

Abstract
Crossbow is a scalable, portable, and automatic cloud computing tool for identifying SNPs from high coverage short read resequencing data. It is built on Apache Hadoop, an implementation of the MapReduce software framework. Hadoop allows Crossbow to distribute read alignment and SNP calling subtasks over a cluster of commodity computers. Two robust tools, Bowtie and SOAPsnp, implement the fundamental alignment and variant calling operations respectively, and have demonstrated capabilities within Crossbow of analyzing approximately one billion short reads per hour on a commodity Hadoop cluster with 320 cores. Through protocol examples, this unit will demonstrate the use of Crossbow for identifying variations in three different operating modes: on a Hadoop cluster, on a single computer, and on the Amazon Elastic MapReduce cloud computing service.
Keywords: short reads, read alignment, SNP calling, cloud computing, hadoop, software package
INTRODUCTION
The production of deep DNA sequencing data is accelerating at a staggering rate. Today, large scale, multi-sample studies can easily produce datasets tens of terabytes in size in a matter of a few weeks and at low cost (Pennisi, 2011). Relying on Moore's law, the doubling of compute capacity approximately every two years, to provide the necessary compute capacity to analyze these datasets today and in the future is no longer a plausible strategy. The output data rate of deep sequencing technology far outpaces computational capacity today and the gap will only widen in the future. The only way to scale compute capacity at a rate comparable to data output is to spread computation across a cluster of machines and parallelize operations (Schatz et al., 2010).
Crossbow (Langmead et al., 2009a) was designed for scalable operation, it not only to meets the needs of researchers today but ensures the ability to analyze the datasets of tomorrow which will undoubtedly grow beyond the capacity of a single computer. Crossbow takes advantage of the well-known MapReduce (Dean and Ghemawat, 2008) processing model initially popularized by Google for web search. A defining characteristic of this model is its ability to take advantage of the locality of data in a distributed computing environment. Data are initially distributed throughout a cluster of compute machines, and whenever possible, data are processed on the node storing those data saving the transfer costs from a traditional shared fileserver model. The MapReduce model has made its way into the open source community in the form of Hadoop, an open source software framework written in Java. Through data replication and intelligent job scheduling Hadoop provides a highly robust platform for distributed computing and is the supporting framework for Crossbow.
Crossbow leverages Hadoop to distribute short read alignment and SNP calling over a cluster of worker nodes. The alignment operation is performed by Bowtie (Langmead et al., 2009b), an ultrafast, in-memory short read aligner that is accurate and efficient. The SNP calling operation is handled by SOAPsnp (Li et al., 2009), which implements a Bayesian model for differentiating true SNPs from sequencing errors. These two programs are run using the streaming module within Hadoop and managed by a collection of scripts that make up the Crossbow framework. This software package has been demonstrated to identify SNPs in deep coverage whole genome resequencing projects at a rate of approximately 1 billion reads per hour and with an accuracy of nearly 99% (Langmead et al., 2009a). As such there is great interest to apply these technologies for other studies and for other genomes.
Prior to job initialization, a reference JAR file containing the necessary Bowtie indexes and SNP data are uploaded to the Hadoop Distributed File System (HDFS). From HDFS, common reference data can be copied to all nodes eliminating the need for inter-machine communication and greatly reducing complexity. The sequencing reads are also copied to HDFS, where they are automatically distributed amongst the compute nodes. Each node is assigned a subset of reads to align, using Bowtie and their local copy of the reference index. After alignment, reads are sorted into genomic bins of 1Mbp regions of the genome, which are distributed throughout the cluster and analyzed simultaneously with a modified version of SOAPsnp. SNP calls from each compute node are then deposited back to HDFS where they can be retrieved using standard Hadoop filesystem commands.
BASIC PROTOCOL 1
RUNNING CROSSBOW ON A LOCAL HADOOP CLUSTER
This protocol uses Crossbow to call SNPs on a small test dataset of mouse chromosome 17 from the study published by Sudbery, Stalker et al. (Sudbery et al., 2009). The reference JAR for chromosome 17 will be built using automated scripts distributed with the Crossbow package. The sequencing data itself will be retrieved from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) during the preprocessing step. The expected output will be a file containing all of the SNPs from this chromosome relative to the reference chromosome sequence.
Necessary Resources
Software
Hadoop 0.20 is recommended and has been tested. Other versions may work but are not supported.
Hardware
Sufficient memory must be available on all Hadoop worker nodes to hold the Bowtie index for a given reference sequence (approximately 3GB for the mouse or human genome). In addition, Hadoop requires some memory for its own operations on the worker nodes, which must be taken into account in order to avoid Java heap out-of-memory exceptions. For a human sized genome, a minimum of 5-6GB of memory per node is recommended.
Shared CROSSBOW_HOME
Crossbow uses Hadoop streaming to distribute the Bowtie and SOAPsnp processes. It makes use of many scripts external to the Hadoop framework to implement this workflow. These external scripts must be accessible on all worker nodes in the cluster. This can be accomplished, for example, by placing the files on a Network File System (NFS) share that is mounted on the workers. Alternately, the files can be copied to each worker using a tool such as rsync.
Running the mouse chromosome 17 example from the command line
1. First, run a test on the installation to ensure basic tools can be found.
$CROSSBOW_HOME/cb_hadoop --test
Ensure all checks pass and that Crossbow can locate the necessary binaries. Fastq-dump will need to be available on all nodes if --preprocess is specified. If Crossbow cannot find fastq-dump, specify the absolute path to the binary using the --fastq-dump flag.
2. Begin by building a reference jar for the mouse genome using the scripts provided with the Crossbow package.
cd CROSSBOW_HOME/reftools ./mm9_chr17_jar
This will use the predefined scripts for preparing the auxiliary files for fast searching and SNP calling in the reference genome mm9. The protocol below will explain how to prepare other reference genomes that do not have a predefined script.
3. Next, copy the mm9 reference jar to the HDFS.
hadoop fs -mkdir /crossbow-refs hadoop fs -put $CROSSBOW_HOME/reftools/mm9_chr17.jar /crossbowrefs/mm9_chr17.jar
These commands create a working directory, crossbow-refs, on the Hadoop Distributed FileSystem (HDFS) and copies the newly prepared JAR file to that directory.
4. Now move the manifest file to the HDFS.
hadoop fs -mkdir /crossbow/example/mouse17 hadoop fs -put $CROSSBOW_HOME/example/mouse17/full.manifest/crossbow/example/mous e17/full.manifest
The manifest file describes where input reads are located. In this case, the reads are located in the NCBI SRA. Crossbow will automatically download the reads to the Hadoop cluster.
5. Run Crossbow using the previously uploaded files.
$CROSSBOW_HOME/cb_hadoop \ --preprocess \ --input=hdfs:///crossbow/example/mouse17/full.manifest \ --output=hdfs:///crossbow/example/mouse17/output_full \ --reference=hdfs:///crossbow-refs/mm9_chr17.jar
This will execute Crossbow on the specified data on the Hadoop cluster. The output can be found in /crossbow/example/mouse17/output_full in HDFS.
6. Download the catalog of identified SNPs
hadoop fs –get /crossbow/example/mouse17/output_full mm17_results
The identified SNPs will be copied to the directory mm17_results on the local filesystem. The output files will be compressed with gzip to facilitate efficient transfer from HDFS to a local filesystem
See Commentary for troubleshooting tips.
BASIC PROTOCOL 2
RUNNING CROSSBOW IN SINGLE-COMPUTER MODE
For users who do not have access to a compute cluster or would like to test Crossbow prior to full-scale deployment, Crossbow can be run on a single computer. The user can request that Crossbow run on multiple processors at once by specifying the --cpus flag.
This protocol will detail the steps necessary to run a small E. coli test dataset through Crossbow using publicly available data from a study performed by Parkhomchuk et al. (Parkhomchuk et al., 2009). The E._coli reference JAR has already been pre-built and is available for download at http://crossbow-refs.s3.amazonaws.com/e_coli.jar. The input for this test will be a dataset from the NCBI's SRA and will be pulled down during the preprocess step immediately following the launch of the Crossbow command.
Necessary Resources
Software
Only the Crossbow software package is required (Hadoop is not required). The server must be running Linux or MacOS. Operating under Cygwin under windows is not supported.
Hardware
Sufficient memory must be available to hold the Bowtie index for a given reference sequence (approximately 3GB for the mouse or human genome). In addition, some extra memory is required for sorting alignments and other memory intensive operations. For a human sized genome, a minimum of 5-6GB of memory is recommended. Runtime will be proportional to the number of cores available, and 48 cores is recommended for mammalian projects.
Running the E. coli example on a single server on the command line
1. Ensure the installation of Crossbow was successful.
cd $CROSSBOW_HOME ./cb_local –test
This will ensure that your build of crossbow is working correctly on your computer.
2. Create a subdirectory crossbow_refs to house the ecoli reference sequence.
mkdir crossbow_refs/e_coli
This specifies the location of the reference genome information.
3. Download the reference jar (e_coli.jar)
wget -o crossbow_refs/e_coli/e_coli.jar \ http://crossbowrefs.s3.amazonaws.com/e_coli.jar
This command download the reference information using the command line tool wget. If this is not available on your server, you can also use other command line tools, e.g. curl, or within your web browser.
4. Unzip the jar
cd crossbow_refs/e_coli & unzip e_coli.jar
This ensures the reference data are accessible to the alignment and analysis tasks.
5. Run Crossbow
cd $CROSSBOW_HOME/example/e_coli \ $CROSSBOW_HOME/cb_local \ --input=small.manifest \ --preprocess \ --reference=$CROSSBOW_HOME/crossbow_refs/e_coli \ --output=output_small \ --all-haploids \ --cpus=<CPUS>
To make Crossbow run on multiple processors in parallel, substitute an integer value greater than one in place of the <CPUS> tag. Otherwise the --cpus option may be omitted. The final output for this job will be deposited into the output directory specified with –output. Because this job is running on the local operating system, the results will be immediately accessible from that directory on the local filesystem without transfer. Note here we used the –all-haploids flag to indicate that the genome is haploid (diploid is the default).
BASIC PROTOCOL 3
RUNNING CROSSBOW ON AMAZON WEB SERVICES VIA THE COMMAND LINE
This section will demonstrate the execution of Crossbow on the Amazon Elastic MapReduce (EMR) service of Amazon Web Services (AWS). Crossbow offers two interfaces to the EMR service. The first is through the Unix command line. This method requires the Amazon elastic-mapreduce script (See Requirements). The second method is through a user-friendly web form and is demonstrated in Alternate Protocol 1.
Necessary Resources
Amazon Account
A valid Amazon EC2 account in necessary. Users should take care to read and understand Amazon's prices and billing policies. Running these examples may incur monetary costs.
Elastic-Mapreduce script (Command line only)
Amazon provides a script to launch Hadoop programs on Elastic MapReduce via the command line. Crossbow uses this script as a conduit to specify Crossbow parameters to Amazon. Installation instructions can be found in the EC2-Installation section of this protocol. You will also need a tool to transfer data from your local computer to the Amazon Simple Scalable Storage (S3) system, such as the s3cmd tool.
Run mm9 example on the Amazon EC2 via the command line
Note that the dataset used in this protocol is the same as is used in the Local-Hadoop cluster protocol above (Basic Protocol 1). The dataset is available via the SRA and will be downloaded during the preprocessing step of the Crossbow job.
1. Ensure Crossbow can find required binaries on your local computer.
cd $CROSSBOW_HOME ./cb_emr –test
2. Begin by building an index of mouse chromosome 17.
./reftools/mm9_chr17_jar
3. The previous command will build the reference jar for chromosome 17. Use the s3 tool to put the reference jar into your s3 bucket.
s3cmd put $CROSSBOW_HOME/mm9_chr17.jar \ s3://<YOUR_BUCKET>/crossbow-refs/
This will copy the reference data into the cloud. See the Amazon S3 documentation for a discussion of how to create and manage a file bucket.
4. In addition to the reference, transfer the manifest file to s3 as well.
s3cmd put $CROSSBOW_HOME/example/mouse17/full.manifest \ s3://<YOUR_BUCKET>/example/mouse17/
5. Run the Elastic Map Reduce job via the cb_emr script in $CROSSBOW_HOME.
cd $CROSSBOW_HOME $CROSSBOW_HOME/cb_emr --name “Crossbow-Mouse17” \ --preprocess \ --input=s3n://<YOUR_BUCKET>/example/mouse17/full.manifest \ --output=s3n://<YOUR_BUCKET>/example/mouse17/output_full \ --reference=s3n://<YOUR_BUCKET>/crossbow-refs/mm9_chr17.jar \ --instances 8
Results will be found in s3n://<YOUR_BUCKET>/example/mouse17/output_full directory. Use the s3 tool to transfer the directory back to your local machine. Here we have specied --instances 8 to indicate that our Hadoop cluster should use 8 worker nodes. And --preprocess to indicate the raw fastq files need to be first preprocessed into the correct format.
ALTERNATE PROTOCOL 1
RUNNING CROSSBOW ON AMAZON WEB SERVICES VIA THE WEB INTERFACE
This section will demonstrate the execution of Crossbow on the Amazon Elastic MapReduce (EMR) service of Amazon Web Services (AWS) using a user-friendly web form.
This protocol will make use of the same dataset from the local-mode Crossbow protocol above (Basic Protocol 2). The dataset is available from the SRA and will be pulled down automatically during the preprocessing step of the Crossbow job.
Necessary Resources
Amazon Account
A valid Amazon EC2 account in necessary. Users should take care to read and understand Amazon's prices and billing policies. Running these examples may incur monetary costs.
Run E. Coli example on Amazon EC2 via the Web Interface
1. Use the s3 tool to upload $CROSSBOW_HOME/example/e_coli/small.manifest to the example/e_coli subdirectory in your s3 bucket.
s3cmd put $CROSSBOW_HOME/example/e_coli/small.manifest \ s3://<YOUR-BUCKET>/example/e_coli/
2. Launch Crossbow using the web interface.
Direct your web browser to the Crossbow web interface (Figure 1).
Figure 1.
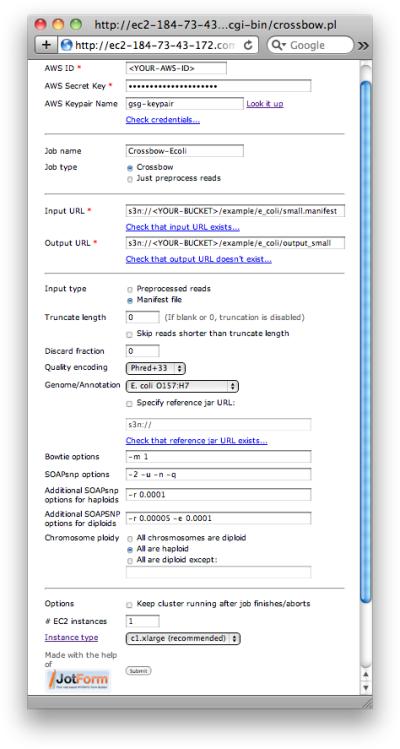
Screenshot of the Crossbow web interface for executing Crossbow on the Amazon cloud entirely from within your web browser.
http://bio-cloud-1449786154.us-east-1.elb.amazonaws.com/cgi-bin/crossbow.pl
3. Then fill in the form as below (substituting for <YOUR-BUCKET>):
For AWS ID, enter your AWS Access Key ID
For AWS Secret Key, enter your AWS Secret Access Key
Optional: For AWS Keypair name, enter the name of your AWS keypair. This is only necessary if you would like to be able to ssh into the EMR cluster while it runs.
Optional: Check that the AWS ID and Secret Key entered are valid by clicking the “Check credentials...” link
For Job name, enter ‘Crossbow-Ecoli’
Make sure that Job type is set to “Crossbow”
For Input URL, enter ‘s3n://<YOUR-BUCKET>/example/e_coli/small.manifest’, substituting for ‘<YOUR-BUCKET>’
Optional: Check that the Input URL exists by clicking the “Check that input URL exists...” link
For Output URL, enter ‘s3n://<YOUR-BUCKET>/example/e_coli/output_small’, substituting for ‘<YOUR-BUCKET>’
Optional: Check that the Output URL does not exist by clicking the “Check that output URL doesn't exist...” link
For Input type, select “Manifest file”
For Genome/Annotation, select “E. coli” from the drop-down menu
For Chromosome ploidy, select “All are haploid”
Click Submit
After submitting the form, the script will initialize a Hadoop cluster on the Amazon cloud using the specified number of computers. This job typically takes about 30 minutes on 1 c1.xlarge node. To download the results, use an S3 tool such as s3cmd to retrieve the contents of the s3n://<YOUR-BUCKET>/example/e_coli/output_small directory.
SUPPORT PROTOCOL 1
OBTAINING AND INSTALLING CROSSBOW
Crossbow is distributed as a software package containing a combination of perl, C, C++, and Ruby software for the various packages. We distribute preconfigured binary packages for some supported operating systems, but we recommend downloading and building the latest version from source.
Necessary Resources
svn
A working version of subversion. Available in the repositories of most linux package managers
bowtie
Bowtie can be downloaded from http://bowtie-bio.sourceforge.net/index.shtml it is recommended that after installation the Bowtie binary be copied to the bin directory in $CROSSBOW_HOME and that $CROSSBOW_HOME/bin be added to $PATH.
Soapsnp
Crossbow requires certain modifications to the SOAPsnp source package that are only available in the Crossbow sourcecode repository.
General Install
It is recommended that the latest version of Crossbow be used. Hadoop is continuously evolving and Crossbow is updated regularly to support new Hadoop releases. To take advantage of the latest version of Crossbow users should checkout the code from the SVN repository.
1. Download the sourcecode from the SVN repository.
svn co https://bowtie-bio.svn.sourceforge.net/svnroot/bowtie-bio bowtie-bio
$CROSSBOW_HOME will now refer to /Path/To/bowtie-bio/crossbow
2. Next build the modified version of soapsnp found in $CROSSBOW_HOME/soapsnp.
cd $CROSSBOW_HOME cd soapsnp & make & cd .. mkdir bin cp -r crossbow bin export PATH=$PATH:$CROSSBOW_HOME/bin
Create a bin subdirectory in the $CROSSBOW_HOME directory and copy the soapsnp directory into the bin directory. Add $CROSSBOW_HOME/bin to $PATH.
3. Ensure crossbow operates properly by running the proper test script for your application.
./cb_hadoop --test ./cb_emr --test ./cb_local --test
Crossbow will attempt to locate all binaries necessary for the job. In Hadoop mode, the paths to these binaries are passed to worker machines; it is therefore recommended that $CROSSBOW_HOME resides on a shared nfs mount accessible by the same path on all nodes.
Download SRA toolkit from NCBI (Optional)
Some examples use data from the SRA housed within .sra files. To extract the raw sequencing data from .sra files, the SRA toolkit is required.
1. Download the SRA toolkit from
http://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?cmd=show\&f=software\&m=software\&s=software
Either install a binary version for your platform or compile the source code. Crossbow needs the binary fastq-dump. Copy this binary into the bin/ directory of $CROSSBOW_HOME to make the Crossbow package portable.
Download and Install Elastic MapReduce Tool (EMR only)
Executing Crossbow with Amazon Elastic MapReduce requires the elastic-mapreduce-ruby package.
1. Download the toolkit from github
https://github.com/tc/elastic-mapreduce-ruby
Add the root of the package (that includes the elastic-mapreduce script) to your $PATH. Crossbow should find the tool during tests
2. Confirm the script is prepared for crossbow
cd $CROSSBOW_HOME ./cb_emr --test
If Crossbow cannot find the elastic-mapreduce script it can also be specified via the --emr-script parameter. The elastic-mapreduce script requires a file called credentials.json containing AWS user information. Create a file called credentials.json in the same directory as the elastic-mapreduce script and populate it with the AWS credentials under which Crossbow will run. Refer to the elastic-mapreduce README file for more information about credentials.json.
SUPPORT PROTOCOL 2
PREPARING MANIFEST FILES WITH SEQUENCE READ INFORMATION
Manifest files describe where the raw read information is available. This is useful because the raw read data for large projects will often exceed 100GB of compressed files, so it is desirable to execute the data transfer using multiple parallel data streams to maximize bandwidth usage and minimize transfer time. A manifest file describes a set of fastq or .sra formatted input files that might be located:
On the local computer
In HDFS
In S3
On an FTP or web server
A manifest file can contain any combination of URLs and local paths from these various types of sources. Fastq files can be gzip or bzip2-compressed (i.e. with .gz or .bz2 file extensions). If .sra files are specified in the manifest and Crossbow is being run in Local-User or Local-Hadoop cluster modes, then the fastq-dump tool must be installed and Crossbow must be able to locate it.
Each line in the manifest file represents either one file, for unpaired input reads, or a pair of files, for paired input reads. For a set of unpaired input reads, the line is formatted:
URL(tab)Optional-MD5
Crossbow will attempt to check the integrity of the file after downloading by comparing the observed MD5 to the MD5 provided in the Optional-MD5 field. To disable this checking, specify ‘0’ in this field.
For a set of paired input reads, the line is formatted:
URL-1(tab)Optional-MD5-1(tab)URL-2(tab)Optional-MD5-2
Where ‘URL-1’ and ‘URL-2’ specify the input files with all of the #1 mates in ‘URL-1’ and all of the #2 mates in ‘URL-2’. The entries in the files must be arranged so that pairs “line up” in parallel. This is commonly the way public paired-end FASTQ datasets, such as those produced by the 1000 Genomes Project, are formatted. Typically these file pairs end in suffixes '_1.fastq.gz and ‘_2.fastq.gz’.
Manifest files may have comment lines, which must start with the hash (#) symbol, and blank lines. Such lines are ignored by Crossbow.
SUPPORT PROTOCOL 3
PREPARING REFERENCE JARS WITH REFERENCE GENOME INFORMATION
All information about a reference sequence needed by Crossbow is encapsulated in a “reference jar” file. A reference jar includes a set of FASTA files (see Appendix 1B) encoding the reference sequences, a Bowtie index of the reference sequence, and a set of files encoding information about known SNPs for the species.
A Crossbow reference jar is organized as:
A ‘sequences’ subdirectory containing one FASTA file per reference sequence.
An ‘index’ subdirectory containing the Bowtie index files for the reference sequences.
A ‘snps’ subdirectory containing all of the SNP description files.
1. Prepare sequences subdirectory
The FASTA files in the ‘sequences’ subdirectory must each be named ‘chrX.fa’, where ‘X’ is the 0-based numeric id of the chromosome or sequence in the file. For example, for a human reference, chromosome 1's FASTA file could be named ‘chr0.fa’, chromosome 2 named ‘chr1.fa’, etc, all the way up to chromosomes 22, X and Y, named ‘chr21.fa’, ‘chr22.fa’ and ‘chr23.fa’. Also, the names of the sequences within the FASTA files must match the number in the file name. I.e., the first line of the FASTA file ‘chr0.fa’ must be ‘>0’.
2. The index files in the ‘index’ subdirectory must have the basename ‘index’. I.e., the index subdirectory must contain these files:
index.1.ebwt
index.2.ebwt
index.3.ebwt
index.4.ebwt
index.rev.1.ebwt
index.rev.2.ebwt
The index must be built using the ‘bowtie-build’ tool distributed with Bowtie. When ‘bowtie-build’ is executed, the FASTA files specified on the command line must be listed in ascending order of numeric id. For instance, for a set of FASTA files encoding human chromosomes 1,2,...,22,X,Y as ‘chr0.fa’, ‘chr1.fa’ ,..., ‘chr21.fa’, ‘chr22.fa’, ‘chr23.fa’, the command for ‘bowtie-build’ must list the FASTA files in that order:
bowtie-build chr0.fa,chr1.fa,...,chr23.fa index
Be sure to use ‘bowtie-build’ version 0.9.8 or newer.
3. The SNP description files in the ‘snps’ subdirectory must also have names that match the corresponding FASTA files in the ‘sequences’ subdirectory, but with extension ‘.snps’. E.g. if the sequence file for human Chromosome 1 is named ‘chr0.fa’, then the SNP description file for Chromosome 1 must be named ‘chr0.snps’. SNP description files may be omitted for some or all chromosomes.
The format of the SNP description files must match the format expected by SOAPsnp's ‘-s’ option. The format consists of 1 SNP per line, with the following tab-separated fields per SNP:
Chromosome ID
1-based offset into chromosome
Whether SNP has allele frequency information (1 = yes, 0 = no)
Whether SNP is validated by experiment (1 = yes, 0 = no)
Whether SNP is actually an indel (1 = yes, 0 = no)
Frequency of A allele, as a decimal number
Frequency of C allele, as a decimal number
Frequency of T allele, as a decimal number
Frequency of G allele, as a decimal number
SNP id (e.g. a dbSNP id such as ‘rs9976767’)
Once these three subdirectories have been created and populated, they can be combined into a single jar file with a command like this:
jar cf ref-XXX.jar sequences snps index
To use ‘ref-XXX.jar’ with Crossbow, you must copy it to a location where it can be downloaded over the internet via HTTP, FTP, or S3. Once it is placed in such a location, make a note if its URL.
Automated Building of Common Reference Jars
The ‘reftools’ subdirectory of the Crossbow package contains scripts that assist in building reference jars, including scripts that handle the entire process for hg18 (UCSC human genome build 18) and mm9 (UCSC mouse genome build 9). The ‘db2ssnp’ script combines SNP and allele frequency information from dbSNP to create a ‘chrX.snps’ file for the ‘snps’ subdirectory of the reference jar. The ‘db2ssnp_*’ scripts executes the ‘db2ssnp’ script for each chromosome in the hg18 and mm9 genomes. The ‘*_jar’ scripts drive the entire reference-jar building process, including downloading reference FASTA files, building a Bowtie index, and using ‘db2ssnp’ to generate the ‘.snp’ files for hg18 and mm9.
GUIDELINES FOR UNDERSTANDING RESULTS
Once a Crossbow job completes successfully, the output is deposited in a ‘crossbow_results’ subdirectory of the specified ‘--output’ directory or URL. Within the ‘crossbow_results’ subdirectory, results are organized as one gzipped result file per chromosome. E.g. if your run was against the hg18 build of the human genome, the output files from your experiment will be named:
<output_url>/crossbow_results/chr1.gz
<output_url>/crossbow_results/chr2.gz
<output_url>/crossbow_results/chr3.gz
...
<output_url>/crossbow_results/chr21.gz
<output_url>/crossbow_results/chr22.gz
<output_url>/crossbow_results/chrX.gz
<output_url>/crossbow_results/chrY.gz
<output_url>/crossbow_results/chrM.gz
Each individual record is in the SOAPsnp output format. SOAPsnp's format consists of 1 SNP per line with several tab-separated fields per SNP. The fields are:
Chromosome ID
1-based offset into chromosome
Reference genotype
Subject genotype
Quality score of subject genotype
Best base
Average quality score of best base
Count of uniquely aligned reads corroborating the best base
Count of all aligned reads corroborating the best base
Second best base
Average quality score of second best base
Count of uniquely aligned reads corroborating second best base
Count of all aligned reads corroborating second best base
Overall sequencing depth at the site
Sequencing depth of just the paired alignments at the site
Rank sum test P-value
Average copy number of nearby region
Whether the site is a known SNP from the file specified with ‘-s’
Note that field 15 was added in Crossbow and is not output by unmodified SOAPsnp. For further details, see the SOAPsnp manual.
COMMENTARY
Background Information
The production cost of second-generation sequencing has decreased by orders of magnitude in recent years. Also, per-instrument sequencing throughput has increased at a rate of about 3-to-5-fold per year over that time, far outpacing improvements in computer speed. Sequencing analysis workloads have grown so large as to present serious issues, especially to smaller laboratories. First, datasets are often so large that, to achieve results in a reasonable amount of time, the computational analysis must run in parallel on many computers and processors. Second, these computations must be fault-tolerant, since the cost of aborting a long-running parallel computation mid-stream (something not unlikely, since it might be caused by a hardware or software failure on a single computer) can be substantial. Third, the cost of the computing resources used must be considered and controlled carefully, since computing costs now often exceed sequencing costs. Finally, upgrades in sequencing throughput necessitate upgrades in computational capacity, sometimes exceeding the laboratory's ability to buy, house and maintain the needed equipment.
MapReduce and cloud computing are two technologies designed to address many of these issues. MapReduce is a parallel programming model and software framework providing many useful services to large, parallel computations. For instance, it provides a redundant and distributed file system, fault tolerant parallel job scheduling, and efficient distributed binning and sorting of data records. The popular Apache Hadoop implementation of MapReduce provides these services in a portable Java package. To benefit from these features, the program must be designed as an alternating series of compute steps and aggregate steps. The aggregate steps take input data and bin and sort the data in a distributed fashion in preparation for the next compute step. A compute step performs a computation on each bin of data, with bins being processed simultaneously in parallel wherever possible. Not all algorithms fit well into this paradigm, but it happens that alignment, variant calling, and other common sequencing analysis tasks do fit it well (Langmead et al., 2010).
Cloud computing is a particular way of organizing and procuring computational resources. At its core, cloud computing is about economies of scale. When huge numbers of computers are brought under one roof, they can be maintained cost effectively. Also, cloud-computing resources are typically virtualized, allowing large computers to be sliced into smaller virtual computers depending on user demand. Virtualization also allows the user to customize the software environment on the computers: operation system version, software versions, security settings, etc. The virtualized computing resources are then rented out on a temporary basis to users on demand. Commercial cloud vendors, such as AWS, charge a fee for resources rented in this way. But some institutional-level clouds or research clouds can be used for free.
Troubleshooting
In Hadoop mode, the $CROSSBOW_HOME path must be accessible from all machines in the cluster. This can be accomplished by placing the files on a Network File System (NFS) share that is mounted at the same path on the workers. In addition, the SRA-Toolkit (Not included in the Crossbow package) must also be accessible from all machines. This can be accomplished by placing the fastq-dump binary in the ‘bin’ subdirectory of the $CROSSBOW_HOME directory on all the workers adding this is to the $PATH environment variable is the recommended procedure. As long as fastq-dump is in $PATH, it will usually be detected automatically, however it is possible to specify an absolute path to the fastq-dump binary using the –fastq-dump flag.
When specifying the Hadoop streaming jar file via --streaming-jar, ensure the streaming jar is named in a format that resembles hadoop-0.20-streaming.jar. Some distributions, such as Cloudera, use a verbose naming scheme for jar files that causes Crossbow to confuse versions. It is best to create a symlink from the streaming jar into $CROSSBOW_HOME and name it according the aforementioned simple version scheme.
A way to find configuration issues prior to running your job is to run the various Crossbow scripts with the --test option (‘cb_emr --test’, ‘cb_hadoop --test’, etc). This will often detect situations where critical software is not installed properly. Also, it is usually helpful to begin with a small example before running Crossbow on larger datasets. For instance, running a job with all but one or two lines in the manifest file commented out using the ‘#’ character can be a helpful test. This reduces the number of input files, making the job smaller, but still allows Crossbow to run through to the end.
When running Crossbow in Hadoop mode, its progress can be monitored via the Hadoop JobTracker. The JobTracker is a web server that provides a point-and-click interface for monitoring the job and reading its output and other log files, including after the job has finished running. If a job fails, the JobTracker interface can often be used to find the relevant error message, which is usually written to the “standard error” filehandle by one the subtasks. Finding the failed subtask and its output usually requires “drilling down” from the “step” level through the “job” level and “task” levels, and finally to the “attempt” level. See the documentation for your version of Hadoop for details.
When running Crossbow in Elastic MapReduce mode, the job can be monitored via the AWS Console. The AWS Console allows you to see: the status for job (e.g. “COMPLETED”, “RUNNING” or “FAILED”), the status for each step of each job, how long a job has been running for and how many “compute units” have been utilized so far, and the exact Hadoop commands used to initiate each job step. The AWS Console also has a useful facility for debugging jobs, accessible via the “Debug” button on the “Elastic MapReduce” tab of the Console. See the Amazon Elastic MapReduce documentation for details.
ACKNOWLEGEMENTS
The authors would like to thank Matthew Titmus, and Hayan Lee of Cold Spring Harbor Laboratory. This work was funded, in part, by the U.S. Department of Energy, Office of Biological and Environmental Research under Contract DE-AC02-06CH11357, NIH R01 HG006677-12, NIH R01 HG006102, NIH P41 HG004059, and NSF IIS- 0844494. We would also like to thank the Amazon Web Services in Education Research Grants.
Footnotes
INTERNET RESOURCES
http://bowtie-bio.sourceforge.net/crossbow
Website where the latest version of the software as well as an extensive manual are available
Website with the Hadoop documentation and software
http://docs.amazonwebservices.com/AWSEC2/latest/GettingStartedGuide/
Describes how to get started using the Amazon web services including the elastic compute cloud (EC2) and the simple storage system (S3).
LITERATURE CITED
- Dean J, Ghemawat S. MapReduce: simplified data processing on large clusters. Commun. ACM. 2008;51(1):107–113. [Google Scholar]
- Langmead B, Schatz MC, Lin J, Pop M, Salzberg SL. Searching for SNPs with cloud computing. Genome Biol. 2009a;10(11):R134. doi: 10.1186/gb-2009-10-11-r134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009b;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Hansen KD, Leek JT. Cloud-scale RNA-sequencing differential expression analysis with Myrna. Genome Biol. 2010;11(8):R83. doi: 10.1186/gb-2010-11-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Fang X, Yang H, Wang J, et al. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19(6):1124–32. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhomchuk D, Amstislavskiy V, Soldatov A, Ogryzko V. Use of high throughput sequencing to observe genome dynamics at a single cell level. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(49):20830–5. doi: 10.1073/pnas.0906681106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi E. Human genome 10th anniversary. Will computers crash genomics? Science. 2011;331(6018):666–8. doi: 10.1126/science.331.6018.666. [DOI] [PubMed] [Google Scholar]
- Schatz MC, Langmead B, Salzberg SL. Cloud computing and the DNA data race. Nat Biotechnol. 2010;28(7):691–3. doi: 10.1038/nbt0710-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudbery I, Stalker J, Simpson JT, Keane T, Rust AG, et al. Deep short-read sequencing of chromosome 17 from the mouse strains A/J and CAST/Ei identifies significant germline variation and candidate genes that regulate liver triglyceride levels. Genome Biology. 2009;10(10):R112. doi: 10.1186/gb-2009-10-10-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]