

Abstract
Mer tyrosine kinase (MerTK) is a major macrophage apoptotic cell (AC) receptor. Its functional impairment promotes autoimmunity and atherosclerosis, while overexpression correlates with poor prognosis in cancer. However, little is known about mechanisms regulating MerTK expression in humans. We found that MerTK expression is heterogenous among macrophage subsets, being mostly restricted to anti-inflammatory “M2c” (CD14+CD16+CD163+CD204+CD206+CD209−) cells, differentiated by M-CSF or glucocorticoids. Small numbers of MerTK+ “M2c-like” cells are also detectable among circulating CD14brightCD16+ monocytes. MerTK expression levels adapt to changing immunological environment, being suppressed in M1 and “M2a” macrophages, and in dendritic cells. Remarkably, while glucocorticoid-induced differentiation is IL-10-independent, M-CSF-driven M2c polarization and related MerTK up-regulation require IL-10. However, neither IL-10 alone nor TGFβ are sufficient to fully differentiate M2c (CD16+CD163+MerTK+) macrophages. M-CSF and IL-10, both released by T lymphocytes, may thus be required together to promote regulatory T cell-mediated induction of anti-inflammatory monocytes-macrophages. MerTK enables M2c macrophages to clear early ACs more efficiently than other macrophage subsets, and mediates AC clearance by CD14brightCD16+ monocytes. Moreover, M2c cells release Gas6, which in turn amplifies IL-10 secretion via MerTK. IL-10-dependent induction of the Gas6/MerTK pathway may, therefore, constitute a positive loop for M2c macrophage homeostasis and a critical checkpoint for maintenance of anti-inflammatory conditions. Our findings give new insight into human macrophage polarization and favor a central role for MerTK in regulation of macrophage functions. Eliciting M2c polarization can have therapeutic utility for diseases such as lupus, in which a defective AC clearance contributes to initiate and perpetuate the pathological process.
INTRODUCTION
The prompt recognition and removal of dead and dying cells is critical for maintenance of immunological tolerance and resolution of inflammation. Physiological mechanisms of apoptotic cell (AC) clearance typically associate with induction of regulatory pathways in phagocytes and release of anti-inflammatory cytokines (1, 2). Such mechanisms have attracted increasing interest over the last decade, and many distinct molecular pathways have been identified. Although there seems to be conspicuous redundancy among these pathways, they differ from each other with regard to several features. These include dependence on specific nuclear transcription factors (3–6), expression under basal conditions vs inducibility by excess numbers of ACs (5–9), recognition of unmodified vs modified phosphatidylserine on ACs, direct AC recognition vs use of bridging molecules, and, importantly, clearance of early vs late (secondarily necrotic) ACs (9–10).
Mer tyrosine kinase (MerTK), a member of the TAM (Tyro3, Axl, Mer) subfamily of receptors, is specifically involved in removal of early ACs, and recognizes unmodified phosphatidylserine through the bridging molecules Protein S and Gas6 (9–12). MerTK is expressed on phagocytes following exposure to ACs and subsequent LXR receptor activation (5). Its functional impairment causes defective AC clearance in the presence of excess ACs (7–9). Furthermore, other critical AC pathways are closely linked with MerTK activity (13–15).
MerTK is expressed in primary and secondary lymphoid organs (7–8), and is key for maintenance of both central and peripheral tolerance through multiple mechanisms: removal of AC-derived potential autoantigens (8, 16); inhibition of TLR-induced production of pro-inflammatory cytokines (17–19); prevention of autoreactive B and T cell expansion (16, 20). The loss of these functions results in lupus-like autoimmunity in MerTK-deficient or LXR-deficient mice (4, 8). In lupus patients, impaired AC clearance is believed to cause the persistence of ACs in various tissues, including lymphoid organs. This may promote the production of autoantibodies against apoptotic material, and may lead to delayed and proinflammatory clearance of secondary necrotic cells mediated by autoantibodies (21–22). In these patients, we recently reported a reduction in plasma levels of the MerTK ligand Protein S (23), which may account for a functional defect in AC clearance. MerTK plays also a protective role in atherosclerosis, where it enables discrete macrophages to phagocytose cholesterol-laden apoptotic macrophages, thereby preventing secondary necrosis, inflammation and plaque instability (24); moreover, MerTK inhibits cholesterol uptake from macrophages themselves (25). In contrast, immunomodulation, cell viability and resistance to apoptosis promoted by MerTK are detrimental in cancer (26), whereby the ligand Gas6, secreted by tumor-associated macrophages, promotes tumor growth and metastasis (27).
Therefore, modulating MerTK activity could be a promising therapeutical approach to various pathological conditions. Surprisingly, little is known about the mechanisms promoting MerTK expression in humans. Both MerTK and Protein S can be up-regulated by steroids (28–30), and Protein S/MerTK-mediated AC clearance is enhanced by these drugs (12). However, steroids induce many other molecules involved in AC clearance (29–30). It is, therefore, unclear whether MerTK up-regulation by glucocorticoids is due to intrinsic and unique properties of these drugs, or to steroid-induced apoptosis, which in turn promotes LXR activation, or to other unidentified mechanisms.
Our study examined how the immunological microenvironment affects MerTK expression, and aimed to identify the human macrophage subsets in which MerTK is clearly expressed and functionally relevant. Macrophages subsets include: “M1”, secreting IL-12 and promoting T helper 1 (TH1) differentiation; “M2a”, secreting IL-4 and related to TH2 polarization; “M2b” and “M2c”, both secreting IL-10 and associated with regulatory T cell (Treg) expansion (31). We found that, in the presence of IL-10, M-CSF differentiates macrophages expressing high levels of MerTK; such macrophages are characterized by an M2c phenotype. Glucocorticoids have analogous effects, with MerTK up-regulation occurring as a consequence of steroid-induced M2c polarization; yet, this process is IL-10-independent. Remarkably, neither M-CSF alone nor IL-10 alone is able to drive full M2c differentiation and up-regulate MerTK significantly. MerTK is also expressed among M2c-like CD14brightCD16+ circulating monocytes and in minor populations of CD14brightCD16+CD163+ cells differentiated among M1 and M2a macrophages. MerTK makes M2c (M-CSF+IL-10) macrophages and M2c-like cells highly capable of clearing ACs, and significantly enhances IL-10 secretion by M-CSF-driven macrophages following Gas6 ligation. Our data support a broad role for MerTK in M2c macrophage homeostasis, and highlight the potential usefulness of polarizing human macrophages with both M-CSF and IL-10 to ensure an optimal, MerTK-mediated, clearance of ACs.
MATERIALS AND METHODS
Cell cultures
Human monocytes from buffy coats of healthy blood donors were isolated by Ficoll-Paque™ Plus gradient (GE Healthcare) and magnetic separation, using a kit for human monocyte enrichment by negative selection (EasySep™, StemCell), according to the manufacturer’s instruction. Purity of CD14+ cells was 89–94%. CD14+ cells were cultured in 24-well plates at 0.8×106 cells/ml for 7–8 days at 37°C in 5% CO2 in complete RPMI 1640 medium containing 10% human AB serum, L-glutamine, penicillin and streptomycin. To prevent formation of clumps induced by autologous serum and to allow optimal differentiation, 10% heat-inactivated FBS was also added. Cells were incubated from day 0 with GM-CSF (100 ng/ml; Peprotech) or M-CSF (50 ng/ml; Peprotech), to differentiate M1 or M2 macrophages, respectively. In some cases, as specified in the text, these or other macrophage polarizing factors were newly added in the last 3 days: for M1, IFNγ (2.5–10 ng/ml; R&D Systems) and/or LPS (1 μg/ml; Sigma-Aldrich); for M2, IL-4 (20 ng/ml; Novus Biologicals), IL-10 (1–100 ng/ml; Peprotech), TGFβ (20 ng/ml; Peprotech) or dexamethasone (1–1000 nM; Sigma-Aldrich). Dendritic cells (DCs) were obtained by culturing cells in the presence of GM-CSF (100 ng/ml) and IL-4 (20 ng/ml) for 7–8 days. For experiments in serum-free conditions, cells were cultured at 0.8×106 cells/ml for 3–4 days in X-Vivo™15 medium (Lonza), and incubated from day 0 with one or more growth/polarizing factors. For inhibiting IL-10 activity, a purified LEAF™ purified mouse anti-human IL-10 antibody was used (Biolegend, clone JES3-9D7).
Prior to participation, all subjects gave informed consent to donate their blood samples. The study was approved by the Institutional Review Boards of Temple University.
Analysis of cell-surface molecules by FACS
Phenotypic analysis by flow cytometry was carried out in freshly isolated and in cultured cells after washing in buffer containing 2% bovine serum albumin. The following mouse monoclonal antibodies were used: anti-CD14 (PE-Cy7), anti-HLA-DR (APC), anti-CD163 (APC or PerCP-Cy5.5), anti-CD206 (APC-Cy7), anti-CD209 (PerCP-Cy5.5), anti-CD1a (APC), anti-CD210/IL-10R (PE) (Biolegend); anti-CD16 (APC-Cy7) (BD Biosciences); anti-CD204 (APC) and anti-MerTK (clone 125518; PE) (R&D Systems). MerTK expression was evaluated using appropriate PE-labeled isotype control (Biolegend). Cells were analyzed using FACSCalibur™ (BD Biosciences) and FlowJo software.
Detection of MerTK expression by Western blot
Cell lysates were obtained in buffer containing 50 mM Hepes, 150 mM NaCl, 10% glycerol, 1% Triton X-100, and freshly added cocktails of protease and phosphatase inhibitors (Sigma-Aldrich). Lysates were resolved on a SDS-PAGE 8% polyacrylamide gel. Proteins, transferred to PVDF membranes (Millipore), were probed with biotinylated goat polyclonal anti-human MerTK (R&D Systems) and rabbit anti-β-actin antibodies (Santa Cruz Biotechnology), followed by horseradish-peroxidase (HRP)-conjugated streptavidin (Biolegend) and secondary goat anti-rabbit antibody (Santa Cruz Biotechnology), respectively. Immunoblots were developed and visualized by enhanced chemiluminescence using Amersham ECL™ reagents (GE Healthcare). Densitometry of bands, normalized to β-actin expression, was calculated using ImageJ software.
Induction of apoptosis and phagocytosis assay
Human neutrophils were isolated from Ficoll-Hypaque pellets through dextran erytrocyte sedimentation and lysis of contaminating erythrocytes by incubation with ice-cold ammonium chloride (0.15 M) and potassium bicarbonate (0.01 M) solution. Neutrophils were resuspended at 1×106 cell/ml in 10% FBS-RPMI, labeled with 2.5 μM CFSE (Sigma-Aldrich), and incubated for 20 hours at 37°C in 5% CO2. The composition of neutrophils routinely obtained after incubation, according to annexin V and propidium iodide (PI) staining, was: 66.0±10.2% early ACs (annexinV+PI−), 3.8±2.2% late ACs (annexinV+PI+), 0.3±0.2% necrotic cells (annexinV−PI+).
Apoptotic neutrophils were added for 60 minutes to cultured monocyte-macrophages, at a 5:1 ratio. Flow cytometry was used to quantify percentages of CD14-labeled macrophages that phagocytosed CFSE-labeled ACs, and to calculate phagocytosis index. In some experiments, as specified in the text, phagocytosis activity was assessed separately on CD14dimCD163− and CD14brightCD163+ macrophage subsets.
For phagocytosis assays on circulating monocytes, PBMCs were mixed with apoptotic neutrophils at a 1:1 ratio, and incubated in BD Falcon™ tubes for 4 hours at 37°C in 5% CO2. Phagocytosis activity was measured on CD14+CD16−, CD14brightCD16+ and CD14dimCD16+ monocyte subsets by flow cytometry.
For inhibition studies, macrophages and circulating monocytes were pre-incubated with a goat anti-human MerTK antibody (R&D Systems) or goat control IgG (SouthernBiotech) for 30 minutes before adding apoptotic neutrophils. The effects of MerTK block during phagocytosis assay was additionally observed using a Leica TCS SP5 confocal laser scanning microscope, 40X/1.25 numerical aperture (NA) oil objective, labeling apoptotic neutrophils with Hoechst 33342 (0.5 μg/ml; Invitrogen) and staining macrophages with mouse anti-CD14 (Biolegend) and goat biotinylated anti-CD163 (R&D Systems) antibodies, followed by secondary APC-conjugated goat anti-mouse antibody and FITC-conjugated streptavidin (Biolegend), respectively.
Gas6, IL-10 and TNFα detection by ELISA
Gas6 levels were measured in supernatants of cell cultures treated with various cytokines for 4 days in serum-free conditions, using sandwich ELISA according to standard procedure (23). Standard curves were prepared with rhGas6 (R&D Systems). Purified goat anti-human Gas6 antibody (R&D Systems) was used for capture. Biotinylated goat polyclonal anti-human Gas6 antibody (R&D Systems), followed by HRP-conjugated streptavidin (Biolegend), was used for detection.
Secretion of IL-10 and TNFα was induced by LPS (50 ng/ml; Sigma-Aldrich) ± rhGas6 (1 μg/ml; R&D Systems), in the presence or absence of goat anti-human MerTK antibody (R&D Systems) or goat control IgG (SouthernBiotech), from cells cultured with or without M-CSF. IL-10 and TNFα levels were measured in supernatants using human IL-10 ELISA MAX™ Standard kit and TNFα ELISA MAX™ Standard kit (Biolegend), following the manufacturer’s instruction.
In Gas6-producing monocyte-macrophages, the potential effect of endogenously produced Gas6 on TNFα production was studied. For this purpose, cells were differentiated in the presence of M-CSF and IL-10, and coincubated with either recombinant MerFc (5 μg/ml; R&D Systems) or a goat anti-human Gas6 antibody (5 μg/ml; R&D Systems) to block Gas6 activity prior to LPS stimulation. Potential variations in TNFα levels were assessed by ELISA.
Statistical analysis
Data are expressed as mean±SEM. Statistical significance among different cell treatments was assessed by Student’s paired t-test, or one-way repeated measures ANOVA if treatment groups were more than two. Statistical significance was defined as P <0.05. Analysis and graphing were performed using GraphPad Prism™ software.
RESULTS
MerTK is up-regulated during monocyte-to-macrophage differentiation, and is further enhanced by M-CSF
Healthy monocytes were sorted from PBMCs through negative selection using magnetic beads, and analyzed for MerTK expression at days 0, 1 and 3 by flow cytometry. Freshly isolated monocytes tended to aggregate with platelets, in variable proportions according to individuals; yet, platelet-monocyte conjugates were poorly detectable after plating cells (Fig. 1A). As platelets can also express MerTK (32), we had to rule out their potential contribution to MerTK detection. Therefore, we used the platelet marker CD42b along with CD14 for analyzing MerTK in monocytes, either isolated or conjugated with platelets. Surprisingly, no MerTK expression was clearly found at day 0 in either case, except for rare cells located in the conjugated fraction (Fig. 1B). In cultured monocytes, MerTK was gradually acquired during monocyte-to-macrophage differentiation, and was evident at intermediate stages (day 3) (Fig. 1C–D). However, in the absence of growth factors (colony stimulating factors, CSFs), MerTK could be detected on the cell surface at variable levels, depending on individual experiments. In contrast, in the presence of M-CSF, MerTK up-regulation was reproducibly enhanced. GM-CSF, instead, decreased MerTK expression levels (Fig. 1E–G).
Figure 1. MerTK is up-regulated during monocyte-to-macrophage differentiation, and is further enhanced by M-CSF.

(A–D) Monocytes were sorted from human healthy PBMCs through negative selection magnetic beads, and analyzed by flow cytometry at day 0, and after 1 and 3 days of culture. Monocytes were gated using an anti-CD14 antibody, and stained with anti-MerTK antibody. An anti-CD42b antibody was also used, to distinguish platelet-monocyte conjugates from isolated monocytes. (E–G) Monocytes were cultured in complete medium in the presence of M-CSF (50 ng/ml) or GM-CSF (100 ng/ml), or in the absence of colony stimulating factors (CSFs), and analyzed for MerTK expression on day 3. Data shown are representative of three independent experiments.
Macrophage MerTK expression promptly adapts to changes of immunological environment
Terminally differentiated macrophages, obtained after 8 days of culture, also up-regulated MerTK in the presence of M-CSF, but not GM-CSF (Fig. 2A). We tested whether M-CSF and GM-CSF effects on MerTK expression were reversible. The addition of GM-CSF at day 5 to M-CSF-differentiated macrophages resulted in significant MerTK down-regulation at day 8 (Fig. 2A–B), while the addition of M-CSF to GM-CSF-driven macrophages tended to increase MerTK expression, yet not significantly (Fig. 2C).
Figure 2. Macrophage MerTK expression promptly adapts to changes of immunological environment.
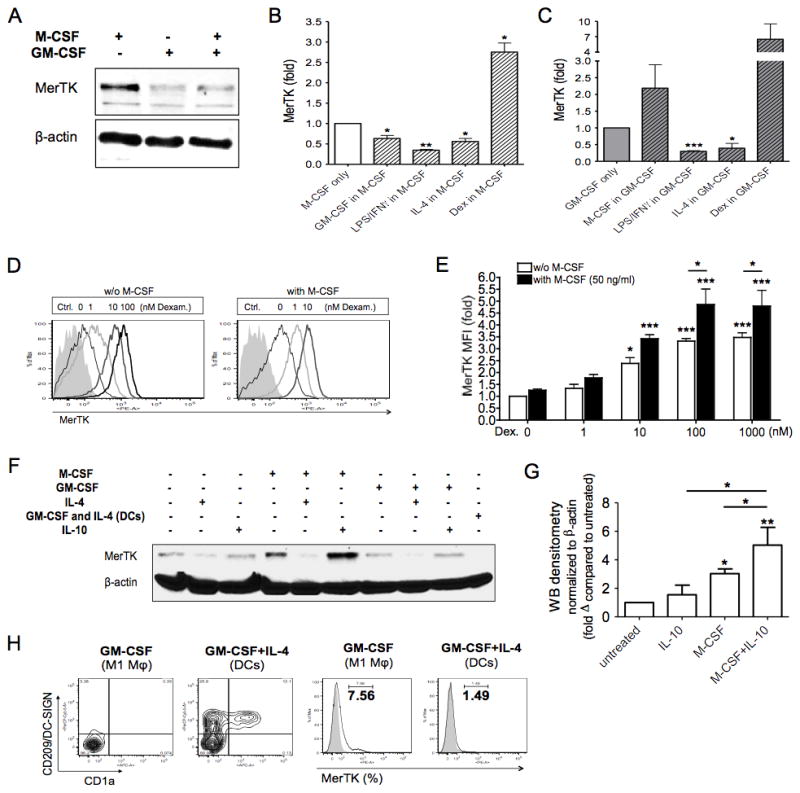
(A–C) CD14+ cells were cultured in complete medium in the presence of M-CSF (50 ng/ml) or GM-CSF (100 ng/ml). On day 5, cells were treated with GM-CSF (100 ng/ml), M-CSF (50 ng/ml), IFNγ (10 ng/ml) + LPS (1 μg/ml), IL-4 (20 ng/ml) or dexamethasone (Dex; 100 nM), for an additional 3 days. MerTK expression was analyzed by Western blot (A), or measured by flow cytometry as MFI fold variation compared to levels obtained with culturing cells with M-CSF alone (B) or GM-CSF alone (C) for 8 days. Data shown are representative of three independent experiments. (D–E) Cells were incubated with dexamethasone (Dex; 1–1000 nM), in the presence or absence of M-CSF (50 ng/ml), for 3 days in serum-free medium. MerTK up-regulation was measured by flow cytometry as MFI fold increase compared to expression levels in untreated cells. Data shown are representative of four independent experiments. (F–G) Cells were cultured in complete medium in the presence of M-CSF (50 ng/ml) or GM-CSF (100 ng/ml), or in the absence of colony stimulating factors. On day 5, cells were treated with IL-4 (20 ng/ml) or IL-10 (50 ng/ml), for an additional 3 days. Dendritic cells (DCs) were differentiated in the presence of GM-CSF and IL-4 from day 0 for 8 days. MerTK expression was analyzed by Western blot (F). Densitometry of Western blots was performed to quantify MerTK expression following cell treatment with IL-10, M-CSF, or both (G). Densitometry values were normalized to β-actin, and are reported as fold variation compared to MerTK expression levels in untreated cells. Data are representative of three independent experiments. (H) Cells were incubated with GM-CSF or GM-CSF + IL-4 for 8 days in complete medium, to differentiate M1 macrophages or DCs, respectively. Cells were stained for CD209 (DC-SIGN), CD1a and MerTK. MerTK+ cells were quantified as percentages of total cells by flow cytometry. *P <0.05; **P <0.01; ***P <0.001.
Because GM-CSF and M-CSF are known to drive M1 or M2 macrophage differentiation, respectively (33–34), we studied the effects of other conventional M1 and M2 stimuli on terminally differentiated macrophages. According to a recent classification, macrophages can be divided into M1 (driven by IFNγ and/or LPS), M2a (driven by IL-4 or IL-13), M2b (driven by immune complexes and LPS) and M2c (driven by IL-10, TGFβ or glucocorticoids) (31). Although M2b and M2c macrophages are elicited by different conditions, both subsets are characterized by IL-10 production, and we focused only on the M2c population, better described in humans (31). In either GM-CSF- or M-CSF-differentiated macrophages, stimulation at day 5 with either M1 or M2a polarizing factors (IFNγ±LPS or IL-4) for 3 days down-regulated MerTK. The relative decreases in MerTK expression were similar in GM-CSF- and M-CSF-differentiated macrophages. The glucocorticoid dexamethasone, instead, increased MerTK in both macrophage subsets, although full statistical significance was only reached in M-CSF-differentiated macrophages (Fig. 2B–C).
Remarkably, M-CSF positively synergized with dexamethasone, and, in the presence of M-CSF, a 10-fold lower dose of dexamethasone (10 nM instead than 100 nM) was sufficient to reach maximal induction of MerTK; moreover, the addition of M-CSF to dexamethasone 100 nM further enhanced MerTK expression, whereas increasing the dose of dexamethasone per se (1 μM) was no longer effective (Fig. 2D–E). IL-10 also synergized with M-CSF in up-regulating MerTK (Fig. 2F–G). In contrast, GM-CSF and IL-4 combined to abrogate MerTK expression in human peripheral monocyte-derived DCs (Fig. 2F and 2H). Therefore, MerTK is not only inducible by AC exposure (6), but is also finely regulated by cytokines and/or growth factors. Loss of MerTK in DCs indicates that in these cells MerTK is dispensable for AC clearance, similarly to what has been previously reported in mice (35–36).
MerTK expression is restricted to (CD163+CD16+CD206+) M2 macrophages
Because MerTK expression was enhanced under M2 conditions, we looked at MerTK expression along with several M2 surface receptors, in order to identify potential markers that could predict the presence of MerTK in human macrophages.
Although CD206 (i.e., mannose receptor) was among the first identified markers for conventional (IL-4-driven) M2 macrophages (37), it is a non-specific M2 marker in humans, being also up-regulated by M-CSF, glucocorticoids (Fig. 3), TGFβ (not shown), and even by GM-CSF, a non-M2 factor. We noted, instead, that CD209 (earlier known as DC-SIGN) was a reliable and specific marker of exposure to IL-4 (M2a polarization), while CD163 (hemoglobin-haptoglobin scavenger receptor) and/or CD16 (FcγRIIIa), in agreement with a recent report (38), were specific markers of exposure to M-CSF, glucocorticoids, IL-10 or TGFβ (M2c polarization). Remarkably, MerTK showed the same expression pattern as CD163. Macrophages that had low levels of CD163 (M1 and M2a) also showed significant MerTK down-regulation. However, CD163 detection was not sufficient to identify MerTK+ cells, as in the case of IL-10-treated cells. What best discriminated the macrophage subset that highly expressed MerTK was the co-expression of CD163 and CD16; alternatively, the same population could be identified by the co-expression of CD163 and CD206. Both M-CSF and dexamethasone were able to induce this specific phenotype (CD163+CD16+CD206+) (Fig. 3). Therefore, MerTK up-regulation by steroids is due to pharmacological induction of M2c polarization, which reproduces the physiological effects of M-CSF differentiation.
Figure 3. MerTK expression is restricted to (CD163+CD16+CD206+) M2 macrophages.

Macrophages were differentiated from peripheral monocytes for 7–8 days in complete medium, in the presence of GM-CSF (100 ng/ml), IFNγ (10 ng/ml), IL-4 (20 ng/ml), M-CSF (50 ng/ml), dexamethasone (100 nM), or IL-10 (50 ng/ml). Cells were stained for MerTK, CD14, CD163, CD204/SR-A1, CD16, CD206, CD209, and analyzed by flow cytometry. Histograms show MFI fold variations compared to levels in untreated cells (basal). Data shown are representative of eight to twelve independent experiments. *P <0.05; **P <0.01; ***P <0.001.
IL-10 is required for M-CSF to induce M2c differentiation and up-regulate MerTK
It was noteworthy that IL-10 could enhance M-CSF effects, although IL-10 per se could not promote further MerTK up-regulation compared to medium (Fig. 2F–G). We wondered, then, if serum-derived bioactive IL-10 present in culture medium (39–40) might account for the basal M-CSF effects observed. Consistent with this hypothesis, M-CSF up-regulated MerTK only in the presence of serum (Fig. 4A). In serum-free medium, the addition of low doses of IL-10 was crucial for M-CSF induction of MerTK expression by the first days of differentiation (Fig. 4B). Conversely, blocking IL-10 in serum-containing medium, through a neutralizing anti-IL-10 antibody, prevented spontaneous increase as well as M-CSF enhancement of MerTK and CD163 expression during differentiation (Fig. 4C). In serum-free conditions, neither M-CSF nor IL-10 alone, but only the combination M-CSF+IL-10 was able to induce MerTK in macrophages, at levels comparable to those reached with dexamethasone. Interestingly, M-CSF+IL-10 gave a modest, but significant, increase in IL-10 receptor (CD210) expression compared to GM-CSF or IL-4. More broadly, we found that the combination M-CSF+IL-10 was critically required for complete M2c differentiation (e.g., CD163 and CD16 up-regulation) in serum-free conditions (Fig. 4D).
Figure 4. IL-10 is required for M-CSF to induce M2c differentiation and up-regulate MerTK.

(A) CD14+ cells were cultured in the presence or absence of M-CSF (50 ng/ml), in either serum-containing or serum-free medium, for 3 days. (B) Cells were incubated with IL-10 (1 ng/ml) and/or increasing doses of M-CSF (0.05 to 50 ng/ml). MerTK up-regulation was measured by flow cytometry as MFI fold increase compared to expression levels in untreated cells. Data shown are representative of three independent experiments. (C) Cells were cultured in serum-containing medium with or without M-CSF (50 ng/ml), in the presence or absence of a neutralizing mouse monoclonal anti-human IL-10 antibody (5 μg/ml; Biolegend, clone JES3-9D7), for 3 days. Cells were stained for CD163 and MerTK, and quantified by flow cytometry as percentages of total cells. (D) Cells were cultured in serum-free medium in the presence of M-CSF (50 ng/ml), IL-10 (50 ng/ml), M-CSF + IL-10, TGFβ (20 ng/ml), dexamethasone (Dex; 100 nM), for 4 days. Cells were stained for MerTK, CD163, CD16 and CD163. MerTK expression is shown as MFI fold variation compared to levels in untreated cells, as well as MerTK+ cell percentages; CD163, CD16 and CD14 MFI fold variations are also reported. Data shown are representative of four independent experiments. The expression of IL-10 receptor (IL-10R, or CD210) was measured after 3-day cytokine stimulations. Data shown are representative of three independent experiments. *P <0.05; **P <0.01; ***P <0.001.
Dexamethasone, instead, up-regulated MerTK and induced M2c macrophages even in serum-free medium (Fig. 4D) or, as previously reported for CD163 (41), in the presence of anti-IL-10 antibody in serum-containing medium (not shown). M2c differentiation was, therefore, IL-10-dependent for M-CSF and IL-10-independent for steroids. M2c (M-CSF+IL-10) macrophages also differed from dexamethasone-treated cells in having significantly higher levels of CD14 (Fig. 4D), a receptor known to be involved not only in LPS recognition, but also in tethering ACs (9).
Although currently classified as an M2c stimulus (31), TGFβ gave a different phenotype, characterized by CD16 induction, but inhibition of MerTK, CD163 and CD14 expression (Fig. 4D). Contrary to IL-10, TGFβ did not up-regulate MerTK even in combination with M-CSF, and tended to reduce the up-regulatory effect of M-CSF+IL-10 (not shown).
MerTK confers to M2c (M-CSF+IL-10) macrophages enhanced ability to clear early ACs
We aimed to assess the functional importance of MerTK up-regulation in M2c macrophages with regard to AC clearance efficiency. For this purpose, we cultured cells for 7 days in complete medium, adding IFNγ (M1 stimulus), IL-4 (M2a stimulus), M-CSF and IL-10 (M2c stimuli) or fresh medium only (“M0”) from day 4. At day 7, we coincubated macrophages with early apoptotic neutrophils (Fig. 5A), at a 1:5 ratio.
Figure 5. MerTK confers to M2c (M-CSF+IL-10) macrophages enhanced ability to clear early ACs.

(A) Early apoptotic cells (ACs) were obtained incubating human neutrophils, isolated from peripheral blood healthy donors, in 10% FCS-RPMI for 20 hours. According to annexin V and propidium iodide (PI) staining, early ACs were around 65–70% of total neutrophils. (B and C) CFSE-labeled apoptotic neutrophils were added for 60 minutes to 7-day differentiated M0 (untreated), M1 (IFNγ, 2.5 ng/ml), M2a (IL-4, 20 ng/ml) and M2c (M-CSF, 50 ng/ml + IL-10, 50 ng/ml) macrophages, labeled with a fluorochrome-conjugated anti-CD14 antibody, at a 5:1 ratio. M2c macrophages showed significantly enhanced ability to clear early ACs, expressed as higher percentages of phagocytic (CSFE+) macrophages. Pre-incubation of M2c macrophages with a goat polyclonal anti-human MerTK antibody (5 μg/ml; R&D Systems) for 30 minutes before addition of apoptotic neutrophils abolished such superiority of M2c cells to phagocytose ACs compared to other macrophage subsets. Blocking MerTK diminished not only the number of CFSE+ macrophages, but also the mean phagocytosis activity per single cell, depicted as CFSE MFI. Altogether, it resulted in a significant decrease of the phagocytosis index, determined by multiplying the percentage of CFSE+ macrophages by the CFSE MFI of phagocytic macrophages. Data shown are representative of three independent experiments. *P <0.05; **P <0.01. (D) By fluorescence microscopy (Leica TCS SP5 confocal laser scanning microscope, 40X/1.25 NA oil objective), M2c macrophages stained for CD14 (red) and CD163 (green) were shown to engulf Hoechst 33342-labeled apoptotic neutrophils (blue) (left panel, yellow arrows). Pre-incubation of M2c macrophages with an anti-MerTK blocking antibody inhibited engulfment, but not the physical interaction between macrophages and apoptotic neutrophils (right panel, white arrows).
We observed a remarkably higher capacity of AC clearance in M2c (M-CSF+IL-10) macrophages, compared to the other subsets. Importantly, such superiority of M2c cells was abrogated in the presence of a blocking anti-MerTK antibody (Fig. 5B–C). Using immunofluorescence microscopy, we could observe that blocking MerTK inhibited AC engulfment by M2c macrophages, but not the physical interactions between macrophages and ACs (Fig. 5D), that typically precede phagocytosis of early ACs (10).
M2c markers universally define MerTK+ macrophages prone to AC clearance
We showed above that IFNγ and IL-4 down-regulate MerTK protein expression, consistently with previous microarray data (42). Nevertheless, after culturing monocytes for 4 days in the presence of IFNγ or IL-4, we were able to detect rare MerTK+ cells even among M1 and M2a macrophages. Looking at the phenotype of these cells, we could observe that they were clearly distinguishable from the other macrophages by the selective expression of M2c markers (CD163, CD16, CD14, CD204) (Fig. 6A–B). In particular, CD204 expression ratio (i.e., percentage of CD204+ cells among MerTK+ macrophages/percentage of CD204+ cells among total macrophages in culture) was significantly higher in M1 conditions, while CD163 and CD16 expression ratios were significantly higher in both M1 and M2a conditions (Fig. 6B). These minor populations of M2c macrophages did not occur as a consequence of IFNγ or IL-4 inefficacy on some cells, as they were less clearly distinguishable among untreated cells; they were, rather, actively induced by treatment. In contrast to MerTK- M2a cells, the MerTK+ M2c macrophages occurring among IL-4-treated cells were CD209 negative and brighter for CD206 (Fig. 6C). Thus, MerTK localizes in macrophage subsets that share the phenotype CD14brightCD16+CD163+CD204+CD206brightCD209null; MerTK+ M2c cells occur even in non-M2c conditions, as small endogenous populations.
Figure 6. M2c markers universally define MerTK+ macrophages prone to AC clearance.

(A–C) CD14+ cells were cultured in serum-free medium in the presence of IFNγ (10 ng/ml; M1), IL-4 (20 ng/ml; M2a), M-CSF (50 ng/ml) + IL-10 (50 ng/ml; M2c), or in the absence of cytokines (M0), for 4 days. Cells were stained for MerTK, CD163, CD14, CD16 and CD204. Co-expression of MerTK and M2c surface markers was studied by flow cytometry (A). For each M2c receptor (CD14, CD16, CD163, CD204), an Expression Ratio was obtained by dividing “percentage of macrophages expressing a given M2c receptor among MerTK+ macrophages” by “percentage of macrophages expressing a given M2c receptor among total macrophages in culture”, after differentiation in M0, M1, M2a or M2c conditions. Frequencies of each M2c receptor (percentages of positive cells) among MerTK+ macrophages and among total macrophages were analyzed for potential significant differences between the two sets of data (B). IL-4-treated cells were also stained for CD209 and CD206 (C). Data shown are representative of four independent experiments. (D–E) CD14+ cells treated with IFNγ or IL-4 for 3 days in serum-free medium were co-incubated with CFSE-labeled apoptotic neutrophils for 1 hour, and then stained for CD14 and CD163. For inhibition studies, cells were pre-incubated with a goat polyclonal anti-human MerTK antibody (2 μg/ml; R&D Systems) or a goat control IgG (2 μg/ml; SouthernBiotech) for 30 minutes before addition of apoptotic neutrophils. Percentages of CFSE+ phagocytic macrophages were determined among the major populations of M1 or M2a cells (CD14dimCD163−) and the minor populations of M2c-like cells (CD14brightCD163+). Data from one representative experiment (D) and three independent experiments (E) are reported. *P <0.05; **P <0.01.
We tested whether such minor populations of M2c cells were more capable of performing AC clearance compared to the major populations of M1 and M2a macrophages. As predicted, CD14brightCD163+ cells showed significantly higher ability to clear apoptotic neutrophils compared to CD14dimCD163− cells. Moreover, phagocytosis of ACs by CD14brightCD163+ cells, but not by CD14dimCD163− cells, was significantly inhibited by blocking MerTK (Fig. 6D–E).
M2c-like CD14brightCD16+ circulating monocytes utilize MerTK to phagocitose ACs
We wanted to determine whether the M2c markers were able to predict MerTK positivity and identify efferocytotic cells even among circulating monocytes. We reported above that freshly isolated monocytes analyzed after negative selection did not express MerTK. Nevertheless, we reasoned that negative selection eliminated CD16+ monocytes along with lymphocytes, and that this minor population of more mature (HLA-DR+) monocytes could represent a counterpart in the circulation of M2c macrophages. Thus, in this set of experiments, we looked at fresh monocytes directly from PBMCs, without magnetic sorting. According to CD14 and CD16 expression, three populations of monocytes were distinguishable: a major subset of CD14+CD16− cells, and small numbers of CD14brightCD16+ and CD14dimCD16+ cells (Fig. 7A–B). We could detect MerTK+ monocytes within the CD14brightCD16+ subset, representing about one third of this population (Fig. 7C). Detection of MerTK was not associated with a higher rate of platelet-monocyte conjugates in this subset (Fig. 7D); rather, MerTK positivity was associated with the expression of the M2c scavenger receptors CD163 and CD204 (Fig. 7E). CD204 is, in fact, known to improve AC clearance by signaling via MerTK (13).
Figure 7. M2c-like CD14brightCD16+ circulating monocytes utilize MerTK to phagocitose ACs.

(A–E) Freshly isolated monocytes were analyzed by flow cytometry directly from PBMCs, without magnetic sorting, in order to include also CD16+ (HLA-DR+) monocytes. On the basis of CD14 and CD16 expression levels, monocytes were divided into 3 categories: CD14brightCD16− (red histograms and peaks), CD14brightCD16+ (blue) and CD14dimCD16+ (green). Platelet-monocyte conjugates were depicted by flow cytometry as events also positive for the platelet marker CD42b in each monocyte subset (D). Percentages in (E) refer to positivity of CD14brightCD16+ cells for the receptors indicated. Data shown are representative of four independent experiments. (F–H) freshly isolated PBMCs were co-incubated with CFSE-labeled apoptotic neutrophils at a 1:1 ratio for 4 hours. For inhibition studies, cells were pre-incubated with a goat polyclonal anti-human MerTK antibody (2 μg/ml; R&D Systems) or a goat control IgG (2 μg/ml; SouthernBiotech) for 30 minutes before addition of apoptotic neutrophils. Percentages of CFSE+ phagocytic monocytes were determined within each monocyte subset. Data from three independent experiments (F–G) and one representative experiment (H) are reported. *P <0.05; **P <0.01; ***P <0.001.
To examine the ability of each monocyte subset to phagocytose ACs, we coincubated PBMCs with apoptotic neutrophils for 4 hours at a 1:1 ratio; then, we gated on monocytes by flow cytometry for analysis. In agreement with a recent study (43), CD16+ monocytes showed higher capacity of AC clearance compared to CD16− monocytes. Differences in efferocytosis rates were statistically significant when CD14brightCD16+ cells were compared with CD16− cells. Although CD14dimCD16+ cells were the most efficient in efferocytosis in 2 of 3 experiments, the difference with CD16− monocytes did not reach full statistical significance (Fig. 7F). Furthermore, only CD14brightCD16+ monocytes were dependent on MerTK activity for phagocytosis of ACs; AC clearance by this subset, in fact, was significantly reduced following pretreatment with a blocking anti-MerTK antibody (Fig 7G–H).
Gas6 is released by M2c macrophages, and amplifies IL-10 secretion via MerTK
Gas6 is a major ligand of MerTK; it is produced by several cell types, including macrophages themselves (36, 42). We examined Gas6 levels in the supernatants of serum-free cell cultures, to investigate how MerTK regulation was related to Gas6 production. We found that Gas6 was released upon cell stimulation with IL-10, dexamethasone or IL-4, but not with GM-CSF or IFNγ, indicating that both M2c and M2a, but not M1, macrophages were Gas6 producers. Similarly to what we observed with MerTK expression, M-CSF alone had no effect on Gas6 production in serum-free conditions, yet enhanced dexamethasone effects. TGFβ was a negative regulator, as shown by its inhibitory effect on IL-4-induced Gas6 secretion (Fig. 8A).
Figure 8. Gas6 is released by M2c macrophages, and amplifies IL-10 production via MerTK.

(A) Gas6 was measured by ELISA in supernatants of CD14+ cells cultured in serum-free medium, in the presence of different treatments (M-CSF, 50 ng/ml; IL-10, 50 ng/ml; dexamethasone, 100 nM; TGFβ, 20 ng/ml; GM-CSF, 100 ng/ml; IFNγ, 10 ng/ml; IL-4, 20 ng/ml), for 4 days. Data shown are representative of four independent experiments. (B–C) CD14+ cells were cultured in serum-free conditions for 3 days in the absence of colony stimulating factors, and stimulated from day 1 with LPS (50 ng/ml) ± rhGas6 (1 μg/ml) for 48 hours; TNFα and IL-10 levels were measured in supernatants by ELISA. Data shown are representative of three independent experiments. (D) Cells were cultured in serum-free conditions for 3 days in the presence of M-CSF (50 ng/ml), and stimulated from day 1 with LPS ± rhGas6 for 48 hours; rhGas6 significantly increased LPS-induced IL-10 release in culture supernatants, as assessed by ELISA. IL-10 increase was prevented by blocking MerTK with a goat polyclonal anti-human MerTK antibody (5 μg/ml; R&D Systems) during LPS + rhGas6 stimulation. Data shown are representative of four independent experiments. (E) Cells were cultured in serum-free conditions for 4 days in the presence or absence of M-CSF (50 ng/ml) ± IL-10 (50 ng/ml). On day 1, a recombinant MerFc (5 μg/ml; R&D Systems) or a goat polyclonal anti-human Gas6 blocking antibody (5 μg/ml; R&D Systems) was added to precipitate Gas6 endogenously produced by cultured cells. From day 2, cells were stimulated with LPS for 48 hours. Neither MerFc or anti-Gas6 antibody were able to restore TNFα secretion, inhibited in M2c (M-CSF+IL-10) cells. Data shown are representative of three independent experiments. *P <0.05; **P <0.01; ***P <0.001.
The Gas6/MerTK pathway is known to inhibit production of proinflammatory cytokines (17–18) and this function can be independent from AC clearance (19). Consistent with previous results (17–18), we found that, in monocyte-macrophages cultured in the absence of colony stimulating factors, Gas6 significantly reduced LPS-induced production of TNFα (Fig. 8B), while tended to increase IL-10 production (Fig. 8C).
Since IL-10 production represents a central functional property of M2c macrophages (31), we hypothesized that Gas6 effects on IL-10 secretion could be more pronounced in cells cultured in the presence of M-CSF. In these conditions, it was possible that Gas6, inducible by IL-10, could signal through MerTK, inducible by M-CSF in an IL-10-dependent manner, to stimulate further IL-10 secretion. We indeed found that rhGas6 significantly increased IL-10 levels in supernatants of M-CSF cultured cells stimulated with low doses of LPS, and such effect was prevented by blocking MerTK activation (Fig. 8D). In the presence of M-CSF, IL-10 and Gas6 were, therefore, able to reciprocally stimulate one another’s production. Gas6 release from MerTK+ M2c cells can, then, be part of an autocrine loop that amplifies IL-10 secretion and positively regulates homeostasis of M2c macrophages.
In the presence of M-CSF, cell secretion of TNFα was instead negligible. We tested whether the endogenous production of Gas6 by M2c (M-CSF+IL-10) cells played a role in inhibiting TNFα production. However, the addition of either recombinant MerFc or a blocking anti-Gas6 antibody to neutralize Gas6 failed to increase TNFα levels in supernatants (Fig. 8E).
DISCUSSION
In the present study, we examine the expression of the key AC receptor MerTK in human populations of monocytes and macrophages, determining for each subset the ability to clear ACs and the functional relevance of MerTK in this process. Furthermore, we make a novel contribution to characterization of human anti-inflammatory macrophages, with respect to their phenotype, the immunological factors promoting their differentiation, and the favoring role of the Gas6/MerTK pathway in mediating IL-10 secretion from these cells.
It has been previously shown that MerTK is regulated by metabolic pathways through LXRs, nuclear sensors activated following macrophage exposure to ACs or to other sources of cholesterol, and through PPARs and RXRs, transcription factors activated during macrophage differentiation (4–6). Here, we show that this molecule is also importantly regulated by specific immunological factors. In fact, MerTK is not homogenously distributed among human macrophage populations, but is mostly restricted to a discrete subset of IL-10-secreting anti-inflammatory M2 macrophages, recently named “M2c” (31). Such subset is distinguishable from “M2a” (IL-4-producing and TH2-related) and “M1” (IL-12-secreting and TH1-related) macrophages for its specific phenotype, here characterized as CD14brightCD16+CD163+CD204+CD206brightCD209null. M2c polarization is closely associated with MerTK up-regulation, and detection of M2c receptors predicts MerTK expression. Remarkably, M2c differentiation is required to obtain macrophages highly capable of clearing ACs. M2c-like cells are also detectable among circulating CD14brightCD16+ monocytes, and occur even in M1 and M2a differentiating conditions as minority populations; consistently, these latter cells are particularly prone to AC clearance, and utilize MerTK for this purpose. M2c macrophages are able to release Gas6, which can in turn amplify IL-10 secretion in an autocrine manner, via MerTK signaling. Taken together, MerTK importantly affects AC clearance activity and is critical for homeostasis of M2c macrophages.
Very recently, Galvan et al. reported that prolonged macrophage exposure to the complement component C1q, another molecule primarily involved in AC clearance, stimulates expression of MerTK and production of Gas6 and C1q itself. Moreover, MerTK was recognized to be essential for C1q-dependent efferocytosis (15). However, the mechanism accounting for C1q induction of MerTK was not defined. In this regard, it will be interesting to determine whether stimulation with C1q is able to elicit the M2c phenotype; alternatively, M2c polarization might promote release of C1q, which in its turn might mediate or amplify MerTK up-regulation.
M2 differentiation is a key process regulating inflammation and fibrosis (33–34, 37). The factors controlling this process are just becoming understood. We performed the first systematic study on the effects of cytokines and growth factors on human macrophage phenotype, MerTK expression and Gas6 secretion. The effects herein observed are summarized in Table 1. Remarkably, MerTK and Gas6 levels follow the expression pattern of CD16 and CD163, molecules identifiable, in agreement with a recent study (38), as specific M2c markers. CD206 is highly expressed in these macrophages; however, CD206 is also up-regulated in M2a (IL-4-treated) and in GM-CSF-differentiated M1 macrophages. M2a cells are selectively characterized by CD209 expression, and down-regulate surface expression of MerTK. Nevertheless, contrary to what was reported by others (27), we found that M2a macrophages secrete Gas6, consistent with IL-4 effects on Gas6 gene induction (42); this suggests that IL-4 might induce other membrane or soluble TAM receptors.
Table I.
Effects of immunological environment on macrophage phenotype markers, MerTK expression and Gas6 production.
| CD206 | CD209 | CD16 | CD163 | CD14 | MerTK | Gas6 | |
|---|---|---|---|---|---|---|---|
| IFNγ | ↓ | = | = | ↓ | =↑ | ↓ | = |
| GM-CSF | ↑ | = | = | ↓ | =↓ | ↓ | = |
| IL-4 | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | ↑ |
| TGFβ | ↑ | =↓ | ↑ | ↓ | ↓ | =↓ | =↓ |
| IL-10 | = | = | = | ↑ | =↑ | = | ↑ |
| M-CSF+IL-10 (or M-CSF+serum) | ↑ | = | ↑ | ↑ | ↑ | ↑ | ↑ |
| Glucocorticoids | ↑ | = | ↑ | ↑ | = | ↑ | ↑ |
Our study gives, in particular, novel insights into mechanisms of M2c polarization in humans. IL-10, TGFβ and glucocorticoids are the factors to date classified as M2c stimuli (31). We show that M2c macrophages are differentiated in the presence of steroids, or in the co-presence of M-CSF and IL-10. These two types of stimulation have comparable effects on macrophage polarization. Steroids are known to increase a broad range of molecules involved in AC clearance, including MerTK, MFG-E8, C1q, Axl, ADORA3, thrombospondin-1 (29–30). We show here that MerTK up-regulation by these drugs is closely related to induction of M2c polarization. Moreover, we report for the first time that steroids stimulate macrophages to secrete Gas6, possibly resulting in enhanced MerTK-dependent clearance independent from Protein S (12, 28). In contrast, we observed that TGFβ gives a different phenotype (CD163−CD16+MerTK−Gas6−), while IL-10 per se gives only a partial phenotype (CD163+CD16−MerTK−Gas6+), that requires M-CSF for its complete expression (CD163+CD16+MerTK+Gas6+).
Incubation of monocytes with M-CSF promotes M2 macrophage differentiation (33–34). We noted that M-CSF synergizes with either IL-10 or glucocorticoids, thereby identifying M-CSF as a specific M2c factor or co-factor rather than a general M2 cytokine. Additive effects between M-CSF and IL-10 were also previously reported. Specifically, IL-10 increased CD16 (FcγRIII) and CD32 (FcγRII) expression in M-CSF-driven macrophages, thus enhancing FcγR-mediated phagocytosis in these cells (44); IL-10 alone, instead, was only able to up-regulate CD64 (FcγRI) (45). Similarly, we found that MerTK expression and MerTK-mediated AC phagocytosis rely on a combined action of M-CSF and IL-10, while IL-10 alone, in agreement with a recent report (27), is only able to induce Gas6.
Importantly, we demonstrated that IL-10 is not only an enhancer of M-CSF effects, but is essential for M-CSF to act as an M2c factor and up-regulate MerTK. We examined the effects of M2c factors in the presence and absence of serum in cell culture medium, as well as in IL-10-blocked serum-containing and IL-10-supplemented serum-free medium. We concluded that M-CSF-driven M2c differentiation is dependent on IL-10 bioactivity, present in serum-containing medium (39–40) or exogenously added in serum-free conditions, whereas glucocorticoid-induced polarization is IL-10-independent.
IL-10 per se did not up-regulate MerTK, consistently with previous microarray studies of IL-10 effects on gene regulation in monocytes-macrophages (46–47). Only Jung and colleagues observed Mer gene up-regulation by IL-10 (48). However, these authors incubated PBMCs rather than purified monocytes in vitro with IL-10, and only afterwards sorted monocytes for microarray analysis. This may have underestimated the contribution of T lymphocytes to MerTK up-regulation; in particular, T cells are a well-known source of M-CSF (49). Indeed, Jung et al. also reported IL-10 induction of CD16 and CD32 (48), for which M-CSF is required (44). Of interest, Tiemessen and colleagues reported that T regulatory cells (Tregs) promote anti-inflammatory monocytes-macrophages expressing CD163 and CD206; however, while CD163 expression was dependent on IL-10 released by Tregs, CD206 induction was suggested to be cytokine-independent (50). Here, we showed that macrophage co-expression of CD163, CD206, and CD16, is inducible by M-CSF, strongly supporting a central role for T-cell derived M-CSF, together with IL-10, in the induction of anti-inflammatory M2c macrophages by Tregs. From this perspective, production of M-CSF by Tregs might counterbalance the well-known production of GM-CSF by proinflammatory T helper 1 and T helper 17 lymphocytes (Th17) (51), with strong and direct repercussions of the Treg vs Th17 balance on M-CSF vs GM-CSF macrophage differentiation.
Interestingly, MerTK+ M2c-like cells could be found even among circulating CD16+ monocytes. CD16+ monocytes represent more mature monocytes, and can phagocytose ACs via CD36 (43). However, only CD14brightCD16+, but not CD14dimCD16+, monocytes express MerTK and the M2c receptors CD163 and CD204; accordingly, their ability to clear ACs is in part MerTK-dependent. That is consistent with previous findings that CD14brightCD16+ monocytes are predominantly anti-inflammatory and secrete IL-10, whereas CD14dimCD16+ are pro-inflammatory and produce TNFα (52–53). A role for TGFβ in the development of CD16+ monocytes was also suggested (54). We reported that M-CSF and IL-10 trigger CD16 expression along with CD14, CD163 and MerTK, whereas TGFβ up-regulates CD16, but not CD14, CD163 or MerTK; therefore, we propose that CD14highCD16+ and CD14dimCD16+ subsets are selectively elicited by different cytokines.
M-CSF-driven macrophages have already been reported by Xu and colleagues to have augmented capability of clearing early ACs; however, these authors attributed such ability to enhanced macropinocytosis activity, independent from IL-10 (55). This conclusion was soon challenged by Krysko et al., who demonstrated a major role for macropinocytosis in removing late, rather than early, ACs (10). In fact, Xu et al. initially showed enhanced AC clearance by M-CSF-driven macrophages even when late ACs were preponderant compared to early ACs, and suggested the involvement of macropinocytosis in clearance of both early ACs and blebs (i.e., late apoptotic debris) (55). Krysko and colleagues also argued that Xu et al. did not provide evidence of co-localization of fluid phase tracers with endosomes at microscopy, which is crucial to definitively prove macropinocytosis (10). Although we do not exclude enhanced macropinocytosis by M-CSF-driven macrophages, we identified MerTK as the major driver of early AC clearance in these cells, dependent on IL-10. In fact, while the great majority of AC clearance receptors preferentially binds to late ACs (10), MerTK specifically clears early membrane-intact ACs (7, 12). Therapeutic induction of M2c polarization may, therefore, ameliorate the defective clearance of early ACs in diseases such as lupus, in which secondary necrotic cells are believed responsible for development of autoimmunity and inflammation (21–22). This may also be beneficial in atherosclerosis, in which secondary necrosis of cholesterol-laden apoptotic macrophages is associated with plaque instability (24).
Additionally, we showed that Gas6 is able to significantly enhance IL-10 secretion via MerTK from M-CSF cultured cells stimulated with LPS. That suggests that M-CSF-driven, IL-10-dependent, M2c macrophages may use this pathway as a positive feedback loop to strengthen and prolongate secretion of IL-10 in the microenvironment, facilitating in this way the re-establishment or the persistence of anti-inflammatory conditions. There are striking parallels, in this regard, with the heme oxygenase 1 (HO-1) pathway, inducible by IL-10 and promoting IL-10 secretion in M-CSF-driven CD163+ (i.e., M2c) macrophages (56). Stimulating Gas6 and MerTK activity in CD163+ macrophages to increase IL-10 production may represent a promising strategy to treat some inflammatory diseases in which CD163+ macrophages and IL-10 production are reduced, such as in atherosclerotic lesions (57–58). Conversely, blocking the Gas6/MerTK pathway in tumor-associated CD163+ macrophages may be critical to reduce IL-10 production and immune tolerance to cancer (59). The prognostic relevance of TAM receptors in cancer is so far uniquely attributed to the aberrant expression of Axl and MerTK in tumor cells, which may promote cell survival and proliferation in response to Gas6 released by tissue macrophages (26–27). Since tumor-associated macrophages are characterized by an M2c phenotype (59), it is tempting to speculate that MerTK is overexpressed also in these non-malignant cells that infiltrate and surround the tumor. In this view, Gas6 might foster tumor growth by also acting on macrophages in an autocrine/paracrine manner; macrophage secretion of anti-inflammatory cytokines mediated by MerTK would ultimately result in suppression of anti-tumor immune responses.
In conclusion, we provide evidence that M2c polarization is desirable to ensure efficient clearance of early ACs by human macrophages and monocytes, due to intense MerTK up-regulation. The presence of both M-CSF and IL-10 is needed to differentiate M2c macrophages, implying that T-cell release of M-CSF, along with IL-10, can be crucial for induction of anti-inflammatory monocytes-macrophages by Tregs. M-CSF and IL-10 may have therapeutic utility in the treatment of autoimmune diseases and atherosclerosis, in which AC clearance is impaired. In addition, modulating MerTK activity in vivo (e.g., through recombinant Gas6 or activatory/inhibitory monoclonal antibodies) may constitute an effective strategy to attenuate or promote innate inflammation related to autoimmune, metabolic and tumoral diseases.
Acknowledgments
We thank Sr. Joanne Manns, PhD, for precious help in collecting blood from volunteers; Michael F. Denny, PhD, for expertise on procedures of neutrophil isolation; and Neelakshi R. Jog, PhD, for useful advice.
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID), grant 5U19AI082726 (Philadelphia Autoimmunity Center of Excellence), and by a bequest from Ms. B. Wicks.
Abbreviations used in this paper
- MerTK
Mer tyrosine kinase
- AC
apoptotic cell
- MFI
mean fluorescence intensity
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–1. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 2.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Majai G, Sarang Z, Csomós K, Zahuczky G, Fésüs L. PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol. 2007;37:1343–54. doi: 10.1002/eji.200636398. [DOI] [PubMed] [Google Scholar]
- 4.Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, Goh YP, Eagle AR, Dunn SE, Awakuni JU, Nguyen KD, Steinman L, Michie SA, Chawla A. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. 2009;15:1266–72. doi: 10.1038/nm.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez A-N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Díaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–58. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roszer T, Menéndez-Gutiérrez MP, Lefterova MI, Alameda D, Núñez V, Lazar MA, Fischer T, Ricote M. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol. 2011;186:621–31. doi: 10.4049/jimmunol.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–11. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 8.Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196:135–40. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregory CD, Devitt A. The macrophage and the apoptotic cell: an innate immune interaction viewed simplistically? Immunology. 2004;113:1–14. doi: 10.1111/j.1365-2567.2004.01959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krysko DV, D’Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis. 2006;11:1709–26. doi: 10.1007/s10495-006-9527-8. [DOI] [PubMed] [Google Scholar]
- 11.Lemke G, Burstyn-Cohen T. TAM receptors and the clearance of apoptotic cells. Ann N Y Acad Sci. 2010;1209:23–9. doi: 10.1111/j.1749-6632.2010.05744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McColl A, Bournazos S, Franz S, Perretti M, Morgan BP, Haslett C, Dransfield I. Glucocorticoids induce protein S-dependent phagocytosis of apoptotic neutrophils by human macrophages. J Immunol. 2009;183:2167–75. doi: 10.4049/jimmunol.0803503. [DOI] [PubMed] [Google Scholar]
- 13.Todt JC, Hu B, Curtis JL. The scavenger receptor SR-A I/II (CD204) signals via the receptor tyrosine kinase Mertk during apoptotic cell uptake by murine macrophages. J Leukoc Biol. 2008;84:510–8. doi: 10.1189/jlb.0307135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci. 2005;118:539–53. doi: 10.1242/jcs.01632. [DOI] [PubMed] [Google Scholar]
- 15.Galvan MD, Foreman DB, Zeng E, Tan JC, Bohlson SS. Complement component C1q regulates macrophage expression of Mer tyrosine kinase to promote clearance of apoptotic cells. J Immunol. 2012;188:3716–23. doi: 10.4049/jimmunol.1102920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shao WH, Kuan AP, Wang C, Abraham V, Waldman MA, Vogelgesang A, Wittenburg G, Choudhury A, Tsao PY, Miwa T, Eisenberg RA, Cohen PL. Disrupted Mer receptor tyrosine kinase expression leads to enhanced MZ B-cell responses. J Autoimmun. 2010;35:368–74. doi: 10.1016/j.jaut.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Camenisch TD, Koller BH, Earp HS, Matsushima GK. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol. 1999;162:3498–503. [PubMed] [Google Scholar]
- 18.Alciato F, Sainaghi PP, Sola D, Castello L, Avanzi GC. TNF-alpha, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J Leukoc Biol. 2010;87:869–75. doi: 10.1189/jlb.0909610. [DOI] [PubMed] [Google Scholar]
- 19.Tibrewal N, Wu Y, D’mello V, Akakura R, George TC, Varnum B, Birge RB. Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-kappaB transcriptional activation. J Biol Chem. 2008;283:3618–27. doi: 10.1074/jbc.M706906200. [DOI] [PubMed] [Google Scholar]
- 20.Wallet MA, Flores RR, Wang Y, Yi Z, Kroger CJ, Mathews CE, Earp HS, Matsushima G, Wang B, Tisch R. MerTK regulates thymic selection of autoreactive T cells. Proc Natl Acad Sci U S A. 2009;106:4810–5. doi: 10.1073/pnas.0900683106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao WH, Cohen PL. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther. 2011;13:202. doi: 10.1186/ar3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muñoz LE, Janko C, Grossmayer GE, Frey B, Voll RE, Kern P, Kalden JR, Schett G, Fietkau R, Herrmann M, Gaipl US. Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1733–42. doi: 10.1002/art.24535. [DOI] [PubMed] [Google Scholar]
- 23.Suh CH, Hilliard B, Li S, Merrill JT, Cohen PL. TAM receptor ligands in lupus: protein S but not Gas6 levels reflect disease activity in systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R146. doi: 10.1186/ar3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe−/− mice. Arterioscler Thromb Vasc Biol. 2008;28:1421–8. doi: 10.1161/ATVBAHA.108.167197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao D, Wang X, Li M, Lin PH, Yao Q, Chen C. Human protein S inhibits the uptake of AcLDL and expression of SR-A through Mer receptor tyrosine kinase in human macrophages. Blood. 2009;113:165–74. doi: 10.1182/blood-2008-05-158048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loges S, Schmidt T, Tjwa M, van Geyte K, Lievens D, Lutgens E, Vanhoutte D, Borgel D, Plaisance S, Hoylaerts M, Luttun A, Dewerchin M, Jonckx B, Carmeliet P. Malignant cells fuel tumor growth by educating infiltrating leukocytes to produce the mitogen Gas6. Blood. 2010;115:2264–73. doi: 10.1182/blood-2009-06-228684. [DOI] [PubMed] [Google Scholar]
- 28.Oner AF, Bay A, Kuru M, Uner A, Arslan S, Caksen H. Effects of high-dose methylprednisolone therapy on coagulation factors in patients with acute immune thrombocytopenic purpura. Clin Appl Thromb Hemost. 2005;11:489–92. doi: 10.1177/107602960501100418. [DOI] [PubMed] [Google Scholar]
- 29.Ehrchen J, Steinmüller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, Nordhues U, Sorg C, Sunderkötter C, Roth J. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood. 2007;109:1265–74. doi: 10.1182/blood-2006-02-001115. [DOI] [PubMed] [Google Scholar]
- 30.Zahuczky G, Kristóf E, Majai G, Fésüs L. Differentiation and glucocorticoid regulated apopto-phagocytic gene expression patterns in human macrophages. Role of Mertk in enhanced phagocytosis. PLoS One. 2011;6:e21349. doi: 10.1371/journal.pone.0021349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 32.Chen C, Li Q, Darrow AL, Wang Y, Derian CK, Yang J, de Garavilla L, Andrade-Gordon P, Damiano BP. Mer receptor tyrosine kinase signaling participates in platelet function. Arterioscler Thromb Vasc Biol. 2004;24:1118–23. doi: 10.1161/01.ATV.0000130662.30537.08. [DOI] [PubMed] [Google Scholar]
- 33.Smith W, Feldmann M, Londei M. Human macrophages induced in vitro by macrophage colony-stimulating factor are deficient in IL-12 production. Eur J Immunol. 1998;28:2498–507. doi: 10.1002/(SICI)1521-4141(199808)28:08<2498::AID-IMMU2498>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 34.Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, Kastelein R, Kolk A, de Waal-Malefyt R, Ottenhoff TH. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci U S A. 2004;101:4560–5. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Behrens EM, Gadue P, Gong SY, Garrett S, Stein PL, Cohen PL. The mer receptor tyrosine kinase: expression and function suggest a role in innate immunity. Eur J Immunol. 2003;33:2160–7. doi: 10.1002/eji.200324076. [DOI] [PubMed] [Google Scholar]
- 36.Seitz HM, Camenisch TD, Lemke G, Earp HS, Matsushima GK. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J Immunol. 2007;178:5635–42. doi: 10.4049/jimmunol.178.9.5635. [DOI] [PubMed] [Google Scholar]
- 37.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 38.Ambarus CA, Krausz S, van Eijk M, Hamann J, Radstake TR, Reedquist KA, Tak PP, Baeten DL. Systematic validation of specific phenotypic markers for in vitro polarized human macrophages. J Immunol Methods. 2011;375:196–206. doi: 10.1016/j.jim.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 39.Hillyer LM, Woodward B. Interleukin-10 concentration determined by sandwich enzyme-linked immunosorbent assay is unrepresentative of bioactivity in murine blood. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1514–9. doi: 10.1152/ajpregu.00378.2003. [DOI] [PubMed] [Google Scholar]
- 40.Malone D, Napolitano LM, Genuit T, Bochicchio GV, Kole K, Scalea TM. Total cytokine immunoassay: a more accurate method of cytokine measurement? J Trauma. 2001;50:821–5. doi: 10.1097/00005373-200105000-00008. [DOI] [PubMed] [Google Scholar]
- 41.Sulahian TH, Högger P, Wahner AE, Wardwell K, Goulding NJ, Sorg C, Droste A, Stehling M, Wallace PK, Morganelli PM, Guyre PM. Human monocytes express CD163, which is upregulated by IL-10 and identical to p155. Cytokine. 2000;12:1312–21. doi: 10.1006/cyto.2000.0720. [DOI] [PubMed] [Google Scholar]
- 42.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177:7303–11. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 43.Mikołajczyk TP, Skrzeczyńska-Moncznik JE, Zarebski MA, Marewicz EA, Wiśniewska AM, Dzieba M, Dobrucki JW, Pryjma JR. Interaction of human peripheral blood monocytes with apoptotic polymorphonuclear cells. Immunology. 2009;128:103–13. doi: 10.1111/j.1365-2567.2009.03087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hashimoto S, Yamada M, Motoyoshi K, Akagawa KS. Enhancement of macrophage colony-stimulating factor-induced growth and differentiation of human monocytes by interleukin-10. Blood. 1997;89:315–21. [PubMed] [Google Scholar]
- 45.Te Velde AA, de Waal Malefijt R, Huijbens RJ, de Vries JE, Figdor CG. IL-10 stimulates monocyte Fc gamma R surface expression and cytotoxic activity. Distinct regulation of antibody-dependent cellular cytotoxicity by IFN-gamma, IL-4, and IL-10. J Immunol. 1992;149:4048–52. [PubMed] [Google Scholar]
- 46.Williams L, Jarai G, Smith A, Finan P. IL-10 expression profiling in human monocytes. J Leukoc Biol. 2002;72:800–9. [PubMed] [Google Scholar]
- 47.Donnelly RP, Dickensheets H, Finbloom DS. The interleukin-10 signal transduction pathway and regulation of gene expression in mononuclear phagocytes. J Interferon Cytokine Res. 1999;19:563–73. doi: 10.1089/107999099313695. [DOI] [PubMed] [Google Scholar]
- 48.Jung M, Sabat R, Krätzschmar J, Seidel H, Wolk K, Schönbein C, Schütt S, Friedrich M, Döcke WD, Asadullah K, Volk HD, Grütz G. Expression profiling of IL-10-regulated genes in human monocytes and peripheral blood mononuclear cells from psoriatic patients during IL-10 therapy. Eur J Immunol. 2004;34:481–93. doi: 10.1002/eji.200324323. [DOI] [PubMed] [Google Scholar]
- 49.Hallet MM, Praloran V, Vié H, Peyrat MA, Wong G, Witek-Giannotti J, Soulillou JP, Moreau JF. Macrophage colony-stimulating factor (CSF-1) gene expression in human T-lymphocyte clones. Blood. 1991;77:780–6. [PubMed] [Google Scholar]
- 50.Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A. 2007;104:19446–51. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–7. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 52.Skrzeczyńska-Moncznik J, Bzowska M, Loseke S, Grage-Griebenow E, Zembala M, Pryjma J. Peripheral blood CD14high CD16+ monocytes are main producers of IL-10. Scand J Immunol. 2008;67:152–9. doi: 10.1111/j.1365-3083.2007.02051.x. [DOI] [PubMed] [Google Scholar]
- 53.Belge KU, Dayyani F, Horelt A, Siedlar M, Frankenberger M, Frankenberger B, Espevik T, Ziegler-Heitbrock L. The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J Immunol. 2002;168:3536–42. doi: 10.4049/jimmunol.168.7.3536. [DOI] [PubMed] [Google Scholar]
- 54.Phillips JH, Chang CW, Lanier LL. Platelet-induced expression of Fc gamma RIII (CD16) on human monocytes. Eur J Immunol. 1991;21:895–9. doi: 10.1002/eji.1830210406. [DOI] [PubMed] [Google Scholar]
- 55.Xu W, Roos A, Schlagwein N, Woltman AM, Daha MR, van Kooten C. IL-10-producing macrophages preferentially clear early apoptotic cells. Blood. 2006;107:4930–7. doi: 10.1182/blood-2005-10-4144. [DOI] [PubMed] [Google Scholar]
- 56.Sierra-Filardi E, Vega MA, Sánchez-Mateos P, Corbí AL, Puig-Kröger A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology. 2010;215:788–95. doi: 10.1016/j.imbio.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 57.Caligiuri G, Rudling M, Ollivier V, Jacob MP, Michel JB, Hansson GK, Nicoletti A. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med. 2003;9:10–7. [PMC free article] [PubMed] [Google Scholar]
- 58.Gleissner CA, Shaked I, Erbel C, Böckler D, Katus HA, Ley K. CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ Res. 2010;106:203–11. doi: 10.1161/CIRCRESAHA.109.199505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–73. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]