inflammatory cells present in the two fat depots is presented in Figure 4. Under basal conditions, nearly 90% of the small number of inflammatory cells in both fat depots were largely represented by Mac-2 negative macrophages and neutrophils were completely absent. The initial infiltration of neutrophils appeared concurrent with the emergence of dead adipocytes (12 hours in mesenteric fat and 48 hours in epididymal fat, Fig. 2C–D and Fig. 4). Neutrophil populations were transient, however, as they quickly diminished. Over time, lymphocyte numbers were also reduced. The vast majority of the inflammatory cells were, at all stages studied, indentified as macrophages. Mac-2 positive macrophages progressively increased in coincidence with an equivalent decrease of Mac-2 negative macrophages and increase of CLS. Based on these histological observations, the time course of inflammation after adipocyte death passes through the following steps: first, neutrophils are recruited, then, macrophages are recruited, and finally, macrophages transition from a Mac-2 negative to Mac-2 positive state CLS are formed. The quality (type of inflammatory cells, and relative quantitative data) and time course was similar in the two visceral depots, even though some subtle differences were observed.
Figure 4.

Quantitative evaluation of inflammatory cell types in mesenteric (A) and epididymal (B) fat depots of wild type (N = 18 samples) and FAT ATTAC mice (N = 18 samples) at 0 (basal), 12, 24, 48, 72 hours and 10 days after dimerizer administration. Density of cell types expressed as percentage of all inflammatory cells. Wild-type values did not change during the time course (basal, 12, 24, 48, 72 hours and 10 days after dimerizer administration) so only the average values are presented. Data are shown as mean ± SEM. # P < 0.05 ## P < 0.01; ### P < 0.001 for comparison between wild type and FAT ATTAC mice at the indicated time points. * P < 0.05 ** P < 0.01; *** P < 0.001 refer to comparison between FAT ATTAC mice at the indicated time points and former preceding FAT ATTAC mice.
We then used electron microscopy to examine the ultrastructural features of adipose depots of FAT-ATTAC mice. The EM features of CLS in both depots confirmed the detailed features previously described [10] and are shown in Figure 3. We then took advantage of the early lack of CLS after 12 and 24 hours of dimerizer treatment in the mesenteric depot. At these time points 10% of adipocytes lacked perilipin labeling enabling electron microscopy of isolated apoptotic adipocytes. We found cytoplasmic structures never found in adipocytes at base line prior to dimerizer treatment or in other control mice (Fig. 5). The main features were: 1-nuclear condensation and very rare nuclear apoptotic bodies; 2-hypertrophic mitochondria; 3-increased electron density (known also as cytoplasmic condensation) of cytoplasm with degenerative changes of organelles; 4-small dense particles mainly at the level of lipid-cytoplasmic interface (consistent with calcium deposits); 5-dilated abundant rough endoplasmic reticulum (rare); 6-numerous small lipid droplets in the peripheral rim of cytoplasm (Fig. 5). The EM features described above are consistent with the features described for apoptosis in general [23] and with features of adipocytes apoptosis in vitro [24, 25].
Figure 5.

Representative electron micrographs showing main EM features of dying adipocytes in mesenteric fat of FAT ATTAC mice treated with dimerizer (12–48 hours): hypertrophic mitochondria (arrow, compare with normal mitochondrial arrowheads) (A); small dense particles (arrows) mainly at the level of lipid-cyoplasmic interface (consistent with calcium deposits) and degenerating organelles (asterisks) in a condensed cytoplasm of a dyeing adipocytes are visible. N = nucleus of a neutrophil leucocyte that is closely associates with dieing adipocytes (B); dilated (arrows) abundant rough endoplasmic reticulum (rare) in mesenteric fat depots (C); numerous small lipid droplets (arrows) in the peripheral rim in mesenteric fat depots (D). Scale Bar: 1 μm for A, B and C; 2.4 μm for D.
DISCUSSION
It is widely accepted that obesity is characterized by a mild chronic inflammation of adipose tissue with macrophage infiltration of fat [1–5]. In a previous paper, we have demonstrated that characteristic structures that we referred to as “crown like structures” (CLS) represent the site where more than 90% of the Mac-2 positive macrophages that infiltrate adipose tissue of obese mice and humans are localized [10]. Based mainly on morphological data, we suggested that CLS represents remnants of dead adipocytes surrounded by lipid-reabsorbing macrophages [10]. The remnant lipid droplet structures of dead adipocytes represent a “foreign body” structure, requiring macrophage intervention for reabsorption [26]. This is further supported by the frequent fusion of macrophages, leading to multinucleated giant cell formation [3, 10].
The FAT ATTAC mouse model lends itself to test this hypothesis directly. Upon systemic administration of a chemical “dimerizer”, adipocytes die because of a forced dimerization of caspase-8 that triggers the activation of the rest of the downstream apoptotic cascade [14, 15]. As expected, we observed that within 12–48 hours post dimerizer exposure of FAT-ATTAC mice, most adipocytes are negative for perilipin. This event precedes the CLS formation. Of note, the epididymal fat seems to respond in a delayed manner. We do not know at this time why but may suggest that the CLS formation in the epididymal fat is partially driven by systemic effects induced by the more rapid apoptosis in the mesenteric fat and then an epiphenomenon. Alternatively, the expression level of the caspase-8 transgene may be lower in these adipocytes, causing a more delayed response. Despite it being delayed, however, the progression eventually mirrors the events in the mesenteric depot, indicating that the tissue-specific events are equivalent. This is the first description of adipocyte apoptosis in vivo at the EM level. The sequence of the most frequent alterations found at cellular level as suggested by the EM studies in the different time points were: 1) appearance of hypertrophic mitochondria; 2) increased density of cytoplasm with progressive loss of morphological features of organelles along with calcium accumulation (“organelle degeneration”); 3) macrophages recruitment with CLS formation.
The immune cell composition at various time points as well as the EM features of dying adipocytes [24, 25] support that CLS represent remnants of dead adipocytes surrounded by activated macrophages taking up residual lipid. In fact, in parallel with the presence of dead adipocytes (perilipin-negative) before the appearance of CLS, we observed EM features consistent with cell stress (dilated endoplasmic reticulum) and death [24, 25] and acute stage inflammatory cell population [27]. Chronic phase inflammatory population progressively replaces neutrophils and lymphocytes. Although lymphocytes are present at baseline and are then reduced progressively during adipocyte loss, we could not study changes in lymphocyte subpopulations further due to technical limitations in delineating these cells by electron microscopy. The progressive increase in the number of active macrophages substituting for the inactive population of macrophages and the increase of the density of crown-like structures further support the histopathological diagnosis of a response to a “foreign body” with respect to the lipid droplet recognition and disposal.
Combined, our morphological, immunohistochemical and morphometric data obtained in this transgenic model support the following conclusions: 1) CLS represent sites of reabsorption of remnants of dead adipocytes; 2) macrophages are recruited to form CLS by remnants of adipocytes; 3) macrophages are progressively recruited and activated during that process.
The histomorphological characterization of adipose tissue filled with dying and dead adipocytes presented here complements our recent analysis of FAT-ATTAC mice in which we characterized the immune cell population of adipose tissue after 2 and more extensively after 14 days of continued exposure to dimerizer. For these studies, we used predominantly qPCR and FACS-based approaches to classify the infiltrating cells based on these markers [28]. Based on this analysis and consistent with the results reported here, we observed CLS formation with lipid resorbing macrophages. In addition, the marked distribution of the macrophages after 14 days of adipocyte ablation was predominantly of the “M2 subtype”, a category of macrophage that is less pro-inflammatory and more involved in remodeling processes. Recent observations [29] suggests that the initial response to adipocyte apoptosis involve a series of macrophages that lean towards the M2 phenotype. Obesity induced phenotypic switch from M2 to M1 of macrophages surronding necrotic adipocytes and is associated with CLS macrophages immunoreactive for TNFα and IL6 [12, 30]. CLS density is higher in diabetic obese db/db mice than in non-diabetic obese ob/ob mice [11]. This suggests a correlation between the number of CLS and insulin resistance. Notably though, the FAT-ATTAC mice display mild glucose intolerance [15] but do not become diabetic after fat ablation despite the large number of CLS present. The CLS-associated macrophages appear to be of the M2 type (in line with the resorbing and remodeling function of CLS macrophages). However, it is possible that the same cells also produce cytokines that are inflammatory. Whether Mac-2 positive macrophages associated with CLS really correspond to M2 macrophages following lipid uptake or whether these cells assume a gene expression pattern reminiscent of M1 macrophages remains to be studied further and will be an interesting topic to be clarified by future reseach.
Acknowledgments
The research leading to these results has received funding from the European Community’s 7th Framework Programme (FP7/2007–2013) under grant agreement n°201608; from Fondazione Cariverona, Grant 2008 to S.C. and from COST ACTION BM0602. PES was supported by NIH grants R01-DK55758, R01-CA112023, RC1-DK086629 and P01-DK088761. JMR was supported by a postdoctoral fellowship from the NIH (F32DK085935-01A1). QAW was supported by a postdoctoral fellowship from the ADA (7–11-MN-47).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008;582:97–105. doi: 10.1016/j.febslet.2007.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 29:415–45. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 3.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 2006;17:4–12. [PubMed] [Google Scholar]
- 5.Zhang J, Wright W, Bernlohr DA, Cushman SW, Chen X. Alterations of the classic pathway of complement in adipose tissue of obesity and insulin resistance. Am J Physiol Endocrinol Metab. 2007;292:E1433–40. doi: 10.1152/ajpendo.00664.2006. [DOI] [PubMed] [Google Scholar]
- 6.Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118:2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrannini E. Is insulin resistance the cause of the metabolic syndrome? Ann Med. 2006;38:42–51. doi: 10.1080/07853890500415358. [DOI] [PubMed] [Google Scholar]
- 9.Bremer AA, Devaraj S, Afify A, Jialal I. Adipose tissue dysregulation in patients with metabolic syndrome. J Clin Endocrinol Metab. 96:E1782–8. doi: 10.1210/jc.2011-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–55. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res. 2008;49:1562–8. doi: 10.1194/jlr.M800019-JLR200. [DOI] [PubMed] [Google Scholar]
- 12.Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW, 2nd, Defuria J, Jick Z, et al. Adipocyte Death, Adipose Tissue Remodeling and Obesity Complications. Diabetes. 2007 doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- 13.Harman-Boehm I, Bluher M, Redel H, Sion-Vardy N, Ovadia S, Avinoach E, et al. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J Clin Endocrinol Metab. 2007;92:2240–7. doi: 10.1210/jc.2006-1811. [DOI] [PubMed] [Google Scholar]
- 14.Trujillo ME, Pajvani UB, Scherer PE. Apoptosis through targeted activation of caspase 8 (“ATTAC-mice”): novel mouse models of inducible and reversible tissue ablation. Cell Cycle. 2005;4:1141–5. doi: 10.4161/cc.4.9.2030. [DOI] [PubMed] [Google Scholar]
- 15.Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, et al. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med. 2005;11:797–803. doi: 10.1038/nm1262. [DOI] [PubMed] [Google Scholar]
- 16.Clackson T, Yang W, Rozamus LW, Hatada M, Amara JF, Rollins CT, et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc Natl Acad Sci U S A. 1998;95:10437–42. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halberg N, Schraw TD, Wang ZV, Kim JY, Yi J, Hamilton MP, et al. Systemic fate of the adipocyte-derived factor adiponectin. Diabetes. 2009;58:1961–70. doi: 10.2337/db08-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. 1981;29:577–80. doi: 10.1177/29.4.6166661. [DOI] [PubMed] [Google Scholar]
- 19.Sano H, Hsu DK, Apgar JR, Yu L, Sharma BB, Kuwabara I, et al. Critical role of galectin-3 in phagocytosis by macrophages. J Clin Invest. 2003;112:389–97. doi: 10.1172/JCI17592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fawcett Don W. Fawcett Ba a textbook of histology. 12. 1994. [Google Scholar]
- 21.Londos C, Brasaemle DL, Schultz CJ, Segrest JP, Kimmel AR. Perilipins, ADRP, and other proteins that associate with intracellular neutral lipid droplets in animal cells. Semin Cell Dev Biol. 1999;10:51–8. doi: 10.1006/scdb.1998.0275. [DOI] [PubMed] [Google Scholar]
- 22.Zhang HH, Souza SC, Muliro KV, Kraemer FB, Obin MS, Greenberg AS. Lipase-selective functional domains of perilipin A differentially regulate constitutive and protein kinase A-stimulated lipolysis. J Biol Chem. 2003;278:51535–42. doi: 10.1074/jbc.M309591200. [DOI] [PubMed] [Google Scholar]
- 23.Kerr JF, Winterford CM, Harmon BV. Cell biology: a laboratory handbook. London: England: Academic Press; 1994. Morphological criteria for identifying apoptosis. [Google Scholar]
- 24.Prins JB, Walker NI, Winterford CM, Cameron DP. Apoptosis of human adipocytes in vitro. Biochem Biophys Res Commun. 1994;201:500–7. doi: 10.1006/bbrc.1994.1730. [DOI] [PubMed] [Google Scholar]
- 25.Prins JB, Walker NI, Winterford CM, Cameron DP. Human adipocyte apoptosis occurs in malignancy. Biochem Biophys Res Commun. 1994;205:625–30. doi: 10.1006/bbrc.1994.2711. [DOI] [PubMed] [Google Scholar]
- 26.Ackerman’s Ra. Surgical Pathology-2 Volume Set. 2004. [Google Scholar]
- 27.Nijhuis J, Rensen SS, Slaats Y, van Dielen FM, Buurman WA, Greve JW. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity (Silver Spring) 2009;17:2014–8. doi: 10.1038/oby.2009.113. [DOI] [PubMed] [Google Scholar]
- 28.Keuper M, Bluher M, Schon MR, Moller P, Dzyakanchuk A, Amrein K, et al. An inflammatory micro-environment promotes human adipocyte apoptosis. Mol Cell Endocrinol. 339:105–13. doi: 10.1016/j.mce.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Fischer-Posovszky P, Wang QA, Asterholm IW, Rutkowski JM, Scherer PE. Targeted Deletion of Adipocytes by Apoptosis Leads to Adipose Tissue Recruitment of Alternatively Activated M2 Macrophages. Endocrinology. doi: 10.1210/en.2011-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 2008;57:3239–46. doi: 10.2337/db08-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]