

Abstract
Mutations in the ATP13A2 gene (PARK9, OMIM 610513) cause autosomal recessive, juvenile-onset Kufor-Rakeb syndrome and early-onset parkinsonism. ATP13A2 is an uncharacterized protein belonging to the P5-type ATPase subfamily that is predicted to regulate the membrane transport of cations. The physiological function of ATP13A2 in the mammalian brain is poorly understood. Here, we demonstrate that ATP13A2 is localized to intracellular acidic vesicular compartments in cultured neurons. In the human brain, ATP13A2 is localized to pyramidal neurons within the cerebral cortex and dopaminergic neurons of the substantia nigra. ATP13A2 protein levels are increased in nigral dopaminergic and cortical pyramidal neurons of Parkinson's disease brains compared with normal control brains. ATP13A2 levels are increased in cortical neurons bearing Lewy bodies (LBs) compared with neurons without LBs. Using short hairpin RNA-mediated silencing or overexpression to explore the function of ATP13A2, we find that modulating the expression of ATP13A2 reduces the neurite outgrowth of cultured midbrain dopaminergic neurons. We also find that silencing of ATP13A2 expression in cortical neurons alters the kinetics of intracellular pH in response to cadmium exposure. Furthermore, modulation of ATP13A2 expression leads to reduced intracellular calcium levels in cortical neurons. Finally, we demonstrate that silencing of ATP13A2 expression induces mitochondrial fragmentation in neurons. Oppositely, overexpression of ATP13A2 delays cadmium-induced mitochondrial fragmentation in neurons consistent with a neuroprotective effect. Collectively, this study reveals a number of intriguing neuronal phenotypes due to the loss- or gain-of-function of ATP13A2 that support a role for this protein in regulating intracellular cation homeostasis and neuronal integrity.
INTRODUCTION
In recent years, a number of genes have been identified that are associated with autosomal recessive forms of parkinsonism including parkin (PARK2), DJ-1 (PARK7), PINK1 (PARK6) and ATP13A2 (PARK9) (1–3). Mutations in the ATP13A2 gene cause Kufor-Rakeb syndrome (KRS), a juvenile-onset, autosomal recessive neurodegenerative disorder characterized by slowly progressive levodopa-responsive parkinsonism together with additional features including supranuclear palsy, pyramidal signs, dystonia and dementia (4–7). Homozygous or compound heterozygous mutations have been identified in KRS subjects that produce frameshift or splicing variants that result in truncated forms of ATP13A2, leading to a loss of function (6,8,9). Numerous missense mutations have recently been identified in subjects with early-onset parkinsonism or Parkinson's disease (PD), suggesting that ATP13A2 mutations may contribute to the development of both KRS and parkinsonism depending on the severity of the mutation (10–13). Neuroimaging studies in KRS subjects with ATP13A2 mutations reveal diffuse brain atrophy with evidence of impaired nigrostriatal dopaminergic function (10,14). Thus, ATP13A2 mutations result in the dysfunction and/or degeneration of the nigrostriatal dopaminergic pathway in addition to other neuronal populations. The definitive confirmation of neuropathology associated with ATP13A2 mutations will await the future post-mortem analyses of affected subjects. The combination of levodopa-responsive parkinsonism and impaired function of the nigrostriatal dopaminergic pathway in KRS and parkinsonism subjects with ATP13A2 mutations supports the selective degeneration of nigrostriatal dopaminergic neurons. Whether ATP13A2-linked neurodegeneration is associated with Lewy body (LB) pathology that is characteristic of idiopathic PD is not yet known. At present, little is understood about how ATP13A2 mutations precipitate neurodegeneration in subjects with KRS or parkinsonism.
Human ATP13A2 is an 1180 amino-acid protein belonging to the P5 subfamily of transport ATPases containing 10 transmembrane domains (15). ATP13A2 is localized, at least in part, to lysosomes where it may participate in the ATP-dependent transport of cations across vesicular membranes (6,8). However, at present, the cation-transporting activity of ATP13A2 has not been directly demonstrated, and the cation selectivity of this transporter is not known. In yeast, deletion of ypk9, an ortholog of ATP13A2, confers sensitivity to growth in the presence of heavy metals, including cadmium, manganese, nickel and selenium, implicating ATP13A2 in the transport or regulation of these metal cations (16,17). A recent study suggests that the overexpression of human ATP13A2 may regulate the intracellular concentration of manganese in mammalian cells and protects from manganese-induced cell death (18). ATP13A2 mRNA is enriched in the mammalian brain where it is widely expressed by many neuronal populations, especially substantia nigra dopaminergic neurons (6,15). ATP13A2 mRNA expression is dramatically up-regulated in surviving nigral dopaminergic neurons of idiopathic PD brains, thus potentially supporting a link between ATP13A2 and idiopathic PD (6). Furthermore, ATP13A2 overexpression was recently shown to confer protection against α-synuclein-induced cytotoxicity in yeast, worm and neuronal models (16). Therefore, ATP13A2 may provide a link between environmental toxins (i.e. manganese) and genetic factors that are implicated in the pathogenesis of PD. At present, the physiological role of ATP13A2 in mammalian neurons is poorly understood and the mechanism through which pathogenic mutations precipitate neuronal degeneration is unclear.
Here, we explore the subcellular localization of ATP13A2 in rodent primary neurons and in human brains from normal and PD subjects. Furthermore, we employ gene silencing or overexpression of ATP13A2 in primary neuronal cultures to model the loss- and gain-of-function effects of ATP13A2. Our data demonstrate that ATP13A2 is normally localized to intracellular acidic vesicular structures within neurons and is up-regulated in neurons from affected regions of PD brains. Furthermore, we demonstrate that ATP13A2 silencing or overexpression regulates neuronal integrity, mitochondrial morphology and intracellular cation flux. The implications of our findings for ATP13A2-linked familial parkinsonism are discussed.
RESULTS
Development of an ATP13A2-specific polyclonal antibody
To begin to explore the function of ATP13A2, we generated an affinity-purified rabbit polyclonal antibody (named LMNR1) raised against a synthetic peptide corresponding to residues 499–511 (LRINLGGKLQLV) of the human protein; a region that is highly conserved with the rat and mouse sequence. This peptide is located within the second intracellular loop of ATP13A2 immediately adjacent to the P-domain phosphorylation motif (DKTGT, 513–517) (6). LMNR1 antibody specifically detects full-length V5-tagged human ATP13A2 and untagged mouse ATP13A2 transiently expressed in HEK-293T cells by western blotting (Fig. 1A). LMNR1 antibody also detects a major protein species in soluble mouse brain extracts with a molecular mass of ∼130 kDa by western blotting, corresponding to the predicted size of endogenous mouse ATP13A2 (Fig. 1B). This ∼130 kDa species is not detected following pre-absorption of the antibody with an excess of peptide antigen (Fig. 1B). To explore the subcellular localization of ATP13A2, we conducted subcellular fractionation of mouse brain tissue. ATP13A2 is enriched within the light membrane (P3, i.e. microsomal) fraction containing lysosomes (LAMP1), Golgi (Giantin) and endoplasmic reticulum (ER) (PDI), but is also detectable albeit at lower levels in the heavy membrane (P2, i.e. mitochondria and crude synaptosomes), synaptosomal membrane (LP1) and synaptic vesicle-enriched (LP2) fractions (Fig. 1C). Notably, ATP13A2 is generally absent from soluble cytosolic fractions (S1, S3, LS1 and LS2), supporting its subcellular localization as a transmembrane-spanning protein. Collectively, our data demonstrate the specificity of the LMNR1 antibody for detecting mammalian ATP13A2 and the enrichment of ATP13A2 within brain microsomal fractions.
Figure 1.

Development of an ATP13A2-specific antibody. (A) HEK-293T cells transiently expressing V5-tagged human ATP13A2 (left panel) or full-length untagged mouse ATP13A2 (right panel) were probed with an antibody to ATP13A2 (LMNR1) to reveal a specific protein band of 130–150 kDa (arrows) in transfected cells (+) that is absent from mock transfected cells (−). (B) Soluble protein extract from whole mouse brain was probed with ATP13A2-specific antibody, LMNR1 (left panel). The LMNR1 antibody detects a major protein species of ∼130 kDa (indicated by arrows), corresponding to the expected mass of endogenous ATP13A2. This protein species was not detected following pre-absorption of the LMNR1 antibody with an excess of peptide antigen (right panels). (C) Subcellular fractionation of whole mouse brain tissue. ATP13A2 (LMNR1 antibody) is enriched in the light membrane (P3, microsomal) fraction, but also detected in heavy (P2), synaptosomal (LP1), synaptic vesicle-enriched (LP2) membrane fractions. The distribution of marker proteins demonstrates the enrichment of lysosomes (LAMP1; P3), mitochondria/heavy membranes (TIM23; P2 and LP1), ER (PDI; P2, P3, LP1 and LP2), Golgi (Giantin; P2 and P3) and synaptosomes/synaptic vesicles (synaptophysin 1; P2, P3, LP1 and LP2). Molecular mass markers are indicated in kilodaltons. Blots are representative of at least two independent experiments.
ATP13A2 localizes to intracellular acidic vesicular compartments in neurons
Exogenously expressed human ATP13A2 has been shown to localize, at least in part, to lysosomal membranes in mammalian cell lines (6). To determine the subcellular localization of ATP13A2 in neurons, we assessed the co-localization of exogenous ATP13A2 with subcellular markers in rat primary cortical neurons by confocal fluorescence microscopy. Cortical neurons were transfected with either V5-tagged human ATP13A2 or green fluorescent protein (GFP)-tagged mouse ATP13A2 alone or together with vesicular fusion proteins (GFP-LC3, RFP-Rab5A, GFP-Rab7A or LAMP1-RFP) and subjected to immunocytochemical analysis. Initially, our ATP13A2 antibody (LMNR1) was not sufficiently sensitive to detect endogenous ATP13A2 in cortical neurons by immunofluorescence (data not shown) but could detect exogenously expressed mouse ATP13A2-GFP (Fig. 2A), which localizes to vesicle membranes within the soma and processes of cortical neurons (Fig. 2A). Exogenous human ATP13A2-V5 adopts a distribution similar to ATP13A2-GFP within the neuronal soma and proximal processes where it co-localizes to a high degree with LAMP1-RFP, a marker of lysosomal membranes, RFP-Rab5A, a marker of early endosomes, and GFP-Rab7A, a marker of late endosomes, but to a smaller degree with the autophagosomal marker, GFP-LC3 (Fig. 2A). Human ATP13A2-V5 also localizes within neuronal processes where it associates closely with markers of the microtubule network including microtubule-associated protein 2 (MAP2; Fig. 2A) and βIII-tubulin (Fig. 2B), suggesting localization of ATP13A2 to vesicles undergoing transport. Mouse ATP13A2-GFP fails to co-localize with the synaptic vesicle marker, synaptophysin-1 (Fig. 2B). Our data suggest that ATP13A2 is localized to a number of acidic vesicular compartments within cortical neurons, particularly lysosomes and endosomes.
Figure 2.
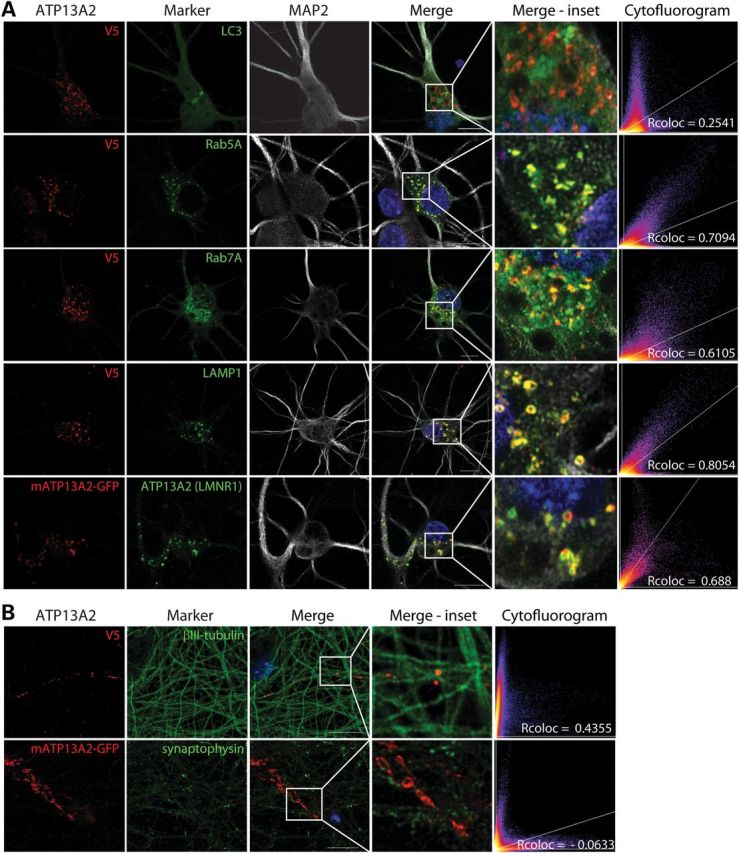
Localization of exogenous ATP13A2 to intracellular acidic vesicles in cortical neurons. (A) Confocal fluorescence microscopy reveals the co-localization of exogenous V5-tagged human ATP13A2 with GFP-LC3 (autophagosomes), RFP-Rab5A (early endosomes), GFP-Rab7A (late endosomes) and LAMP1-RFP (lysosomes) in neuronal soma and processes. Exogenous GFP-tagged mouse ATP13A2 is specifically labeled by the ATP13A2 antibody (LMNR1). (B) ATP13A2-V5 localizes to punctate structures located upon βIII-tubulin-positive neuronal processes, whereas ATP13A2-GFP fails to co-localize with endogenous synaptophysin-1, a marker of synaptic vesicles. Cytofluorograms and correlation coefficients (Rcoloc) indicate the extent of co-localization between exogenous ATP13A2 and each marker. Confocal images are representative of at least two independent cultures. Scale bar: 10 µm.
ATP13A2 protein levels are increased in human PD/dementia with Lewy body brains
To explore the expression of ATP13A2 in the human brain, we conducted western blot analysis with our LMNR1 antibody on detergent-soluble extracts derived from the striatum and cerebral cortex (medial frontal gyrus) of human brains from normal control subjects or subjects with PD and dementia with Lewy bodies (DLB). ATP13A2 is detected in human brain extracts as a major ∼130 kDa protein species (Fig. 3A). Furthermore, ATP13A2 levels are markedly increased in PD/DLB brains compared with normal control brains (Fig. 3A and B). To evaluate the localization of ATP13A2 in human brain, immunohistochemical analysis was conducted on brain sections from control and PD subjects. ATP13A2 localizes specifically to cytoplasmic punctate structures in pyramidal neurons located throughout the cingulate cortex of control and PD subjects with an increase in immunoreactivity in PD brains (Supplementary Material, Fig. S1). In the substantia nigra, ATP13A2 localizes selectively within the cytoplasmic compartment of neuromelanin-positive dopaminergic neurons, and we observe a marked increase of ATP13A2 immunoreactivity in surviving dopaminergic neurons of PD brains compared with control brains (Fig. 3C and D). The increase of ATP13A2 immunoreactivity in surviving dopaminergic neurons and cortical pyramidal neurons of PD brains compared with control brains could be confirmed using two additional antibodies to ATP13A2 (A9732, residues 1162–1180 and A3361, residues 804–821) raised against distinct peptide epitopes (Supplementary Material, Fig. S2). To determine whether ATP13A2 co-localizes with LB pathology, confocal fluorescence microscopy was conducted on cingulate cortex from DLB subjects using antibodies to ATP13A2 (LMNR1) and phospho-serine-129 α-synuclein, a pathological species localized to the LB halo. We do not observe direct co-localization of ATP13A2 with LBs, but we do observe strong ATP13A2-positive cytoplasmic punctate structures surrounding intracellular LBs (Fig. 3E). Initial quantitation reveals that LB-positive neurons frequently contained ATP13A2-positive puncta (70.8 ± 4.2% of LB + neurons). Furthermore, the intensity of ATP13A2-positive immunofluorescence is significantly increased in LB-positive neurons compared with LB-negative neurons, suggesting an up-regulation of ATP13A2 levels in response to LB biogenesis (Fig. 3F). Our data demonstrate that ATP13A2 protein is localized to neurons in the human brain where it is up-regulated in affected brain regions of PD and DLB subjects.
Figure 3.
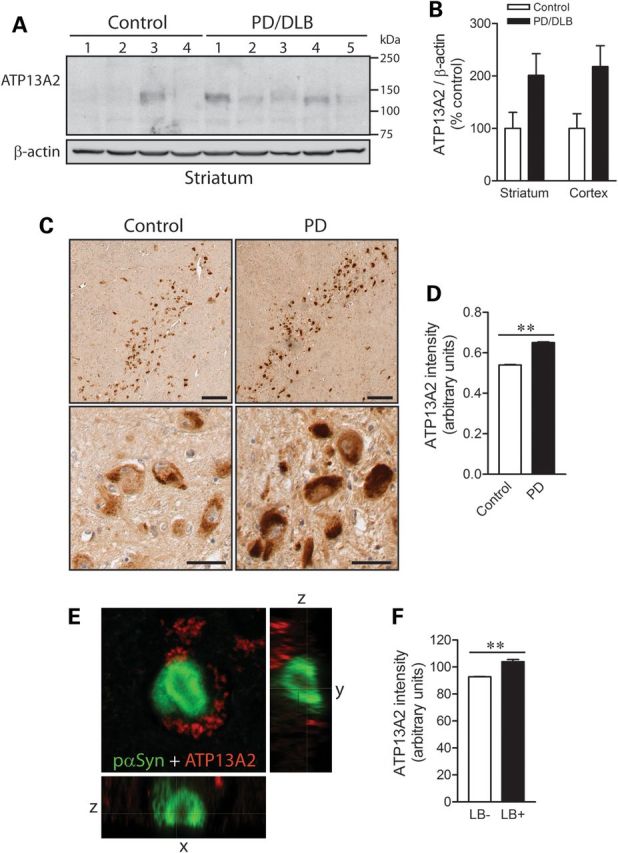
Increased levels of ATP13A2 in PD/DLB brains. (A) Soluble extracts derived from the striatum of normal control (nos 1–4) or PD/DLB (nos 1–5) subjects probed with ATP13A2 antibody (LMNR1), and β-actin antibody as a control for protein loading. (B) Densitometric analysis of western blots revealing increased levels of ATP13A2 in soluble extracts from striatum or medial frontal gyrus of PD/DLB subjects compared with normal control subjects. Data represent the mean ± SEM levels of ATP13A2 normalized to β-actin and are expressed as a percent of control levels (n = 4 for control or n = 5 for PD/DLB subjects). (C) Representative DAB immunostaining with ATP13A2 antibody (LMNR1) in the substantia nigra pars compacta from normal control or PD subjects. Staining indicates ATP13A2 localized to characteristic neuromelanin-positive dopaminergic neurons. Scale bar: 100 µm (upper panel) or 20 µm (lower panel). (D) Semi-quantitative analysis of DAB intensity corresponding to ATP13A2 levels in individual substantia nigra dopaminergic neurons from control or PD subjects. Data represent the mean ± SEM intensity of ATP13A2 staining in individual dopaminergic neurons (n = 1449/control or 1257/PD neurons) taken from at least three subjects per group. (E) Representative confocal fluorescent image of an intraneuronal LB from the cingulate cortex of a DLB subject (no. 8) immunolabeled with phospho-S129-α-synuclein (pαSyn, green) and ATP13A2 (LMNR1, red) antibodies. (F) Semi-quantitative analysis of ATP13A2 fluorescence intensity in cortical pyramidal neurons with (LB+, n = 86 neurons) or without (LB−, n = 2990 neurons) LBs. Data represent the mean ± SEM fluorescence intensity of ATP13A2 in cortical pyramidal neurons from the cingulate cortex of a single DLB subject. **P < 0.001 analyzed by Student's t-test.
Modulation of ATP13A2 expression compromises dopaminergic neuronal integrity
Recessive mutations in ATP13A2 compromise the nigrostriatal dopaminergic pathway in subjects with KRS or early-onset parkinsonism, which may reflect the degeneration of nigral dopaminergic neurons (10,14). However, post-mortem neuropathological data have not been reported from subjects with ATP13A2 mutations in order to confirm dopaminergic neuronal loss. To directly explore the impact of ATP13A2 loss of function on the viability and integrity of dopaminergic neurons, we employed mir-30-adapted short hairpin RNA (shRNA) constructs to silence the expression of endogenous ATP13A2. To validate shRNA constructs, we demonstrate by western blot analysis the successful knockdown of full-length mouse ATP13A2 transiently expressed in HEK-293T cells using shRNA constructs targeting rodent ATP13A2 but not with a non-silencing control shRNA (Fig. 4A). We also demonstrate the knockdown of endogenous ATP13A2 by western blot analysis in cultured rat primary cortical neurons using an ATP13A2-specific shRNA construct (Fig. 4B). To explore the consequences of shRNA-mediated silencing of endogenous ATP13A2 expression in neurons, we employed rat primary midbrain cultures that typically consist of 6–8% tyrosine hydroxylase (TH)-positive dopaminergic neurons (data not shown). Midbrain cultures were co-transfected at days-in-vitro (DIV) 3 with shRNA and GFP constructs at a molar ratio of 10:1 to label transfected neurons, and cultures were fixed at DIV 7 and immunostained with anti-TH antibody to identify dopaminergic neurons (Fig. 4C). The number of GFP-labeled dopaminergic neurons is similar following expression of control and ATP13A2-specific shRNAs, suggesting that ATP13A2 knockdown does not induce overt neuronal cell death (data not shown). We could confirm the absence of neuronal cell death following shRNA-mediated silencing of ATP13A2 using TUNEL labeling (data not shown). However, silencing of ATP13A2 impairs neuronal integrity as reflected by a marked reduction in the length of GFP-labeled dopaminergic neuritic processes compared with control shRNA (Fig. 4C and D). The overexpression of ATP13A2 orthologs in yeast, worm and primary neuronal models was previously shown to protect against α-synuclein-induced cell death (16). To evaluate the potentially neuroprotective effects of ATP13A2 overexpression, primary midbrain neurons were co-transfected with V5-tagged human ATP13A2 and GFP at a 10:1 molar ratio. Unexpectedly, overexpression of human ATP13A2 induces a marked reduction in neurite length of GFP-labeled dopaminergic neurons, similar to the effects of ATP13A2 knockdown (Fig. 4C and E). To assess whether the effects of ATP13A2 silencing are specific to dopaminergic neurons, we transfected rat primary cortical neurons with GIPZ plasmids co-expressing mir-30-adapted shRNA and enhanced GFP (EGFP) at DIV 3, and the length of GFP-positive neurites was assessed at DIV 7. Silencing of ATP13A2 expression does not influence the length of MAP2-positive cortical neurites compared with control shRNA (Supplementary Material, Fig. S3). Furthermore, the shRNA-mediated silencing of ATP13A2 does not promote apoptotic cell death as revealed by TUNEL labeling of cortical neurons (Supplementary Material, Fig. S3). Taken together, our data suggest that ATP13A2 is involved in maintaining the integrity of midbrain dopaminergic neurons. Our data further suggest that ATP13A2 overexpression may compromise dopaminergic neuronal integrity and may not therefore provide a promising neuroprotective strategy.
Figure 4.

Modulation of ATP13A2 expression impairs neurite outgrowth of midbrain dopaminergic neurons. (A) Soluble extracts from HEK-293T cells transiently co-expressing untagged mouse ATP13A2 and rodent-specific ATP13A2 (sh-ATP13A2) or non-silencing control (sh-control) shRNA plasmids were probed with an antibody to ATP13A2 (LMNR1) to demonstrate shRNA-mediated knockdown of ATP13A2. β-tubulin indicates equivalent protein loading. (B) Soluble extracts from rat primary cortical neurons transiently expressing control or ATP13A2-specific shRNAs, or untagged mouse ATP13A2, were probed with antibodies to ATP13A2 (LMNR1) or β-tubulin as a control for protein loading. ATP13A2-specific shRNA reduces the levels of endogenous ATP13A2 compared with control shRNA, whereas overexpression of mouse ATP13A2 is also detected. (C) Rat primary midbrain dopaminergic neurons co-transfected at DIV 3 with shRNA (control or ATP13A2-specific) or V5-tagged human ATP13A2 (or control empty vector) and GFP plasmids at a 10:1 molar ratio. Cultures were fixed at DIV 7. Representative fluorescent micrographs reveal the co-labeling of dopaminergic neurons with GFP and TH for each condition. Axonal processes are indicated by arrows. Scale bar: 100 µm. (D and E) Quantitative analysis of the length of GFP+/TH+ dopaminergic neurites reveals a significant shortening of axonal processes due to (D) the shRNA-mediated silencing of ATP13A2 or (E) the overexpression of human ATP13A2, compared with control neurons (sh-control or empty vector). Bars represent the mean ± SEM length of TH+ neurites in micrometers (n = 39–42 neurons for shRNAs or n = 28–39 neurons for hATP13A2) sampled across four independent cultures. Histograms indicate the mean frequency distribution of neurite length for each condition (n = 4 experiments). *P < 0.05 or **P < 0.01 compared with control plasmids assessed by unpaired Student's t-test.
Modulation of ATP13A2 expression induces alterations in autophagy
ATP13A2 co-localizes with intracellular acidic vesicular compartments in primary cortical neurons, particularly lysosomes and endosomes and partially with autophagosomes. Autophagy forms the major pathway for the transport of cytoplasmic materials to lysosomes for their degradation and has been implicated in neurodegenerative diseases, including PD (19–23). To explore the potential relationship between ATP13A2 and the autophagy pathway, the effect of ATP13A2 silencing and overexpression on autophagic activation was assessed. Primary cortical neurons were co-transfected with shRNAs or V5-tagged ATP13A2 and GFP-LC3 constructs at a 10:1 molar ratio followed by western blot analysis of neuronal extracts. LC3 exists as a cytosolic form (LC3-I, 18 kDa) that undergoes conversion to a membrane-associated form (LC3-II, 16 kDa) localized specifically to autophagosomes. The ratio of LC3-I and -II isoforms can be used to assess the level of autophagic activation (24). ATP13A2 knockdown or overexpression in cortical neurons fails to alter the ratio of LC3-II to LC3-I, but instead leads to a marked decrease in the steady-state levels of total GFP-LC3 (Fig. 5A and B). These data suggest that modulating the expression of ATP13A2 does not influence autophagic activation, but may enhance the turnover of GFP-LC3 most likely through lysosomal degradation. Next, we explored the impact of ATP13A2 expression on GFP-LC3-positive autophagosomes in cortical neurons by confocal microscopy. The overexpression of human ATP13A2 produces a marked increase in the diameter and number of GFP-LC3-positive puncta per neuron (Fig. 5C and D), whereas silencing of ATP13A2 expression causes a reduction in the diameter of GFP-LC3-positive puncta but without a significant alteration in their number (Fig. 5E and F). In general, however, ATP13A2 silencing or overexpression results in fewer GFP-LC3-positive neurons with reduced fluorescence per neuron compared with control neurons (data not shown) consistent with western blotting data (Fig. 5A and B). Collectively, our data suggest that ATP13A2 modestly regulates the number and size of autophagosomes.
Figure 5.

ATP13A2 modulates the size and number of LC3-positive autophagosomes in neurons. (A) Soluble extracts from rat primary cortical neurons co-transfected with control or ATP13A2-specific shRNAs or V5-tagged human ATP13A2 and GFP-LC3 plasmids at a 10:1 molar ratio were probed with antibodies to GFP to reveal LC3-I and LC3-II isoforms or β-tubulin as a control for protein loading. (B) Quantitative analysis of western blots revealing a normal ratio of LC3-II to LC3-I (left), but reduced steady-state levels of total GFP-LC3 (right) due to ATP13A2 silencing or overexpression in cortical neurons. Bars represent the mean ± SEM (n = 4 experiments). (C and E) Representative confocal fluorescent images indicating GFP-LC3-positive puncta in cortical neurons following (C) overexpression of human ATP13A2 or (E) shRNA-mediated silencing of ATP13A2, compared with control plasmids (empty vector or control shRNA). Scale bar: 5 µm. (D and F) Quantitative analysis of GFP-LC3-positive puncta indicates (D) the increased length and number of puncta due to ATP13A2 overexpression or (F) the reduced length of puncta due to ATP13A2 silencing. Bars represent the mean ± SEM length of puncta (micrometers) or percent particles/cell compared with the control condition (n = 9 neurons/condition from three experiments). Histograms indicate the mean frequency distribution of GFP-LC3-positive puncta length in micrometers for each condition (n = 9 neurons/condition). *P < 0.05, **P < 0.01 or ***P < 0.005 compared with control assessed by unpaired Student's t-test.
ATP13A2 silencing modulates the kinetics of intracellular pH in neurons
ATP13A2 is predicted to mediate the transport of heavy metal cations, including cadmium, nickel, manganese and selenium, since yeast cells with a deletion of the ykp9 gene (an ortholog of ATP13A2) exhibit impaired growth in the presence of these metals (16,17). However, the ability of ATP13A2 or Ykp9 to transport cations has not yet been directly demonstrated. Since ATP13A2 is localized predominantly to acidic vesicular compartments in neurons, we reasoned that modulation of ATP13A2 expression may influence intracellular pH. Alteration of intracellular pH also serves as an indirect measure of cation transport. Primary cortical neurons were co-transfected with V5-tagged human ATP13A2 and GFP constructs at a 10:1 molar ratio to label transfected cells, and intracellular pH was monitored in individual GFP-labeled neurons by live-cell confocal microscopy using the ratiometric fluorescent pH indicator, SNARF-1 (Fig. 6A–C). The overexpression of ATP13A2 fails to alter the basal intracellular pH of neurons (Fig. 6C). Next, we monitored alterations in intracellular pH in real time following acute exposure to 1 mm cadmium (Fig. 6B). Cadmium induces a rapid intracellular acidification measured over a period of 400 s in cortical neurons, although overexpression of ATP13A2 does not influence the rate of acidification compared with control neurons expressing GFP alone (Fig. 6B and C). Next, we assessed the effects of ATP13A2 silencing on intracellular pH in cortical neurons by co-transfecting cultures with shRNA and GFP plasmids at a 10:1 ratio (Fig. 6D–F). Silencing of ATP13A2 expression similarly fails to alter the basal intracellular pH of neurons (Fig. 6F). ATP13A2 silencing does, however, lead to an increased rate of acidification in response to cadmium treatment compared with neurons expressing control shRNA (Fig. 6E and F). Equivalent exposure to manganese fails to elicit alterations in intracellular pH in cortical neurons (data not shown), and it was not therefore possible to assess the effects of ATP13A2 expression. Our data suggest that ATP13A2 silencing modestly regulates the kinetics of intracellular pH in neurons in response to cadmium exposure rather than influencing basal pH, which supports a role for ATP13A2 in regulating intracellular cation transport. Furthermore, ATP13A2 may play a specific role in regulating the transport of cadmium ions.
Figure 6.
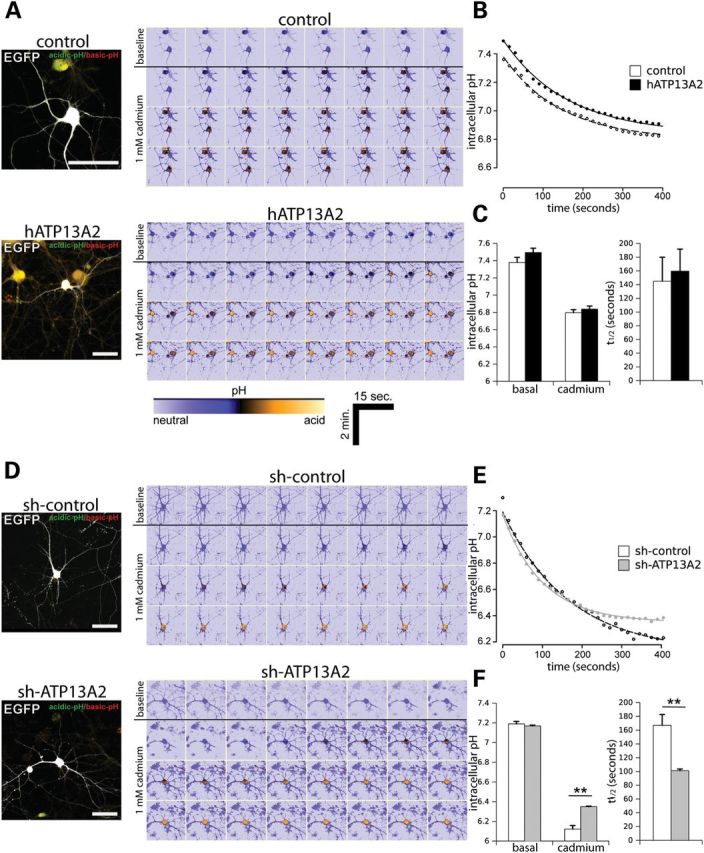
Silencing of ATP13A2 expression regulates the kinetics of intracellular pH in neurons. (A) Rat primary cortical neurons were co-transfected with V5-tagged human ATP13A2 (or control empty vector) and EGFP plasmids at a 10:1 molar ratio at DIV 9 to label transfected neurons. At DIV 12, neurons were loaded with SNARF-1 fluorescent dye to monitor the intracellular pH of individual EGFP-positive neurons by time-lapse, live-cell confocal microscopy. Following time-lapse imaging of baseline pH over a 120 s period, neurons were exposed to 1 mm cadmium and EGFP-positive neurons were imaged over a 360 s period. Representative images of EGFP-positive neurons loaded with SNARF-1 dye are shown in the left image, and pseudo-color encoded mosaics indicating basal (top row) and progressive cadmium-induced acidification (bottom rows) with time are shown in the right image. Warmer tones indicate acidic pH as indicated by lower color scale bar. Scale bar: 50 µm. (B) Average time course of intracellular pH induced by acute cadmium exposure fitted to a first-order kinetic model (AR3). Cadmium was added at time point 0 s. (C) Fitted parameters of basal and peak intracellular pH after cadmium exposure (left graph) and the rate of pH change following cadmium exposure (t1/2 in seconds; right graph) are indicated. (D–F) Similar experiments were conducted on primary cortical neurons co-transfected with non-silencing control shRNA or ATP13A2-specific shRNA and EGFP plasmids at a 10:1 molar ratio. (D) Representative confocal images of EGFP-positive cortical neurons loaded with SNARF-1 under basal conditions or induced by acute cadmium exposure. (E) Average time course of intracellular pH following cadmium treatment (fitted to AR3 model) and (F) basal and peak pH (left) or rate (right) induced by cadmium exposure. Data represent n = 15 neurons/condition sampled from five independent experiments. **P < 0.01 compared with control condition assessed by unpaired Student's t-test.
Modulation of ATP13A2 expression in neurons reduces intracellular calcium levels
The regulation of intracellular pH by ATP13A2 most likely reflects contributions from many cations in addition to H+ ions. A previous report demonstrated that overexpression of ATP13A4, a related member of the P5 ATPase subfamily, caused an increase in intracellular calcium levels in COS-7 cells (25). Therefore, we decided to explore the effects of modulating ATP13A2 expression on intracellular calcium levels. Primary cortical neurons were co-transfected with V5-tagged human ATP13A2 and DsRed2 constructs at a 10:1 molar ratio to label transfected cells, and intracellular calcium levels were monitored in individual DsRed2-labeled neurons by live-cell confocal microscopy using the fluorescent calcium indicator, Fluo-4 (Fig. 7A–J). Overexpression of ATP13A2 leads to a marked decrease in basal intracellular calcium levels in cortical neurons compared with control neurons expressing DsRed2 alone (Fig. 7A and B). We next monitored alterations in intracellular calcium levels in real time following acute exposure to 1 mm cadmium. Cadmium induces a marked increase in intracellular calcium levels measured over 200 s in control neurons, whereas neurons overexpressing ATP13A2 display a smaller increase in calcium levels (Fig. 7B and C). ATP13A2 overexpression does not alter the rate or maximum levels of cadmium-induced calcium release in cortical neurons (Fig. 7C–E). We also assessed the effects of ATP13A2 silencing on intracellular calcium release in cortical neurons (Fig. 7F–J). Similar to ATP13A2 overexpression, ATP13A2 silencing reduces basal and cadmium-induced intracellular calcium levels (Fig. 7F and G), but does not influence the rate or maximum levels of cadmium-induced calcium release (Fig. 7H–J). Our initial experiments demonstrate that cadmium induces the release of calcium from intracellular stores, rather than as a result of increased extracellular calcium uptake (data not shown). Manganese exposure fails to alter intracellular calcium levels in neurons, and therefore, it was not possible to assess the effects of ATP13A2 expression (data not shown). Our data suggest that ATP13A2 regulates basal and cadmium-induced intracellular calcium levels in neurons, thus further supporting a role for ATP13A2 in regulating intracellular cation transport.
Figure 7.
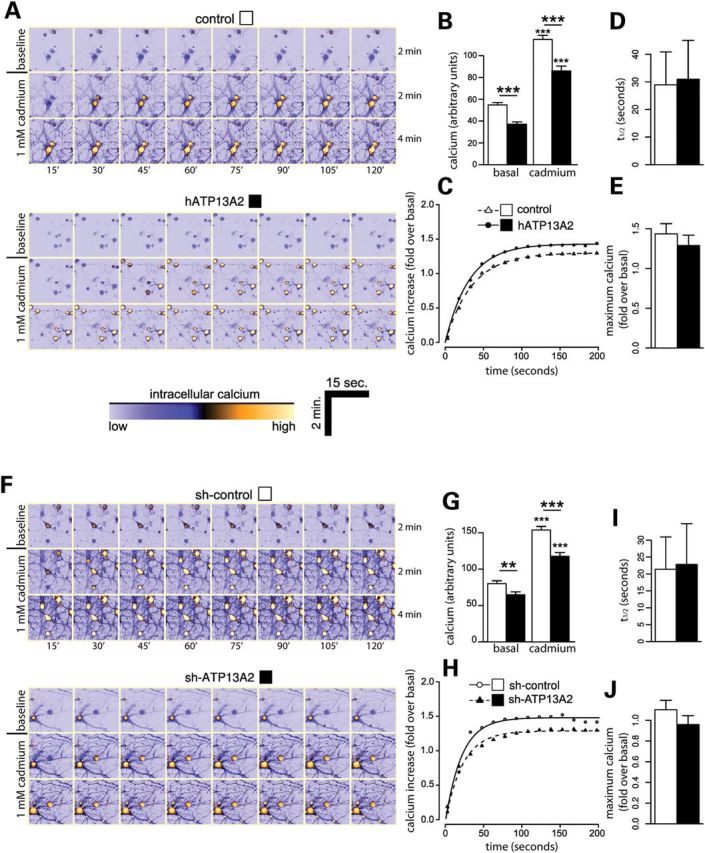
Modulation of ATP13A2 expression reduces basal intracellular calcium concentration. (A) Rat primary cortical neurons were co-transfected with V5-tagged human ATP13A2 (or control empty vector) and DsRed plasmids at a 10:1 molar ratio at DIV 9 to label transfected neurons. At DIV 12, neurons were loaded with Fluo-4 AM fluorescent dye to monitor the intracellular calcium levels of individual DsRed-positive neurons by time-lapse, live-cell confocal microscopy. Representative time-lapse series of baseline intracellular calcium levels (over 120 s) and following acute cadmium exposure (over 240 s) in DsRed-positive neurons. Warmer tones indicate higher intracellular calcium concentrations as indicated by lower color scale bar. (B) ATP13A2 overexpression reduces the levels of basal and cadmium-induced intracellular calcium in neurons. (C) Time course of average intracellular calcium levels induced by cadmium exposure (at time 0 s) expressed as increased calcium levels over initial basal levels for each neuron. Data were fitted to a first-order kinetic model, and the mean (D) rate (half-life, t1/2) and (E) maximum peak calcium levels were determined. (F–J) Similar experiments were conducted on primary cortical neurons co-transfected with non-silencing control shRNA or ATP13A2-specific shRNA and DsRed plasmids at a 10:1 molar ratio. (F) Representative time-lapse series of baseline and cadmium-induced intracellular calcium levels. (G) ATP13A2 silencing reduces the levels of basal and cadmium-induced intracellular calcium in neurons. (H) Time course of intracellular calcium levels induced by cadmium exposure and determination of mean (I) rate and (J) maximum calcium levels. Data represent n = 15 neurons/condition sampled from five independent experiments. **P < 0.01 or ***P < 0.005 compared with the appropriate control condition as indicated by lines, or by comparing basal versus cadmium conditions, assessed by unpaired Student's t-test.
Silencing of ATP13A2 expression induces mitochondrial fragmentation in neurons
Mitochondria are important for buffering intracellular calcium and for maintaining neuronal viability (26). Since manipulating ATP13A2 expression reduces intracellular calcium levels and disrupts dopaminergic neuronal integrity, we assessed the impact of modulating ATP13A2 expression on mitochondrial health. Mitochondria normally undergo dynamic cycles of fission and fusion, but following dysfunction or damage, mitochondria can become fragmented which often precedes neuronal cell death (27). To examine the impact of ATP13A2 overexpression on mitochondrial integrity, primary cortical neurons were co-transfected with V5-tagged human ATP13A2 and mito-DsRed2 constructs at a 10:1 molar ratio, and the morphology of DsRed2-labeled mitochondria was assessed in individual neurons by live-cell confocal microscopy (Fig. 8A–C). The overexpression of ATP13A2 does not influence mitochondrial length under basal conditions or following cadmium exposure (Fig. 8A and C). Acute exposure of cortical neurons to 1 mm cadmium induces a further decrease in mitochondrial length monitored over a period of 300 s (Fig. 8B). ATP13A2 overexpression leads to a reduced rate of mitochondrial fragmentation in response to cadmium exposure in cortical neurons (Fig. 8B and C). The delayed mitochondrial fragmentation induced by overexpression of wild-type ATP13A2 was prevented by introduction of the disease-associated missense mutation, F182L (Supplementary Material, Fig. S4), a putative loss-of-function homozygous mutation identified in a Japanese subject with early-onset parkinsonism (13). To examine the impact of ATP13A2 silencing on mitochondrial integrity, primary cortical neurons were co-transfected with shRNA and mito-DsRed2 constructs at a 10:1 molar ratio as described earlier (Fig. 8D–F). Silencing of ATP13A2 expression induces a marked decrease in mitochondrial length consistent with a fragmented phenotype compared with control shRNA (Fig. 8D and F). ATP13A2 silencing further leads to an increased rate of mitochondrial fragmentation in response to cadmium exposure in cortical neurons (Fig. 8E and F). The effect of ATP13A2 silencing on mitochondrial integrity was also assessed in primary midbrain dopaminergic neurons. Midbrain cultures were co-transfected at DIV 3 with shRNA and mito-GFP constructs at a 10:1 molar ratio to label transfected neurons, fixed at DIV 7 and immunostained with anti-TH antibody to identify dopaminergic neurons (Supplementary Material, Fig. S5). ATP13A2 silencing promotes the fragmentation of mitochondria in dopaminergic neurons compared with control shRNA (Supplementary Material, Fig. S5). Together, our data suggest that silencing of ATP13A2 expression in cortical and dopaminergic neurons promotes basal mitochondrial fragmentation and sensitizes to the effects of cadmium exposure, whereas oppositely, overexpression of ATP13A2 in cortical neurons delays cadmium-induced mitochondrial fragmentation.
Figure 8.

Silencing of ATP13A2 expression induces mitochondrial fragmentation in neurons. (A) Rat primary cortical neurons were co-transfected with V5-tagged human ATP13A2 (or control empty vector) and mito-DsRed2 plasmids at a 10:1 molar ratio at DIV 9 to label transfected neurons. At DIV 12, mito-DsRed2-labeled mitochondria in individual neurons were monitored by time-lapse, live-cell confocal microscopy. Shown are representative time-lapse image series of mitochondria at baseline (over 120 s) and following acute cadmium exposure (over 240 s) in neurons. (B) Time course of average mitochondrial length induced by acute cadmium exposure (at time 0 s) indicating delayed cadmium-induced fragmentation in neurons overexpressing human ATP13A2. Data were fitted to a second-order kinetic model. (C) ATP13A2 overexpression does not alter mean basal or cadmium-induced (peak) mitochondrial length (left graph), but increases the half-life (t1/2) of cadmium-induced mitochondrial fragmentation (right graph). (D–F) Similar experiments were conducted on primary cortical neurons co-transfected with non-silencing control shRNA or ATP13A2-specific shRNA and mito-DsRed2 plasmids at a 10:1 molar ratio. (D) Representative time-lapse image series of mitochondria at baseline and following cadmium treatment. (E) Time course of average mitochondrial length induced by cadmium exposure (at time 0 s) and determination of (F) mean basal and cadmium-induced (peak) length or the rate of mitochondrial fragmentation. ATP13A2 silencing reduces basal and cadmium-induced mitochondrial length (left graph) and reduces the half-life of cadmium-induced fragmentation (right graph). Data represent n = 15 neurons/condition sampled from five independent experiments. *P < 0.05 or ***P < 0.005 compared with the appropriate control condition as indicated by lines, or by comparing basal versus cadmium conditions, assessed by unpaired Student's t-test.
DISCUSSION
Mutations in the ATP13A2 gene cause KRS and early-onset parkinsonism (4,6,10–12,28). The normal function of ATP13A2 and the pathogenic mechanism(s) by which recessive mutations precipitate neurodegeneration are poorly understood. Here, we demonstrate that endogenous ATP13A2 is enriched in the microsomal fraction from mouse brain, and exogenous ATP13A2 localizes to intracellular acidic vesicles within mammalian neurons, particularly lysosomes and endosomes, and to a smaller extent autophagosomes. We further demonstrate that ATP13A2 protein is localized to neuronal populations of the human cerebral cortex and substantia nigra where this protein is increased in PD and DLB brains. The overexpression or silencing of ATP13A2 compromises the integrity of midbrain dopaminergic neurons, leading to reduced neurite length but without evidence of overt cell death. Modulation of ATP13A2 expression in cortical neurons modestly alters the size and number of LC3-positive autophagosomes but without an obvious effect on autophagic activation. Consistent with a potential role in regulating vesicular cation transport, we find that silencing of endogenous ATP13A2 in cortical neurons modulates the kinetics of intracellular pH following cadmium-induced acidification and reduces intracellular calcium levels. Overexpression of human ATP13A2 similarly reduces intracellular calcium levels but without influencing pH. Finally, we show that silencing of ATP13A2 in neurons induces mitochondrial fragmentation, whereas ATP13A2 overexpression delays mitochondrial fragmentation induced by cadmium exposure. Collectively, our study reveals a number of intriguing phenotypes owing to loss- or gain-of-function of ATP13A2 and suggests a role for ATP13A2 in regulating vesicular cation transport and neuronal integrity. Furthermore, our data potentially implicate a role for ATP13A2 in idiopathic PD.
Using a specific polyclonal antibody to ATP13A2, we demonstrate the enrichment of ATP13A2 in the microsomal fraction from mouse brain which contains lysosomes, the Golgi complex and ER. We further demonstrate the localization of exogenous ATP13A2 to intracellular vesicular compartments within cortical neurons, particularly lysosomes, early and late endosomes, microtubule-associated vesicles and to a smaller extent autophagosomes. Previous studies examining exogenous ATP13A2 have demonstrated its localization largely to lysosomes in mammalian cell lines (6,8,9,18). Therefore, the function of ATP13A2 may be broader than first anticipated with a preference for vesicular compartments that are known to be acidic. This might suggest a role for ATP13A2 in regulating the acidity of such vesicles, similar to the vesicular vacuolar-type H+-ATPase, through the active transport of cations across vesicular membranes in an ATP-dependent manner. In yeast, an ortholog of ATP13A2 termed Ykp9 is localized to the vacuole where it is implicated in regulating the transport of heavy metal ions, including cadmium, nickel, manganese and selenium (16,17). Of note, however, human ATP13A2 is unable to complement the phenotype of ykp9 null yeast, indicating a lack of functional conservation between Ykp9 and ATP13A2 (16). The transport of heavy metal ions by mammalian ATP13A2 has not yet been directly demonstrated, and consequently, the cation selectivity of this transporter is not known. A recent study has shown that the overexpression of human ATP13A2 regulates intracellular manganese concentration and protects from manganese-induced toxicity in mammalian cell lines (18). Whether or not these effects relate to the direct ATP-driven transport of manganese ions across membranes by ATP13A2 remains to be determined, and whether ATP13A2 can similarly regulate the homeostasis of other heavy metal ions is not clear.
In the human brain, we demonstrate the localization of ATP13A2 to various neuronal populations, including pyramidal neurons throughout the cerebral cortex and dopaminergic neurons of the substantia nigra. Furthermore, we detect ATP13A2 protein migrating as an ∼130 kDa species in extracts of human striatum and medial frontal gyrus. A previous study has reported the expression of ATP13A2 mRNA throughout the human and mouse brain where it appears to adopt a neuronal localization with enrichment in the ventral midbrain and expression in individual dopaminergic neurons (6). We can confirm that ATP13A2 protein is similarly localized to neurons, including nigral dopaminergic neurons, throughout the mouse (data not shown) and human brain. We further demonstrate that ATP13A2 protein levels are increased in surviving substantia nigra dopaminergic neurons and cortical pyramidal neurons of PD brains compared with normal control brains. These findings are similar to a previous study which estimated a 10-fold increase in ATP13A2 mRNA in individual nigral dopaminergic neurons from PD brains compared with control brains (6). The smaller increase in protein levels compared with mRNA levels of ATP13A2 in PD brains could relate to inefficient translation of mRNA, reduced mRNA stability or to differences in post-mortem brain tissue. Nevertheless, these data collectively indicate that ATP13A2 protein levels are increased in vulnerable neurons from the substantia nigra, striatum and cerebral cortex of PD brains. The reason for this increase is unclear, but could relate to a compensatory neuroprotective response in neurons exposed to PD and/or DLB pathogenic processes that potentially correlates with up-regulation of the autophagy–endosomal–lysosomal pathways. We have previously shown that the autophagosomal marker LC3 is increased in neurons in vulnerable areas of DLB brains (29). Moreover, we demonstrate that ATP13A2 protein is commonly detected in neurons harboring LB pathology and that the levels of cytoplasmic ATP13A2 are increased in LB-positive neurons compared with neurons without LBs. While ATP13A2 does not co-localize with LBs directly, this might suggest a compensatory up-regulation of ATP13A2 or acidic vesicles in response to LBs. It is unlikely that increased ATP13A2 levels directly impair neuronal viability since (i) this protein is increased in ‘surviving’ nigral dopaminergic neurons of PD brains, (ii) the overexpression of ATP13A2 is reported to protect from neuronal toxicity induced by α-synuclein and manganese (16,18) and (iii) loss of function of ATP13A2 due to recessive mutations induces neurodegeneration (4,6,7), consistent with a normal role for ATP13A2 in neuronal maintenance and/or neuroprotection in humans.
In attempting to model the pathogenic effects of recessive, loss-of-function ATP13A2 mutations in primary midbrain dopaminergic neurons, we find that silencing of endogenous ATP13A2 expression reduces neurite outgrowth but without causing neuronal cell death. This effect is somewhat selective since silencing of ATP13A2 in primary cortical neurons does not impair neurite outgrowth or induce cell death. Surprisingly, the overexpression of human ATP13A2 also markedly reduces neurite outgrowth of dopaminergic neurons. Therefore, it appears that there is a critical range of ATP13A2 levels required for the proper maintenance of dopaminergic neuronal processes. Conceivably, ATP13A2-mediated cation transport across vesicular membranes is a tightly regulated process, and dysregulation of this process could potentially impact the normal function of acidic vesicles, axonal transport and the subsequent maintenance of neuronal processes. Recent studies have shown that the overexpression of ATP13A2 orthologs protects from toxicity induced by mutant α-synuclein in yeast, worm and cultured primary dopaminergic neuronal models (16). Although we have not been able to show a detrimental effect of ATP13A2 overexpression on neuronal viability, we find instead that the integrity of neuronal processes is compromised. Therefore, a potential note of caution is warranted for neuroprotective strategies that seek to increase ATP13A2 levels or activity in pre-clinical models of PD. It is interesting to note that the lentiviral-mediated overexpression of ATP13A2 in cultured dopaminergic neurons appears to cause mild neuronal loss (16). However, in our hands, using single-cell analysis, we do not observe obvious neuronal loss or apoptotic cell death. The functional implications of reduced neurite length induced by silencing or overexpression of ATP13A2 in dopaminergic neurons are unclear at present and will await the future development and analysis of knockout or knockdown mice. Similarly, the reported protective effects of ATP13A2 overexpression against α-synuclein-induced toxicity will require confirmation in animal models of PD (16).
Modulation of ATP13A2 expression in cultured primary cortical neurons produces a range of intracellular phenotypes. ATP13A2 appears to modestly influence the number and size of LC3-positive autophagosomes, but does not appreciably alter autophagic activation based on the levels of the autophagosomal marker, LC3-II. Similar experiments conducted in human neural cell lines did not reveal a consistent effect of modulating ATP13A2 expression on autophagic activation or flux (data not shown). Instead, in cortical neurons, we observe a reduction in steady-state total GFP-LC3 levels following ATP13A2 silencing or overexpression which may indicate increased lysosomal turnover. We also demonstrate that ATP13A2 silencing modestly increases the rate of intracellular acidification induced by acute cadmium exposure but without an effect on basal pH. These data may suggest that lysosomes and related acidic vesicles exhibit less tolerance to alterations in pH following ATP13A2 silencing, indicating that this protein may play a selective role in maintaining the homeostasis of H+ ions under conditions of cellular stress. It is not clear whether vesicular pH is similarly affected by ATP13A2 silencing. Yeast cells lacking ykp9 appear most sensitive to growth on media containing cadmium when compared with other heavy metals (17). Our data tend to support a functional connection between ATP13A2 and cadmium, whereas manganese exposure failed to influence intracellular pH in these assays, thus preventing a similar functional assessment.
We also find that both silencing and overexpression of ATP13A2 reduce the basal levels of intracellular calcium ions, an effect that is maintained following cadmium-induced calcium release. While these observations do not support a direct relationship between ATP13A2 and cadmium in these assays, they instead suggest that ATP13A2 may play a role in regulating calcium homeostasis. Whether or not this effect is through direct ATP13A2-mediated transport and sequestration of calcium ions into acidic vesicles is unclear at present, but it could also result from a general impairment of cation homeostasis in neurons which might indirectly influence calcium storage and/or availability. The ability to release intracellular calcium induced by cadmium exposure is still intact in neurons following ATP13A2 silencing, suggesting a modest but not essential role for ATP13A2 in the sequestration of calcium into intracellular stores. Such a role for ATP13A2 may potentially relate to an indirect effect on the general ionic homeostasis of neurons due to the dysregulation of acidic vesicles. ATP13A4, a related member of the P5-type ATPase subfamily, is localized to the ER where its overexpression in COS-7 cells increases intracellular calcium levels (25). Therefore, two P5-type ATPases are potentially involved in calcium regulation albeit within distinct subcellular locations, i.e. ATP13A4 in the ER and ATP13A2 in lysosomes and related acidic vesicles. It will be interesting to determine whether additional P5-type ATPases are also involved in regulating intracellular calcium levels. We further show that ATP13A2 influences mitochondrial morphology potentially as a precursor to mitochondrial damage. Similar to the effects on intracellular calcium, silencing of ATP13A2 expression induces basal mitochondrial fragmentation and increases the rate of mitochondrial fragmentation induced by cadmium exposure. The overexpression of ATP13A2 did not influence mitochondrial morphology, but instead delayed cadmium-induced mitochondrial fragmentation consistent with a neuroprotective effect in this paradigm. ATP13A2 harboring an F182L mutation, identified as a homozygous variant in a Japanese subject with early-onset parkinsonism (13), failed to similarly delay mitochondrial fragmentation, suggesting that this disease variant causes a loss of function. The effects of modulating ATP13A2 expression on mitochondrial fragmentation do not initially appear to correlate with their effects on intracellular calcium levels and pH. Furthermore, ATP13A2 fails to co-localize with mitochondria in cortical neurons (data not shown), suggesting an indirect effect on mitochondria potentially through altered vesicular cation transport and cation homeostasis.
Our data clearly demonstrate that silencing of ATP13A2 expression, which is anticipated to model recessive disease, is consistently detrimental to neurons by manifesting dopaminergic neurite shortening and by altering the regulation of intracellular pH, reducing intracellular calcium levels and inducing mitochondrial fragmentation in primary cortical neurons. Therefore, neuronal phenotypes due to ATP13A2 silencing may provide potential insight into the molecular pathways underlying neurodegeneration due to recessive ATP13A2 mutations in KRS and early-onset parkinsonism. However, for reasons that are not yet clear, the overexpression of human ATP13A2 produces varied phenotypes including dopaminergic neurite shortening and reduced intracellular calcium levels, protection from cadmium-induced mitochondrial fragmentation, but no effect on intracellular pH regulation. Thus, ATP13A2 overexpression appears to produce both detrimental and protective effects on neurons, depending on the specific phenotype assessed. It will be important to determine in future experiments whether or not ATP13A2 overexpression produces an overall beneficial neuroprotective effect in cultures and in vivo, consistent with previous studies reporting a protective effect against cytotoxicity induced by α-synuclein overexpression and manganese exposure (16,18). Collectively, our data support a role for ATP13A2 in the maintenance of neuronal integrity and the regulation of intracellular cation homeostasis, autophagy and mitochondrial health. These broad cellular and subcellular effects may relate to the localization of ATP13A2 to intracellular vesicular compartments within neurons. Silencing of ATP13A2 in primary neurons may provide a useful model for understanding the normal function of this protein and for elucidating the molecular mechanisms underlying neurodegeneration due to recessive ATP13A2 mutations.
MATERIALS AND METHODS
Animals
Rats and mice were housed and treated in strict accordance with the Swiss Legislation (Canton de Vaud, Animal Authorization No. 2293) and the European Community Council directive (2010/63/EU) for the care and use of laboratory animals. Animals were maintained in a pathogen-free barrier facility and exposed to a 12 h light/dark cycle with food and water provided ad libitum. Pregnant female Sprague-Dawley rats were obtained from Charles River Laboratories (L'Arbresle Cedex, France), and resulting P0 rats were used for the preparation of post-natal primary neuronal cultures.
Expression plasmids, antibodies and reagents
A mammalian expression plasmid containing full-length V5-tagged human ATP13A2 was kindly provided by Dr Christian Kubisch (University of Cologne, Germany) (6). A disease-associated missense mutation (F182L) was introduced into V5-tagged ATP13A2 by site-directed mutagenesis using the Stratagene QuickChange II XL kit (Agilent Technologies, La Jolla, CA, USA), according to manufacturer's instructions. The integrity of this construct was confirmed by DNA sequence analysis. A plasmid containing full-length untagged mouse ATP13A2 in pCMV-SPORT6 was obtained from Open Biosystems (Huntsville, AL, USA). Mouse ATP13A2-EGFP was generated by polymerase chain reaction from plasmid pCMV-SPORT6-mATP13A2 using forward (5′-TACTCGAGGCGAGGAGCCGAAATGAGCGC-3′) and reverse (5′-TAACCGGTCCCCTCACGGAGCCAACGGG CA-3′) primers and cloned in-frame into 5′ XhoI and 3′ AgeI sites of plasmid pEGFP-N1 (Clontech, Mountain View, CA, USA). Mir-30-adapted shRNA sequences in retroviral plasmid pSM2c targeting rodent ATP13A2 (sh-ATP13A2, V2MM_84755) or a non-silencing control (sh-control, V2MM_84756) were obtained from Open Biosystems. GIPZ lentiviral plasmids co-expressing mir-30-adapted shRNAs targeting rodent ATP13A2 (sh-ATP13A2 #1, V2LMM_84755; sh-ATP13A2 #2, V3LMM_461110) or a non-silencing control (sh-control, V3LMM_461109) and turbo GFP were obtained from Open Biosystems. As plasmid controls, a pEGFP-N1 plasmid was obtained from Clontech, a pDsRed-Max-N1 plasmid was from Addgene [plasmid no. 21718, Cambridge, MA, (30)] and a pcDNA3.1 plasmid was obtained from Invitrogen (Basel, Switzerland). Expression plasmids containing human GFP-LC3B [plasmid no. 11546, (31)], human RFP-Rab5A [plasmid no. 14437, (32)], human GFP-Rab7A [plasmid no. 12605, (33)] and rat LAMP1-RFP [plasmid no. 1817, (34)] were obtained from Addgene. Mito-DsRed2, mito-GFP and mito-RFP plasmids were kindly provided by Dr Manuel Rojo (Université Victor Segalen, France). The following antibodies were employed: mouse monoclonal anti-MAP2 (clone HM-2), anti-TH (clone TH-2) and anti-β-tubulin (clone TUB 2.1) and rabbit polyclonal anti-ATP13A2 (A3361, human residues 804–821 and A9762, human residues 1162–1180), anti-MAP2, anti-βIII-tubulin and anti-β-actin (Sigma-Aldrich, Buchs, Switzerland); mouse monoclonal anti-synaptophysin 1 (Synaptic Systems, Göttingen, Germany); mouse monoclonal anti-LAMP1 (clone LY1C6; EMD Millipore, Billerica, MA, USA); rabbit polyclonal anti-Giantin (ab24586; Abcam, Cambridge, UK); rabbit monoclonal anti-PDI (clone C81H6; Cell Signaling Technology, Danvers, MA, USA); mouse monoclonal anti-TIM23 (clone 32; BD Biosciences, Allschwil, Switzerland); mouse monoclonal anti-phospho-Ser129-α-synuclein (Wako Chemicals GmbH, Neuss, Germany); peroxidase-coupled anti-mouse and anti-rabbit IgG, light chain-specific secondary antibodies (Jackson ImmunoResearch, Inc., West Grove, PA, USA) and anti-rabbit IgG and anti-mouse IgG coupled to AlexaFluor-488, -546 and -633 (Invitrogen). Fluo-4 AM and SNARF-1 AM fluorescent indicators were obtained from Invitrogen. Cadmium chloride (CdCl2) and manganese chloride (MnCl2) were obtained from Sigma.
Generation of ATP13A2 antibodies
A synthetic peptide (CLRINLGGKLQLV) corresponding to residues 499–511 of human ATP13A2 was used for antibody generation. This peptide sequence shares high conservation with rat and mouse ATP13A2. An N-terminal cysteine residue (underlined) was added to facilitate conjugation to keyhole limpet hemocyanin. Immunization of rabbits and affinity purification of antibodies on columns containing peptide-immobilized Sulfolink gel matrix (Pierce Biotechnology, Rockford, IL, USA) were performed as described previously (35–37).
Cell culture and transient transfection
HEK-293T cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen), supplemented with 10% fetal bovine serum and 1× penicillin/streptomycin at 37°C in a 5% CO2 atmosphere. Cells were transfected with plasmid DNAs using FuGENE HD reagent (Roche Applied Sciences, Basel, Switzerland), according to manufacturer's recommendations. For biochemical assays, cells were routinely harvested at 48–72 h post-transfection.
Primary neuronal cultures and transfection
Primary cortical and midbrain neurons were prepared from rats, as described previously (38). Whole brains were dissected from Sprague-Dawley P0 rats, and the cerebral cortices and ventral midbrain (containing the substantia nigra and ventral tegmental area) were isolated stereoscopically and dissociated by digestion in media containing papain (20 U/ml; Sigma-Aldrich) and mechanical trituration. Cells were plated in 35 mm dishes onto 10 mm glass cover slips or in 35 mm IBIDI micro-dishes coated with poly-d-lysine (20 ng/ml; BD Biosciences) and mouse laminin (33 μg/ml; Invitrogen) and cultured in Neurobasal media containing B27 supplement (2% w/v), l-glutamine (500 μm) and penicillin/streptomycin (100 U/ml) (Invitrogen). At DIV 3, cortical and midbrain cultures were treated with cytosine β-arabinofuranoside (AraC, 10 μm) to inhibit glial cell division. Primary cortical or midbrain neurons were transfected with plasmid DNAs using Lipofectamine 2000 reagent (Invitrogen), according to manufacturer's instructions to a maximum of 5 μg total DNA per 35 mm dish of cells. Cultures were normally maintained up to DIV 12 without replacement of growth media.
Immunocytochemistry and confocal microscopy on primary neurons
For co-localization of ATP13A2 and subcellular markers, primary cortical cultures transiently co-expressing human ATP13A2-V5 and GFP-LC3, RFP-Rab5A, GFP-Rab7A or LAMP1-RFP fusion proteins were fixed in 4% paraformaldehyde (PFA) containing 4% sucrose in phosphate-buffered saline (PBS), permeabilized with 0.3% Triton X-100 and sequentially incubated with mouse anti-V5 antibody (Invitrogen) and either rabbit anti-MAP2 (Sigma) or anti-βIII-tubulin (Sigma) antibodies. Cortical neurons expressing mouse ATP13A2-GFP were processed for immunocytochemistry with rabbit anti-ATP13A2 (LMNR1), mouse anti-MAP2 (Sigma) or mouse anti-synaptophysin 1 (Synaptic Systems). After washing, sections were incubated with the appropriate AlexaFluor-conjugated secondary antibodies (Invitrogen) and mounted on glass slides with Vectashield mounting medium containing DAPI (Vector Laboratories, Burlingame, CA). Fluorescent images were acquired using a Zeiss LSM 700 inverted confocal microscope (Carl Zeiss AG, Feldbach, Switzerland) with a Plan-Apochromat 63×/1.40 oil objective in x-, y- and z-planes. Image analysis was performed using Fiji software (Image Processing and Analysis in Java) or Imaris software (Bitplane AG, Zurich, Switzerland). Images were subjected to deconvolution using HuygensPro software (Scientific Volume Imaging, Hilversum, The Netherlands). Pearson's correlation coefficients (Rcoloc) were calculated in Fiji using z-stacks between the indicated fluorescent channels. Representative images are taken from a single z-plane at a thickness of 0.1 µm.
Cell fractionation and western blotting
For western blotting, HEK-293T cells maintained in growth medium in six well plates (250 000 cells/well) were transiently transfected with 2 μg of each plasmid DNA. After 48 h, cells were harvested in media, resuspended in lysis buffer [1× PBS pH 7.4, 1% Triton X-100, Complete Mini protease inhibitor cocktail (Roche Applied Sciences)] and rotated at 4°C for 1 h. Lysates were centrifuged at 17 500g for 10 min at 4°C, and the resulting supernatant (Triton-soluble) fraction was collected. Triton-soluble fractions were combined 1:1 with 2× Laemmli sample buffer (Bio-Rad AG, Reinach, Switzerland) containing 5% 2-mercaptoethanol and resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (7.5%), transferred to Protran nitrocellulose (0.2 μm; Perkin Elmer, Schwerzenbach, Switzerland) and subjected to western blot analysis. Nitrocellulose membranes were probed with anti-ATP13A2 antibody (LMNR1), anti-β-tubulin or anti-β-actin antibodies (Sigma-Aldrich) and peroxidase-coupled anti-mouse or anti-rabbit IgG (Jackson ImmunoResearch) antibodies. Proteins were visualized by enhanced chemiluminescence (GE Healthcare, Glattbrugg, Switzerland) on a FujiFilm LAS-4000 Luminescent Image Analysis system. Quantitation of protein levels by densitometry was conducted on acquired images using LabImage 1D software (Kapelan Bio-Imaging Solutions, Leipzig, Germany).
Human brain tissue
Post-mortem human brain tissue was obtained through the brain donation program of the Morris K. Udall Parkinson's Disease Research Center of Excellence at Johns Hopkins Medical Institutions in compliance with local Institutional Review Board and Health Insurance Portability and Accountability Act regulations. Fresh-frozen putamen or medial frontal gyrus tissue from four normal control brains and five PD/DLB brains was utilized for western blot analysis. Formalin-fixed, paraffin-embedded tissue sections (10 µm thick) containing midbrain or cingulate cortex tissue from three normal control brains and six PD/DLB brains were used for immunohistochemistry. Table 1 shows the clinical details of human subjects used in this study.
Table 1.
Clinical details of human brain tissue
| Subject | Diagnosis | Brain region | PMD (h) | Age (years) | Use |
|---|---|---|---|---|---|
| 1 | Normal/control | PUT, MFG | 12 | 66 | WB |
| 2 | Normal/control | PUT, MFG | 4 | 74 | WB |
| 3 | Normal/control | PUT, MFG, CING, MB | 8 | 91 | WB, IHC |
| 4 | Normal/control | PUT, MFG | 16 | 79 | WB |
| 5 | Normal/control | MB | 8 | 80 | IHC |
| 6 | Normal/control | CING, MB | 12 | 48 | IHC |
| 1 | PD/DLB | PUT, MFG | 16 | 86 | WB |
| 2 | PD/DLB | PUT, MFG | 19.5 | 88 | WB |
| 3 | PD/dementia | PUT, MFG | 6 | 75 | WB |
| 4 | PD/dementia | PUT, MFG | 17 | 76 | WB |
| 5 | PD/dementia | PUT, MFG | 24 | 71 | WB |
| 6 | PD | CING | 12 | 45 | IHC |
| 7 | PD/AD/LB variant | CING | 18 | 73 | IHC |
| 8 | DLB/AD | CING | 7 | 89 | IHC |
| 9 | PD | MB | 22 | 71 | IHC |
| 10 | PD | CING, MB | 36 | 77 | IHC |
| 11 | PD/AD/DLB | CING | 24 | 63 | IHC |
AD, Alzheimer's disease; CING, cingulate cortex; IHC, immunohistochemistry; MB, midbrain; MFG, medial frontal gyrus; PMD, post-mortem delay; PUT, putamen; WB, western blot.
Fractionation of brain tissue
Detergent-soluble protein fractions were prepared from human brain tissue (putamen and medial frontal gyrus) or whole mouse brain by homogenization of samples in TNE buffer [10 mm Tris–HCl pH 7.4, 150 mm NaCl, 5 mm EDTA, 0.5% NP-40, 1× Complete protease inhibitor cocktail (Roche)]. The homogenate was centrifuged at 100 000g for 20 min at 4°C, and the resulting supernatant fraction was collected. Protein fractions were quantitated using the bicinchoninic acid (BCA) kit (Pierce Biotechnology) with bovine serum albumin standards. Proteins (20–30 µg/lane) were resolved by SDS–PAGE and analyzed by western blotting with anti-ATP13A2 (LMNR1) and anti-β-actin antibodies. For antibody pre-absorption controls, ATP13A2 antibody (LMNR1) was pre-incubated with an excess of peptide antigen (200 µg/ml) in Tris-buffered saline overnight at 4°C prior to western blot analysis of mouse brain extracts.
Subcellular fractionation of brain tissue
Subcellular fractionation was conducted as described previously (35,39) using whole brain tissue from adult C57BL/6J mice. Briefly, mouse brain homogenates were subjected to centrifugation at 800g for 10 min to generate pellet (P1, nuclear/whole cell) and soluble (S1, cytosolic) fractions. S1 fractions were centrifuged at 9200g for 15 min to produce P2 (heavy and crude synaptosomal membranes) and S2 (soluble cytosolic) fractions. The P2 fraction was solubilized and centrifuged at 25 000g for 20 min to produce LP1 (synaptosomal membranes) and LS1 (synaptosomal cytosolic) fractions. The LS1 fraction was further fractionated by ultracentrifugation at 165 000g for 2 h to produce LP2 (synaptic vesicle-enriched) and LS2 (synaptic vesicle cytosolic) fractions. The S2 fraction was subjected to ultracentrifugation at 165 000g for 2 h to produce P3 (light membranes/microsomes) and S3 (soluble cytosolic) fractions. Protein concentrations were determined by BCA assay (Pierce Biotechnology), and equal quantities of each fraction were validated by western blotting with specific antibodies labeling mitochondria (TIM23; S1, P2 and LP1), ER (PDI; P2, P3, LP1 and LP2), Golgi complex (Giantin; P2 and P3), synaptosomes/synaptic vesicles (synaptophysin 1; P2, P3, LP1 and LP2) and lysosome (LAMP1; P3 and S3) subcellular compartments, in addition to ATP13A2 (LMNR1 antibody).
Immunohistochemistry and immunofluorescence on human brains
Human midbrain and cingulate cortex tissue sections were subjected to paraffin removal at 60°C for 30 min, xylene treatment and rehydration through a graded ethanol series. Antigen unmasking was performed by heating at 100°C for 20 min in 10 mm sodium citrate buffer pH 6.0. DAB immunohistochemistry was performed on all sections using rabbit anti-ATP13A2 antibodies (LMNR1, A9762/Sigma or A3361/Sigma). Sections were counterstained with Nissl and dopaminergic neurons were identified by neuromelanin content, whereas cortical layers were identified by characteristic laminar cytoarchitecture. Slides were imaged on an Olympus AX70 motorized microscope using a UPLAN FL 4×/0.13 or 20×/0.5 objective, and mosaic images comprising the substantia nigra and cortex were assembled for three PD and three control cases (for LMNR1 antibody only), comprising 1257 and 1449 neurons, respectively. The intensity of DAB immunostaining within these neurons was subjected to semi-quantitative morphometric analysis using NIH ImageJ software as described previously (29,40) and expressed in arbitrary units. For the cortex, at least two different areas separated by at least 5 mm were analyzed. For the co-localization analysis of ATP13A2 with LBs, following antigen unmasking, the sections were sequentially incubated with rabbit anti-ATP13A2 antibody (LMNR1) and mouse anti-phospho-Ser129-α-synuclein (Wako Chemicals) antibody. High resolution z-stacks were acquired on a Leica SP2 confocal microscope and were subjected to deconvolution with HuygensPro software. Six distinct regions of cingulate cortex were analyzed from a single DLB subject (no. 8), encompassing 2179 ATP13A2-positive neurons and 2630 LBs. All image processing was performed in ImageJ, and data were statistically post-processed in R software.
Neurite length assays
Primary midbrain cultures at DIV 3 were co-transfected with V5-tagged human ATP13A2 (or control empty vector) or mir-30-adapted shRNAs (sh-control or sh-ATP13A2 in retroviral vector pSM2c) and EGFP plasmids at a 10:1 DNA molar ratio to mark transfected neurons. At DIV 7, cultures were fixed with 4% PFA and subjected to immunocytochemistry with mouse anti-TH antibody (Sigma-Aldrich) and anti-mouse IgG-AlexaFluor-546 antibody (Invitrogen). Fluorescent images were acquired using an EVOS inverted fluorescence digital microscope (Advanced Microscopy Group, Bothell, WA, USA) with a 10× objective. Images were assembled in ImageJ using a 2D/3D Stitching plugin developed by Stephan Preibisch (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany) to construct the entire neuritic tree of each EGFP+/TH+ neuron. The length of EGFP+/TH+ dopaminergic neurites was measured using the line tool function of ImageJ by an investigator blinded to each condition. Only neurons that had extended neurites were measured, whereas neurons without processes were excluded from the analysis. The longest EGFP+ neurite (i.e. the axon) was measured and used for comparison among groups. We measured neurites from every EGFP+/TH+ dopaminergic neuron identified across five cover slips per experiment from at least four independent cultures (n = 39–42 neurons for shRNAs, or n = 28–39 neurons for hATP13A2/empty vector). For primary cortical neurons, cultures at DIV 3 were transfected with GIPZ constructs co-expressing mir-30-adapted shRNAs and turbo-EGFP (sh-control, sh-ATP13A2 #1 or sh-ATP13A2 #2) or with control empty vector and EGFP plasmids at a 10:1 molar ratio. Cortical neurons were fixed at DIV 7 and subjected to immunocytochemistry with mouse anti-MAP2 antibody (Sigma-Aldrich) and anti-mouse IgG-AlexaFluor-633 antibody (Invitrogen). Fluorescent images were acquired, and EGFP+/MAP+ merged images were pseudo-colored using ICA1 in ImageJ to improve the contrast of neuritic processes and used for neurite length measurements. The length of EGFP+/MAP2+ cortical neurites was determined using ImageJ as described earlier. In each experiment, neuronal processes from cortical neurons randomly sampled across five cover slips from three independent cultures were measured (n = 111–128 neurons/condition).
TUNEL labeling
Primary cortical neurons at DIV 3 were transfected with GIPZ-shRNA-EGFP constructs (sh-control, sh-ATP13A2 #1 or sh-ATP13A2 #2) and fixed with 4% PFA at DIV 7. TUNEL staining was conducted using the In Situ Cell Death Detection Kit (Roche Applied Sciences) containing tetramethylrhodamine red-labeled dUTP as per the manufacturer's instructions. Cultures were further subjected to immunocytochemistry with mouse anti-MAP2 antibody (Sigma-Aldrich) and anti-mouse IgG-AlexaFluor-633 antibody (Invitrogen). Fluorescent microscopic images were acquired of double-positive (GFP+/MAP2+) cortical neurons, and the proportion of TUNEL-positive nuclei was scored. In each experiment, the number of TUNEL-positive nuclei from GFP+/MAP+ neurons (n = 199–222/condition) randomly sampled across two cover slips from three independent experiments was measured. Data represent TUNEL-positive neurons as a percent of total GFP+/MAP2+ neurons (mean ± SEM) for each condition.
Primary midbrain cultures at DIV 3 were co-transfected with shRNAs (sh-control or sh-ATP13A2 in vector pSM2c) and EGFP plasmids at a 10:1 DNA molar ratio to mark transfected neurons. At DIV 7, cultures were fixed with 4% PFA and subjected sequentially to TUNEL staining as described earlier and immunocytochemistry with mouse anti-TH antibody (Sigma-Aldrich) and anti-mouse IgG-AlexaFluor-633 antibody (Invitrogen). Fluorescent microscopic images were acquired of double-positive (GFP+/TH+) dopaminergic neurons, and the proportion of TUNEL-positive nuclei was scored. In each experiment, the number of TUNEL-positive nuclei from GFP+/TH+ neurons (n = 15–18/condition) randomly sampled across three cover slips from two independent experiments was measured.
Analysis of intracellular pH
Live-cell imaging was performed on a Leica SP2 laser scanning confocal microscope equipped with a CO2 environmental chamber and temperature control. Primary cortical neuronal cultures seeded on IBIDI micro-dishes were co-transfected at DIV 9 with V5-tagged human ATP13A2 (or control empty vector) or shRNAs (sh-control or sh-ATP13A2) and EGFP at a 10:1 DNA molar ratio to mark transfected neurons. At DIV 12, neurons were incubated with 1 µm SNARF-1 AM for 20–30 min in media without Phenol red. Imaging was performed on EGFP+ neurons. SNARF-1 was excited with the 561 nm HeNe laser line and simultaneously recorded at 590/10 and 650/20 nm. Time-lapse high-resolution confocal z-stacks were acquired, and the F590/F650 ratio was computed for each voxel. A baseline of 120 s was acquired for each neuron prior to treatment with 1 mm cadmium and imaging for a further 360 s. Intracellular pH was calculated using the equation pH = pKa × log10(R−RB/RA−R) × FB590/FA650, where R is the ratio of fluorescence intensities after subtracting the background, the subscripts A and B the limiting values at acidic and basic endpoints (RA = 2.4 and RB= 0.05), pKa the experimental acid dissociation constant (3.16 × 10−8) and FB590/FA650 the ratio of fluorescence at 650 nm in basic and acid endpoints (0.8). In situ calibration was performed with the potassium ionophore nigericin (10 µm). Neurons were manually segmented into cytosolic soma, nucleus and neurites. Average intracellular pH was calculated for each area.
Intracellular calcium levels
Primary cortical neurons seeded on IBIDI micro-dishes were co-transfected at DIV 9 with V5-tagged human ATP13A2 (or control empty vector) or shRNAs (sh-control or sh-ATP13A2) and DsRed plasmids at a 10:1 DNA molar ratio to mark transfected neurons. At DIV 12, neurons were incubated with 1 µm Fluo-4 AM for 20–30 min in media without Phenol red. Imaging was performed on DsRed+ neurons. Fluo-4 was excited with the 488 nm Argon laser line and recorded at 510/10 nm. Time-lapse high-resolution confocal z-stacks were acquired for each neuron and were manually segmented into cytosolic soma, nucleus and neurites. Average intracellular Fluo-4 fluorescence was calculated for each area, and data were expressed as raw fluorescence values. A baseline of 120 s was acquired for each DsRed+ neuron prior to treatment with 1 mm cadmium and imaging for a further 240 s.
Mitochondrial morphology
Primary cortical neurons seeded on IBIDI micro-dishes were co-transfected at DIV 9 with V5-tagged human ATP13A2 (or control empty vector) or shRNAs (sh-control or sh-ATP13A2) and mito-DsRed2 plasmids at a 10:1 DNA molar ratio to mark transfected neurons. Mito-DsRed2+ neurons were imaged at DIV 12 with a Leica SP2 inverted confocal microscope. A baseline of 120 s was acquired for each neuron prior to treatment with 1 mm cadmium and imaging for a further 240 s. Time-lapse high-resolution confocal z-stacks were subjected to deconvolution with HuygensPro software, and 3D image reconstruction was conducted using ImageJ. Three-dimensional morphometry measures (area, ellipticity, sphericity and volume) were conducted with a 3D Object Counter plugin for ImageJ developed by Fabrice Cordelieres (Institut Curie, Orsay, France) and Jonathan Jackson (UCL Institute of Neurology, London, UK).
Mitochondrial fragmentation in dopaminergic neurons
Primary midbrain neurons at DIV 3 were co-transfected with shRNAs (sh-control or sh-ATP13A2 in vector pSM2c) and mito-GFP plasmids at a 10:1 DNA molar ratio to mark transfected neurons. At DIV 7, cultures were fixed with 4% PFA and subjected to immunocytochemistry with mouse anti-TH antibody (Sigma-Aldrich) and anti-mouse IgG-AlexaFluor-546 antibody (Invitrogen). Confocal microscopic analysis was conducted on a Zeiss LSM 700 inverted confocal microscope (Carl Zeiss AG) with a Plan-Apochromat 63×/1.40 oil objective in x-, y- and z-planes. Mitochondrial morphology was assessed in individual GFP+/TH+ dopaminergic neurons using mito-GFP fluorescence. Mitochondria were classified as tubular (normal), intermediate (partially fragmented plus tubular network) or fragmented (fully fragmented without tubular network), according to previously reported criteria (41). Mitochondrial morphology was scored from dopaminergic neurons expressing shRNA/mito-GFP (n = 24–27) sampled from five cover slips from two independent cultures. Mitochondrial morphological subclasses were expressed as a percent of the total number of mitochondria for each condition.
Autophagosome morphology analysis
Primary cortical neurons were co-transfected at DIV 9 with V5-tagged human ATP13A2 (or control empty vector) or shRNAs (sh-control or sh-ATP13A2) and EGFP-LC3 plasmids at a 10:1 DNA molar ratio to mark transfected neurons. At DIV 12, high-resolution confocal z-stacks were acquired for EGFP-LC3+ neurons. Confocal stacks were subjected to deconvolution with HuygensPro software. The size and number of EGFP-LC3+ puncta were quantified from 3D stacks using ImageJ.
Statistical analysis
All quantified experiments correspond to at least three independent primary cultures. Data are plotted as mean ± SEM. For comparison between conditions, two-tailed unpaired Student's t-test was used, and P < 0.05 was considered significant. Statistical analyses were performed with R software using the CRAN public library for population distribution, non-lineal fittings and kinetics (The R Project for Statistical Computing; http://www.r-project.org).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by funding from the Michael J. Fox Foundation for Parkinson's Research (D.J.M.), Swiss National Science Foundation (grant no. 310030_127478 to D.J.M.), Ecole Polytechnique Fédérale de Lausanne (D.J.M.), the Johns Hopkins University Morris K. Udall Parkinson's Disease Research Center of Excellence (NINDS P50NS38377; J.C.T.) and the JHU Alzheimer's Disease Research Center (NIA P50AG05146; J.C.T.).
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank members of the EPFL Histology and BioImaging and Optics Core facilities for technical assistance.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Gasser T. Mendelian forms of Parkinson's disease. Biochim. Biophys. Acta. 2009;1792:587–596. doi: 10.1016/j.bbadis.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J., Cai H., Cookson M.R., Gwinn-Hardy K., Singleton A. Genetics of Parkinson's disease and parkinsonism. Ann. Neurol. 2006;60:389–398. doi: 10.1002/ana.21022. doi:10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- 3.Moore D.J., West A.B., Dawson V.L., Dawson T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. doi:10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 4.Behrens M.I., Bruggemann N., Chana P., Venegas P., Kagi M., Parrao T., Orellana P., Garrido C., Rojas C.V., Hauke J., et al. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov. Disord. 2010;25:1929–1937. doi: 10.1002/mds.22996. doi:10.1002/mds.22996. [DOI] [PubMed] [Google Scholar]
- 5.Najim al-Din A.S., Wriekat A., Mubaidin A., Dasouki M., Hiari M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol. Scand. 1994;89:347–352. doi: 10.1111/j.1600-0404.1994.tb02645.x. [DOI] [PubMed] [Google Scholar]
- 6.Ramirez A., Heimbach A., Grundemann J., Stiller B., Hampshire D., Cid L.P., Goebel I., Mubaidin A.F., Wriekat A.L., Roeper J., et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. doi:10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 7.Williams D.R., Hadeed A., al-Din A.S., Wreikat A.L., Lees A.J. Kufor Rakeb disease: autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov. Disord. 2005;20:1264–1271. doi: 10.1002/mds.20511. doi:10.1002/mds.20511. [DOI] [PubMed] [Google Scholar]
- 8.Ugolino J., Fang S., Kubisch C., Monteiro M.J. Mutant Atp13a2 proteins involved in parkinsonism are degraded by ER-associated degradation and sensitize cells to ER-stress induced cell death. Hum. Mol. Genet. 2011;20:3565–3577. doi: 10.1093/hmg/ddr274. doi:10.1093/hmg/ddr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park J.S., Mehta P., Cooper A.A., Veivers D., Heimbach A., Stiller B., Kubisch C., Fung V.S., Krainc D., Mackay-Sim A., et al. Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor-Rakeb syndrome, a form of early-onset parkinsonism. Hum. Mutat. 2011;32:956–964. doi: 10.1002/humu.21527. doi:10.1002/humu.21527. [DOI] [PubMed] [Google Scholar]
- 10.Crosiers D., Ceulemans B., Meeus B., Nuytemans K., Pals P., Van Broeckhoven C., Cras P., Theuns J. Juvenile dystonia-parkinsonism and dementia caused by a novel ATP13A2 frameshift mutation. Parkinsonism Relat. Disord. 2011;17:135–138. doi: 10.1016/j.parkreldis.2010.10.011. doi:10.1016/j.parkreldis.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 11.Djarmati A., Hagenah J., Reetz K., Winkler S., Behrens M.I., Pawlack H., Lohmann K., Ramirez A., Tadic V., Bruggemann N., et al. ATP13A2 variants in early-onset Parkinson's disease patients and controls. Mov. Disord. 2009;24:2104–2111. doi: 10.1002/mds.22728. doi:10.1002/mds.22728. [DOI] [PubMed] [Google Scholar]
- 12.Lin C.H., Tan E.K., Chen M.L., Tan L.C., Lim H.Q., Chen G.S., Wu R.M. Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology. 2008;71:1727–1732. doi: 10.1212/01.wnl.0000335167.72412.68. doi:10.1212/01.wnl.0000335167.72412.68. [DOI] [PubMed] [Google Scholar]
- 13.Ning Y.P., Kanai K., Tomiyama H., Li Y., Funayama M., Yoshino H., Sato S., Asahina M., Kuwabara S., Takeda A., et al. PARK9-linked parkinsonism in eastern Asia: mutation detection in ATP13A2 and clinical phenotype. Neurology. 2008;70:1491–1493. doi: 10.1212/01.wnl.0000310427.72236.68. doi:10.1212/01.wnl.0000310427.72236.68. [DOI] [PubMed] [Google Scholar]
- 14.Santoro L., Breedveld G.J., Manganelli F., Iodice R., Pisciotta C., Nolano M., Punzo F., Quarantelli M., Pappata S., Di Fonzo A., et al. Novel ATP13A2 (PARK9) homozygous mutation in a family with marked phenotype variability. Neurogenetics. 2011;12:33–39. doi: 10.1007/s10048-010-0259-0. doi:10.1007/s10048-010-0259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schultheis P.J., Hagen T.T., O'Toole K.K., Tachibana A., Burke C.R., McGill D.L., Okunade G.W., Shull G.E. Characterization of the P5 subfamily of P-type transport ATPases in mice. Biochem. Biophys. Res. Commun. 2004;323:731–738. doi: 10.1016/j.bbrc.2004.08.156. doi:10.1016/j.bbrc.2004.08.156. [DOI] [PubMed] [Google Scholar]
- 16.Gitler A.D., Chesi A., Geddie M.L., Strathearn K.E., Hamamichi S., Hill K.J., Caldwell K.A., Caldwell G.A., Cooper A.A., Rochet J.C., et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009;41:308–315. doi: 10.1038/ng.300. doi:10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt K., Wolfe D.M., Stiller B., Pearce D.A. Cd2+, Mn2+, Ni2+ and Se2+ toxicity to Saccharomyces cerevisiae lacking YPK9p the orthologue of human ATP13A2. Biochem. Biophys. Res. Commun. 2009;383:198–202. doi: 10.1016/j.bbrc.2009.03.151. doi:10.1016/j.bbrc.2009.03.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan J., Zhang T., Jiang L., Chi J., Hu D., Pan Q., Wang D., Zhang Z. Regulation of intracellular manganese hemeostasis by kufor-rakeb syndrome associated ATP13A2. J. Biol. Chem. 2011;286:29654–29662. doi: 10.1074/jbc.M111.233874. doi:10.1074/jbc.M111.233874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong E., Cuervo A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010;13:805–811. doi: 10.1038/nn.2575. doi:10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cuervo A.M., Stefanis L., Fredenburg R., Lansbury P.T., Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. doi:10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Vicente M., Talloczy Z., Kaushik S., Massey A.C., Mazzulli J., Mosharov E.V., Hodara R., Fredenburg R., Wu D.C., Follenzi A., et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravikumar B., Sarkar S., Davies J.E., Futter M., Garcia-Arencibia M., Green-Thompson Z.W., Jimenez-Sanchez M., Korolchuk V.I., Lichtenberg M., Luo S., et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. doi:10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 23.Winslow A.R., Chen C.W., Corrochano S., Acevedo-Arozena A., Gordon D.E., Peden A.A., Lichtenberg M., Menzies F.M., Ravikumar B., Imarisio S., et al. alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. J. Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. doi:10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N., Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 25.Vallipuram J., Grenville J., Crawford D.A. The E646D-ATP13A4 mutation associated with autism reveals a defect in calcium regulation. Cell. Mol. Neurobiol. 2010;30:233–246. doi: 10.1007/s10571-009-9445-8. doi:10.1007/s10571-009-9445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z., Okamoto K., Hayashi Y., Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. doi:10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Detmer S.A., Chan D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. doi:10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 28.Schneider S.A., Paisan-Ruiz C., Quinn N.P., Lees A.J., Houlden H., Hardy J., Bhatia K.P. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov. Disord. 2010;25:979–984. doi: 10.1002/mds.22947. doi:10.1002/mds.22947. [DOI] [PubMed] [Google Scholar]
- 29.Higashi S., Moore D.J., Minegishi M., Kasanuki K., Fujishiro H., Kabuta T., Togo T., Katsuse O., Uchikado H., Furukawa Y., et al. Localization of MAP1-LC3 in vulnerable neurons and Lewy bodies in brains of patients with dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 2011;70:264–280. doi: 10.1097/NEN.0b013e318211c86a. doi:10.1097/NEN.0b013e318211c86a. [DOI] [PubMed] [Google Scholar]
- 30.Strack R.L., Strongin D.E., Bhattacharyya D., Tao W., Berman A., Broxmeyer H.E., Keenan R.J., Glick B.S. A noncytotoxic DsRed variant for whole-cell labeling. Nat. Methods. 2008;5:955–957. doi: 10.1038/nmeth.1264. doi:10.1038/nmeth.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jackson W.T., Giddings T.H.,, Jr, Taylor M.P., Mulinyawe S., Rabinovitch M., Kopito R.R., Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. doi:10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vonderheit A., Helenius A. Rab7 associates with early endosomes to mediate sorting and transport of Semliki forest virus to late endosomes. PLoS Biol. 2005;3:e233. doi: 10.1371/journal.pbio.0030233. doi:10.1371/journal.pbio.0030233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choudhury A., Dominguez M., Puri V., Sharma D.K., Narita K., Wheatley C.L., Marks D.L., Pagano R.E. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Invest. 2002;109:1541–1550. doi: 10.1172/JCI15420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sherer N.M., Lehmann M.J., Jimenez-Soto L.F., Ingmundson A., Horner S.M., Cicchetti G., Allen P.G., Pypaert M., Cunningham J.M., Mothes W. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic. 2003;4:785–801. doi: 10.1034/j.1600-0854.2003.00135.x. doi:10.1034/j.1600-0854.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- 35.Biskup S., Moore D.J., Celsi F., Higashi S., West A.B., Andrabi S.A., Kurkinen K., Yu S.W., Savitt J.M., Waldvogel H.J., et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. doi:10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- 36.Moore D.J., Zhang L., Dawson T.M., Dawson V.L. A missense mutation (L166P) in DJ-1, linked to familial Parkinson's disease, confers reduced protein stability and impairs homo-oligomerization. J. Neurochem. 2003;87:1558–1567. doi: 10.1111/j.1471-4159.2003.02265.x. doi:10.1111/j.1471-4159.2003.02265.x. [DOI] [PubMed] [Google Scholar]
- 37.West A.B., Moore D.J., Biskup S., Bugayenko A., Smith W.W., Ross C.A., Dawson V.L., Dawson T.M. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl Acad. Sci. USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. doi:10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramonet D., Daher J.P., Lin B.M., Stafa K., Kim J., Banerjee R., Westerlund M., Pletnikova O., Glauser L., Yang L., et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One. 2011;6:e18568. doi: 10.1371/journal.pone.0018568. doi:10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hallett P.J., Collins T.L., Standaert D.G., Dunah A.W. Biochemical fractionation of brain tissue for studies of receptor distribution and trafficking. Curr. Protoc. Neurosci. 2008;Chapter 1:Unit 1.16. doi: 10.1002/0471142301.ns0116s42. [DOI] [PubMed] [Google Scholar]
- 40.Higashi S., Moore D.J., Yamamoto R., Minegishi M., Sato K., Togo T., Katsuse O., Uchikado H., Furukawa Y., Hino H., et al. Abnormal localization of leucine-rich repeat kinase 2 to the endosomal-lysosomal compartment in lewy body disease. J. Neuropathol. Exp. Neurol. 2009;68:994–1005. doi: 10.1097/NEN.0b013e3181b44ed8. doi:10.1097/NEN.0b013e3181b44ed8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakamura K., Nemani V.M., Azarbal F., Skibinski G., Levy J.M., Egami K., Munishkina L., Zhang J., Gardner B., Wakabayashi J., et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011;286:20710–20726. doi: 10.1074/jbc.M110.213538. doi:10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.