

Abstract
Cytochalasin B (CB) and forskolin (FSK) inhibit GLUT1-mediated sugar transport in red cells by binding at or close to the GLUT1 endofacial sugar binding site. Paradoxically, very low concentrations of each of these inhibitors produce a modest stimulation of sugar transport (Cloherty, E. K., Levine, K. B., & Carruthers, A. (2001). The red blood cell glucose transporter presents multiple, nucleotide-sensitive sugar exit sites. Biochemistry, 40(51), 15549–15561). This result is consistent with the hypothesis that the glucose transporter contains multiple, interacting, endofacial binding sites for CB and FSK. The present study tests this hypothesis directly and, by screening a library of cytochalasin and forskolin analogs, asks what structural features of endofacial site ligands determine binding site affinity and cooperativity. Like CB, FSK competitively inhibits exchange 3-O-methylglucose transport (sugar uptake in cells containing intracellular sugar) but non-competitively inhibits sugar uptake into cells lacking sugar at 4°C. This refutes the hypothesis that FSK binds at GLUT1 endofacial and exofacial sugar binding sites. Some forskolin derivatives and cytochalasins inhibit equilibrium [3H]-CB binding to red cell membranes depleted of peripheral proteins at 4°C. Others produce a moderate stimulation of [3H]-CB binding when introduced at low concentrations but inhibit binding as their concentration is increased. Yet other analogs modestly stimulate [3H]-CB binding at all inhibitor concentrations applied. These findings are explained by a carrier that presents at least two interacting endofacial binding sites for CB or FSK. We discuss this result within the context of models for GLUT1-mediated sugar transport and GLUT1 quaternary structure and we evaluate the major determinants of ligand binding affinity and cooperativity.
Cellular exchange of nutrients, ions and metabolites proceeds via membrane-spanning proteins called channels and carriers (1). The Major Facilitator Superfamily of carriers is responsible for the largest portion of nutrient transport in cells (2) and among these carriers, the sugar porter sub-family is one of the oldest and largest family classifications.
Sugar porters catalyze both cellular sugar import and export but net sugar transport always proceeds from high to low sugar concentration. The first human sugar transporter to be isolated was the erythrocyte membrane protein 1 GLUT1 (3, 4). GLUT1 is primarily expressed in the cardiovascular system, in astrocytes of the central nervous system and mediates glucose transport across blood-tissue barriers (5). Any one of several mutations in GLUT1 results in GLUT1 Deficiency Syndrome (GLUT1DS) in which reduced glucose transport into the brain leads to developmental defects and seizures (6).
Hydropathy analysis, scanning glycosylation mutagenesis and proteolytic digestion studies confirm that GLUT1 (a 55 kDa protein) contains twelve alpha-helical transmembrane domains (7). Each GLUT1 polypeptide is thought to function as a simple carrier (8), presenting either a sugar uptake (exofacial) or a sugar exit (endofacial) site at any given moment. However, this proposed transport mechanism does not explain the behavior of GLUT1 in human red cells (9) where GLUT1 monomers self-associate into cooperative oligomers simultaneously exposing exofacial and endofacial binding sites and displaying a litany of catalytic behaviors incompatible with the simple carrier mechanism (10).
Scanning cysteine mutagenesis analysis (11) suggests that the GLUT1 sugar uptake site involves portions of alpha-helical, transmembrane spanning regions 1, 5, 7, 8 and 11. Peptide mapping studies of affinity labeled GLUT1 suggest that the exit site contains a subdomain of membrane spanning regions 10 and 11 (12). However, specific GLUT1 residues contacting glucose in GLUT1 exofacial (e2) and endofacial (e1) conformations are unknown.
The present study characterizes the GLUT1 export conformation by analysis of inhibitor binding to the e1 conformer. Comprehensive analysis of GLUT1 interaction with a library of inhibitors may reveal details of the complementary relationship between ligand and binding pocket structures. We selected GLUT1 endofacial site inhibitors and their derivatives for this analysis. Cytochalasin B (CB) is a cell-permeable alkaloid that disrupts actin filaments and inhibits glucose transport (13). Forskolin (FSK) is a cell-permeable diterpenoid that inhibits GLUT1 and activates adenylate cyclase (14). Both CB and FSK are thought to bind to the endofacial orientation of GLUT1 where they act as noncompetitive inhibitors of erythrocyte glucose uptake and competitive inhibitors of exit (14, 15). These endofacial inhibitors have also been docked to an homology-based, theoretical GLUT1 structure where they are proposed to bind to cytoplasmic domains of the carrier (16). Our findings confirm that endofacial, export-site inhibitors inhibit ligand binding by two mechanisms - direct competition and cooperative inhibition - and provide new insights into the molecular determinants of each type of inhibition.
MATERIALS AND METHODS
Solutions
Kaline consisted of 150 mM KCl, 5 mM MgCl2, 5 mM EGTA, 5mM HEPES, pH 7.4. Lysis buffer contained 10 mM Tris-HCl, 2mM EDTA, pH 8.0. Stripping solution contained 2 mM EDTA, 15.4 mM NaOH, pH 12. Sugar-stop solution consisted of ice-cold Kaline containing 20 μM CB and 200 μM phloretin.
Materials
[3H]-3-O-methylglucose, [3H]-cytochalasin B, and [3H]-forskolin were purchased from Sigma Chemicals. Human blood was purchased from Biological Specialties Cooperation. Forskolin derivatives were synthesized by A.N. Appleyard (2001, PhD thesis, ‘Elucidation of protein-antibiotic complexes by novel chemical and NMR approaches’; Astbury Center for Structural Molecular Biology, Leeds University, U.K.). Other reagents were purchased from Sigma Chemicals.
Red Cells
Red cells were isolated from whole human blood by centrifugation as described previously (9).
Red Cell Membranes
Red cell membranes depleted of peripheral membrane proteins (including the cytochalasin B binding protein actin) were prepared as described in (17).
3-O-methylglucose uptake
Zero trans 3MG uptake (3MG uptake into cells lacking intracellular sugar) and equilibrium exchange 3MG sugar uptake (unidirectional [3H]-3MG uptake in cells where intracellular [3MG] = extracellular [3MG]) were measured at 4 °C as described previously (9, 18).
Forskolin or cytochalasin B Inhibition of 3OMG transport
3MG uptake was measured as described above in the absence and presence of cytochalasin B, forskolin or their derivatives. Inhibitor concentrations ranged from 10−9 -10−4 M using ethanol or dimethylsulfoxide as solvents. Solvent concentration never exceeded 0.1% (vol:vol) and is without effect on sugar transport rates. Cells were preincubated with inhibitor for 15 minutes on ice before starting uptake measurements. When transport inhibition was profound (> 75%), the uptake interval was extended to permit more accurate determinations of uptake.
Ligand binding competition assay
Stripped ghosts (peripheral protein-depleted membranes) were diluted to 2 mg/ml in 50 mM Tris medium, pH 7.4 at 4°C. Ligand binding assay solutions comprised Tris medium plus 50 nM unlabeled CB, 14 μCi [3H]-CB plus a range of inhibitor (I) concentrations (0 – 100 μM). Some ligand binding assays substituted 50 nM forskolin and 34 μCi [3H]-forskolin for cytochalasin B. Binding assays were as described previously (19). Membranes (50 μL) and ligand binding solutions (50 μL) were mixed in a microcentrifuge tube and, following 15 minutes of inversion at 4°C, two 10 μL aliquots were sampled (Total dpms). The tubes were centrifuged at 14,000 rpm for 5 minutes, and two 10 μL aliquots of the supernatant were sampled (Free dpms). ‘Bound’ dpms were calculated as the difference between ‘total’ and ‘free’ dpms. Each inhibitor concentration was tested three times in triplicate.
LIGAND BINDING TO GLUT1 - THEORY
Assay by ligand depletion
The nature of the binding assay adds a significant complication to the analysis. This is a ligand depletion assay in which [GLUT1] ≈ 1 to 2 μM, radiolabeled CB or FSK (Ltotal) is 50 nM and competing (unlabeled ligand, I) ranges from 0 to 50 μM. Thus at low [I]total, the concentration of [I]free < [I]total due to binding to GLUT1. [L]free and [L]bound are always measured directly. Errors in determination of KI, β and γ derive from the assumption [I]total = [I]free when KI ≤ [GLUT1].
Even after allowing for ligand depletion as above, equilibrium cytochalasin B and forskolin binding to GLUT1 display non-Michaelis kinetics at low ligand concentrations (20–22). The simplest model that accounts for this behavior (see Scheme 1) assumes that the glucose transporter complex presents two interacting binding sites for endofacial ligands. This could be explained either by two sites on one transport protein or two interacting transport proteins each with one site. When presented with a binary mixture of ligands (e.g radio-ligand L plus inhibitor I), the transporter, e, may form several liganded states (see scheme 1): e (unliganded transporter), 2(I. e), 2(L.e), 2(I.e.L), L.e.L and I.e.I.
Scheme 1.

When measuring CB binding in the presence of FSK, binding of the first CB molecule to unoccupied GLUT1 is described by the dissociation constant KL and binding of the second CB molecule is described by αKL where α is a dimensionless constant greater than zero. Binding of the first FSK molecule to unoccupied transporter is described by K and binding of the second FSK molecule is described by βKI (β is a dimensionless constant greater than zero). When heterocomplexes are formed, binding of the first molecule (CB or FSK) is described by KL or KI and binding of the second molecule (FSK or CB) is described by γ KL or γ KI where γ is a dimensionless constant greater than zero. This model explains enhanced CB binding in the presence of at least 11 of the 20 “antagonists” tested in the present study. Cooperative interactions between binding sites (positive or negative cooperativity) may depend on whether the first and second ligands to bind are identical or different. For example, the ligand CCB might reduce binding of the remaining site for CCB (α > 1; negative homo-cooperativity) but increase the affinity of the remaining site for inhibitor I (γ < 1; positive hetero-cooperativity) or vice versa.
Equilibrium ligand binding (Lb) to GLUT1 in the presence of a competing e1 inhibitor I is described by:
| Eqn 1 |
where Bmax is given by:
| Eqn 2 |
Kd(app) is given by:
| Eqn 3 |
when I=0,
| Eqn 4 |
| Eqn 5 |
where KL, KI, α, β and γ are shown in scheme 1. Since Bmax does not include I terms, inhibitor I does not affect Bmax for L binding to GLUT1. The ratio CB binding in the presence of inhibitor: CB binding in the absence of inhibitor (bi/b) is thus given by:
| Eqn 6 |
When cytochalasin B (I) inhibition of [3H]-cytochalasin B (L) binding (or forskolin (I)inhibition of [3H]- forskolin (L)) is measured, the analysis simplifies considerably because α = β = γ and KL = Ki. Equation 6 simplifies to:
| Eqn 7 |
I and L are measured directly from the raw binding data, so the homo-inhibition experiment allow the unambiguous calculation of KL and α for cytochalasin B and forskolin (Table 1).
Table 1.
Modulation of GLUT1 Cytochalasin B binding by cytochalasins and forskolins
| a Ligand | b KL nM | cα | d Ki nM | eΔG° kcal/mol | fΔΔG° kcal/mol | gβ | hγ | i R2 | j Inhibition | kβ KI nM | lγ Ki μM | mβ*γ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCB | 98 ± 08.5 | 6.284 ±3.849 | 98 ± 08.5 | −8.88 | 6.284 ± 3.849 | 6.284 ± 3.849 | 0.997 | 1 | 616 | 616 | 39 | |
| CCA | 815 ± 96 | −7.72 | 1.17 | 1.11 ± 0.17 | 0.374 ± 0.052 | 1.00 | 2 | 905 | 302 | 0.42 | ||
| CCH | 98 ±3 | −8.88 | 0.00 | 249 ± 45 | 0.449 ± 0.019 | 0.94 | 2 | 24,402 | 44 | 110 | ||
| CCJ | 12 ±1 | −10.04 | −1.16 | 6399 ± 347 | 0.635 ± 0.013 | 0.99 | 3 | 76,788 | 8 | 4063 | ||
| CCD | 1969 ± 98 | −7.23 | 1.65 | 101 ± 61 | 0.557 ± 0.019 | 0.98 | 3 | 198,869 | 1097 | 51 | ||
| CCE | 99 ± 8 | −8.88 | 0.01 | 7917 ± 2935 | 0.555 ± 0.016 | 0.96 | 3 | 783,783 | 55 | 4394 | ||
| CCC | 1591 ± 431 | −7.35 | 1.53 | ∞ | 0.433 ± 0.003 | 0.99 | 4 | ∞ | 689 | ∞ | ||
| FSK | 2750 ± 787 | 2.43 ± 0.23 | 3282 ± 960 | −6.95 | 1.54 ± 0.73 | 0.930 ± 0.103 | 0.93 | 1 | 5,054 | 3052 | 1 | |
| 14.15DiHFSK | 29.1 ± 17 | −9.55 | −2.60 | 242.7 ± 140 | 0.571 ± 0.015 | 0.99 | 1 | 7,042 | 17 | 139 | ||
| 6AFSK | 949 ± 164 | −7.63 | −0.68 | 9.84 ± 0.68 | 0.416 ± 0.019 | 0.95 | 2 | 9,338 | 389 | 4 | ||
| 1,6DiAFSK | 11 ± 2 | −6.28 | 0.67 | 1480 ± 268 | 0.489 ± 0.017 | 0.94 | 2 | 16,280 | 5 | 724 | ||
| 1DOFSK | 1.0 ± 0.7 | −11.41 | −4.46 | 22179 ± 5104 | 0.438 ± 0.017 | 0.97 | 2 | 22,179 | 0.4 | 9714 | ||
| 7FAFSK | 629 ±.644 | −7.86 | −0.91 | 45.3 ± 5.0 | 0.475 ± 0.010 | 0.97 | 2 | 28,494 | 296 | 22 | ||
| 1AFSK | 3 ± 2 | −10.80 | −3.85 | 11923 ± 8441 | 0.440 ± 0.008 | 0.86 | 2 | 35,769 | 1 | 5246 | ||
| 9DOFSK | 430 ± 75 | −8.07 | −1.12 | 120.3 ± 12.3 | 0.464 ± 0.016 | 0.75 | 2 | 51,729 | 198 | 56 | ||
| 7FPAFSK | 142 ± 78 | −4.88 | 2.07 | 1115 ± 218 | 0.508 ± 0.012 | 0.65 | 3 | 158,330 | 71 | 566 | ||
| 7FPPNeaFSK | 25 ± 8.4 | −9.63 | −2.68 | 40416 ± 1646 | 0.430 ± 0.086 | 0.74 | 4 | 1,010,400 | 11 | 17379 | ||
| 7DeAFSK | 2 ± 2 | −11.02 | −4.07 | ∞ | 0.450 ± 0.008 | 0.86 | 4 | ∞ | 1 | ∞ | ||
| 6A7DeAFSK | o NA | o NA | o NA | o NA | 5 | |||||||
| 7FPPFSK | o NA | o NA | o NA | o NA | 5 | |||||||
| nmean | 0.493 ± 0.056 |
Modulation of cytochalasin B binding to red cell membranes by cytochalasins and forskolins - summary of findings. The results of analyses such as those in Figures 3, 4 and 5 are summarized for the cytochalasins and the forskolins. The concentration of radioligand ([3H]-CB) was 50 nM in all experiments.
The total concentration of unlabeled competing ligand was varied from 0 to 100 μM. Analysis of binding inhibition was carried out as described in Figures 2 and 3 using equations 6 and 7 and the iterative analysis procedure outlined in Methods.
KL (nM) and
α for CB and FSK binding were computed in studies of CB inhibition of [3H]-CB binding and FSK inhibition of [3H]-FSK binding respectively and assume that α = β = γ.
KI (nM) represents the affinity of unoccupied GLUT1 for ligand.
ΔG° was calculated as RT ln(1/KI).
ΔΔG° was calculated as the change in ΔG° using the lowest KI for cytochalasins or foskolins as the baseline.
Analysis according to equation 6 also permits computation of the cooperativity parameters β and γ and yields a correlation coefficient R2.
Types of inhibition are: 1, Michaelis like inhibition (curves a & e, Figure 1); 2, stimulation followed by inhibition (curves c & f, Figure 1); 3, high and low affinity inhibition (curve d, Figure 1); 4, stimulation (curve g, Figure 1) and, 5, no effect.
These types of inhibition are unrelated to the products βKI, γKI or β γ.
While β parameters are ligand dependent, γ parameters appear to be invariant and are averaged for the cytochalasins and forskolins (mean ± SEM).
No effect on binding was observed. In all experiments, at least three (normally 4) complete dose responses were performed with each data point in triplicate. The 9 or 12 data points were then averaged at each concentration to perform the final analysis.
This analysis assumes that all binding and dissociation steps are rapid relative to the duration of experimental measurement and that free and bound ligand achieve true equilibrium. These assumptions are most likely satisfied because cytochalasin B binding to GLUT1 occurs with a time constant of 1 sec at 4°C (10, 23) whereas binding was measured over a period of 15 minutes (> 1,000 half-lives). The model allows for several possible effects of inhibitor (I) on ligand (L) binding.
1 Binding is not cooperative
When binding of L and I at any single site is mutually exclusive and lacks hetero-cooperativity (γ = 1; Figure 1, curve a), the inhibitor I will serve as a simple competitive inhibitor of L binding.
Figure 1.

Simulations of effects of inhibitors on cytochalasin B binding to GLUT1 when each transporter complex contains two interacting binding sites. Ordinate: CB binding in the presence of an inhibitor/CB binding in the absence of inhibitor (calculated using equation 5). Abscissa: inhibitor concentration in μM (note the log scale). Seven (a–g) scenarios were simulated. In each instance KL for CB binding, Ki for inhibitor binding and α (cooperativity between CB binding sites when only the CB is present) were set at 0.25 μM, 1 μM and 1 respectively. The curves show: a, β = γ = 1; b, β =1, γ = 10; c β = 1, γ = 0.1; d, β = 0.1, γ = 1; e, β = 10, γ = 1; f, β = γ = 0.1; g, β = 100, γ = 0.1.
2 Binding is negatively cooperative
When binding of L and I at any single site is mutually exclusive and displays negative hetero-cooperativity (γ > 1; Figure 1, curve b), the inhibitor I will appear to serve as a simple competitive inhibitor of L binding although the inhibition dose response is shifted to the left.
3 Binding is positively cooperative
When binding of L and I at any single site is mutually exclusive but displays positive hetero-cooperativity between sites (γ < 1; Figure 1, curve c), the inhibitor I will enhance L binding at low [I] where I and L bind at adjacent sites. When [I] is increased further such that I and L compete for binding to the same site, L binding is inhibited.
4 Zero hetero-cooperativity and positive or negative homo-cooperativity
When binding of L and I at any single site is mutually exclusive but displays zero hetero-cooperativity but positive homo-cooperativity (i.e. β < 1 and γ = 1; Figure 1, curve d), the inhibitor I will inhibit L binding more effectively (the inhibition curve is left-shifted). If the inhibitor I displays negative homo-cooperativity, I will be a less effective inhibitor and maximum inhibition will be reduced (i.e. β > 1 and γ =1; Figure 1, curve e).
5 Positive hetero-cooperativity and positive or negative homo-cooperativity
When binding of L and I at any single site is mutually exclusive but displays positive-hetero-cooperativity and positive homo-cooperativity (i.e. β ≪ 1 and γ ≪1; Figure 1, curve f), the inhibitor I will enhance L binding at low [I] and inhibit binding at greater [I]. If the inhibitor I displays negative homo-cooperativity (β ≫1) but strong positive hetero-cooperativity (γ ≪ 1), I will enhance ligand binding at all [I] (Figure 1, curve g).
Data Analysis
Having measured KL and α from analyses of CB inhibition of [3H]-CB binding or forskolin inhibition of [3H]-forskolin binding (Eqn 7), the analytical cycle is:
Compute initial estimates of KI, β and γ by nonlinear regression analysis of bi/b (experimental) vs [I]total using equation 6.
Using these parameter estimates, compute [I]free using Berkeley Madonna (version 8.3.22) - a general purpose differential equation solver that assumes [I] ≈ [GLUT1] and thus allows [I]free to fall as I interacts with GLUT1. The details of the model (differential equations describing Scheme 1) are available in the Supplementary Material.
Using these estimates of [I]free, re-compute KI, β and γ by nonlinear regression of bi/b (experimental) vs computed [I]free using equation 6.
Using these values of newly computed KI, β and γ, compare plots of bi/b (theoretical) vs [I]total to bi/b (experimental) vs [I]total by calculating the sum of the squares of deviations of theoretical data from experimental data.
Repeat steps 2 through 4 until the sum of the squares of deviations of bi/b (theoretical) vs [I]total from bi/b (experimental) vs [I]total is minimized.
RESULTS
Cytochalasin B is a competitive inhibitor of equilibrium exchange transport in erythrocytes (24) and a noncompetitive inhibitor of net sugar uptake into red cells lacking sugar (15). Equilibrium exchange transport is a condition in which intra- and extracellular [sugar] are identical and unidirectional fluxes are monitored through use of tracer sugar. Competitive inhibition by cytochalasin B indicates that extra- or intracellular sugar compete with cytochalasin B for binding to the transporter. Noncompetitive inhibition of uptake into cells lacking sugar indicates the absence of competition between extracellular sugar and inhibitor at the uptake site. This aggregate behavior is characteristic of an inhibitor that binds at (or whose binding site is mutually exclusive with) the endofacial sugar-binding site (15).
Forskolin is a competitive inhibitor of cytochalasin B binding to the human erythrocyte sugar transporter (25) suggesting that forskolin, like cytochalasin B binds at or close to the transporter endofacial site. In support of this hypothesis, we observe that forskolin acts as a noncompetitive inhibitor of net sugar uptake (Figure 2A) and as a competitive inhibitor of exchange sugar uptake (Figure 2B) in human erythrocytes. Forskolin reduces Vmax for sugar uptake by sugar-depleted cells but increases Km(app) for equilibrium exchange transport. Assuming noncompetitive and competitive inhibition of 3MG zero-trans and equilibrium exchange transport respectively, Ki(app) for FSK inhibition of net and exchange transport are 1.9 ± 0.4 μM and 1.2 ± 0.6 μM respectively.
Figure 2.

Inhibition of erythrocyte sugar transport by forskolin. Panels A & B, Ordinate: initial rate of 3MG uptake (mmol/L cell water/min). Abscissa: [3MG] mM. Results are shown as mean ± SEM of at least 3 experiments made in quadruplicate. Uptake was measured in the absence (○) and presence (●) of 2 μM FSK. Curves drawn through the points were computed by nonlinear regression assuming that 3MG uptake follows simple Michaelis-Menten kinetics. A Zero-trans 3MG uptake (cells lack intracellular sugar). Control: Kmapp = 1.51 ± 0.22 mM; Vmax = 0.114 ± 0.009 mM/L/min; R2 = 0.995. FSK: Kmapp = 1.09 ± 0.15 mM; Vmax = 0.055 ± 0.004 mM/L/min., R2 = 0.994. B Equilibrium-exchange 3MG uptake (intracellular [sugar]= extracellular [sugar]). Control: Kmapp = 17.8 ± 4.7 mM; Vmax = 6.5 ± 0.7 mM/L/min, R2 = 0.981. FSK: Kmapp = 48.5 ± 7.3 mM; Vmax = 8.3 ± 0.7 mM/L/min, R2 = 0.997.
We screened the ability of forskolin and its derivatives to displace cytochalasin B from GLUT1 by measuring forskolin inhibition of equilibrium 3H-cytochalasin B binding to red cell membranes depleted of peripheral membrane proteins. Cytochalasin B binding to peripheral membrane protein-depleted human red cell membranes, is competitively displaced by D-glucose and other GLUT1 substrates and is quantitatively accounted for by ligand binding to GLUT1 (26, 27).
Quantitation of inhibition of nM levels of 3H-cytochalasin B binding to μM concentrations of GLUT1 by mM levels of sugars is quite straightforward. However, the ligand binding assay employed in the current study is a “radioligand depletion” assay (see Materials and Methods). Specifically, we measure total [radioligand] in a suspension of GLUT1 proteoliposomes and, following sedimentation of GLUT1 proteoliposomes by centrifugation, we measure free[radioligand] remaining in the supernatant. Bound [radioligand] is thus total-free [radioligand]. By necessity, the concentration of GLUT1 used in these studies must deplete free [radioligand] sufficiently to produce a measurable difference between total and free [radioligand]. If a non-radiolabeled inhibitor also binds to GLUT1 to deplete free [inhibitor], then plots of equilibrium bound [radioligand] versus total [inhibitor] do not reflect the free [inhibitor] that produces inhibition of radioligand binding. Since radiolabeled inhibitors are, in most instances, unavailable, it is often impossible to measure free [inhibitor] directly. The net effect is that the use of radioligand depletion assays, in which bound [radioligand] is expressed as a function of total [inhibitor], inevitably overestimate KI for inhibition of radioligand binding by inhibitor. This is illustrated in measurements of inhibition of [3H]-CB binding to GLUT1 by nonradiolabeled CB (Figure 3A) and by measurements of inhibition of [3H]-forskolin binding to GLUT1 by nonradiolabeled forskolin (Figure 3B).
Figure 3.

Modulation of [3H]-CB binding to red cell membranes by CB (A) and modulation of [3H]-FSK binding to red cell membranes by FSK (B). Ordinate: The ratio radioligand binding in the presence of inhibitor to radioligand binding in the absence of inhibitor. Abscissa: [inhibitor], μM (note the log scale). Results are shown as mean ± SEM of at least 3 separate measurements made in triplicate. Radioligand binding was measured at 50 nM total [3H]-CB or [3H]-FSK. Results are shown for inhibition versus total [inhibitor] (○), inhibition versus measured free [inhibitor] (●) and inhibition versus computed free [inhibitor] (△). Curves drawn through the points were computed by nonlinear regression using equation 7. A The results for CB inhibition of [3H]-CB binding are, Kl = 0.408 ± 0.178 (total CB), 0.098 ± 0.014 (measured free CB) and 0.101 ± 0.009 (simulated free CB) μM; α = 10.0 ± 1.0 (total CB), 5.441 ± 0.558 (measured free CB) and 7.01 ± 0.53 (simulated free CB). The black curve shows the computed best fit of bi/b vs measured free [CB]. The red curve shows the computed best fit of bi/b vs simulated free [CB]. The blue curve shows the computed best fit of bi/b vs total [CB] resulting from the simulations. The green curve shows the computed best fit of bi/b vs free [CB] assuming that free [CB] = total [CB]. B The results for FSK inhibition of [3H]-FSK binding are, Kl = 3.329 ± 0.893 (total FSK), 2.833 ± 0.754 (measured free FSK) and 2.750 ± 0.787 (simulated free FSK) μM; α = 2.383 ± 2.116 (total FSK), 2.400 ± 0.212 (measured free FSK) and 2.434 ± 0.235 (simulated free FSK). The black curve shows the computed best fit of bi/b vs measured free [CB]. The red curve shows the computed best fit of bi/b vs simulated free [CB]. The green curve shows the computed best fit of bi/b vs free [CB] assuming that free [CB] = total [CB]. Note the black and red curves are almost superimposable.
CB binding to GLUT1 is a relatively high affinity process with Kd(app) ≈ 100 nM. CB is also available in radiolabeled ([3H]-CB) and nonradiolabeled ([1H]-CB) forms. This permits direct measurements of bound and free [CB] as outlined above. Thus when CB binding is measured as the ratio of binding in the presence of unlabeled CB to binding in the absence of unlabeled CB, or bi/b, and is expressed as a function of total or free [CB], two observations are apparent. 1) The curve describing bi/b versus [CB] is significantly right-shifted when total [CB] is used versus free [CB]. 2) The curve fit is significantly better when using free [CB].
A third data set is shown in Figure 3A. This data set shows measured bi/b versus simulated free [CB]. Simulated free[CB] was obtained through a multi-step process: 1) We computed binding parameters (KL and α see equation 7) by using the bi/b versus total [CB] data. 2) Using these parameters, we then simulated CB binding to GLUT1 under conditions where total [CB] and [GLUT1] are identical to those used experimentally. Our simulations used a general purpose differential equation solver (Berkeley Madonna) that recognizes [CB] ≈ [GLUT1] and thus allows [CB]free to fall as CB interacts with GLUT1. The details of the model (differential equations describing Scheme 1) are available in the Supplementary Material. The output includes equilibrium CB binding and free [CB] at each total [CB] employed. 3) Measured CB binding is then plotted versus simulated free [CB] and KL and α are recalculated. 4) We calculate the deviation between measured and simulated binding at each total [CB], then square and sum these deviations. 5) Steps 2–4 are repeated until the sum of the square of the deviations no longer changes significantly. The result is two binding isotherms which accurately reproduce the plots of bi/b versus free [CB] and bi/b versus total [CB]. Similar results are obtained with assays of forskolin inhibition of [3H]-forskolin binding to GLUT1 (Figure 3B) although the binding measurements show more experimental variation because Kd for forskolin binding to GLUT1 is at least 20-fold greater than Kd for CB binding. A second consequence of the higher Kd for forskolin binding is that the curves are significantly less left-shifted when bi/b is plotted as a function of measured or simulated free [forskolin] versus total [forskolin].
These computations provide an iterative process for accurately estimating free [inhibitor] when a radiolabeled form of inhibitor is unavailable. Tests for internal consistency mandate that studies of unlabeled forskolin inhibition of [3H]-forskolin binding to GLUT1 (forskolin homo-inhibition) provide binding constants that are indistinguishable from analogous constants for forskolin-modulation of [3H]-cytochalasin B binding (hetero-inhibition, see Figure 4 and Table 1). As we will show, this is observed experimentally.
Figure 4.

Modulation of [3H]-CB binding to red cell membranes by forskolin and its derivatives. Ordinate: The ratio CB binding in the presence of forskolin to CB binding in the absence of forskolin. Abscissa: [forskolin], μM (note the log scale). Results are shown as mean ± SEM of at least 3 separate measurements made in triplicate. CB binding was measured at 50 nM [3H]-CB. Results are shown for inhibition by FSK (○), 7DAFSK (●) or 1DOFSK (△). Curves drawn through the points were computed by nonlinear regression using equation 6. Based on CB inhibition of [3H]-CB, Kl and α were set at 0.098 ± 0.085 μM and 6.284 ± 0.558 respectively. The results are: FSK, Ki = 3.28 ± 0.98 μM, β = 1.54 ± 0.73, γ = 0.93 ± 0.10, R2 = 0.93. 7DeA-FSK, Ki = 2 ± 2 nM, β = ∞, γ = 0.45 ± 0.01, R2 = 0.86. 1DO-FSK, Ki = 1.0 ± 0.7 nM, β = 22,179 ± 5,104, γ = 0.438 ± 0.017, R2 = 0.97.
Forskolin and its derivatives modify [3H]-cytochalasin B (50 nM) binding to GLUT1 in several interesting ways (Figure 4 and Table 1). Some forskolins appear to function as simple inhibitors of cytochalasin B binding in which inhibition is a saturable function in [forskolin]. Other forskolins (see Figure 4 and Table 1) first enhance then inhibit cytochalasin B binding as the concentration of the inhibitor is raised. Yet other inhibitor analogs simply increase cytochalasin B binding (see Figure 4 and Table 1) while others appear to be without effect.
These behaviors are predicted by a simple binding model (Scheme 1) in which each transporter complex presents two cytochalasin B or forskolin binding sites that interact in ways that are dependent upon the nature of the bound ligands. For example, if binding of a forskolin derivative at the first site increases the affinity of the second site for cytochalasin B or the forskolin derivative, cytochalasin B binding may be enhanced at low [forskolin] then inhibited as [forskolin] is raised and competes more effectively with cytochalasin B for binding at the second site. Other permutations are possible (see Scheme 1 and Figure 1) and, according to the model, the net effect of any [forskolin] on cytochalasin B binding is determined by 5 parameters: KL (Kd for cytochalasin B binding), KI (Kd for forskolin derivative binding), α (a cooperativity factor indicating how cytochalasin B binding at the first site impacts Kd(app) for cytochalasin B binding at the second site), β (a cooperativity factor indicating how forskolin binding at the first site impacts Kd(app) for forskolin binding at the second site), and γ (a cooperativity factor indicating how cytochalasin B binding at the first site impacts Kd(app) for forskolin binding at the second site. In homo-inhibition studies where, for example, the radioligand and unlabeled competing ligand are the same species (e.g. cytochalasin B or forskolin), KL = KI and α = β = γ thereby simplifying analysis of experimental data to the resolution of two affinity parameters - KL and α. These values are then inserted into binding equation 6 thereby simplifying non-linear curve fitting of forskolin-modulation of cytochalasin B binding (hetero-inhibition) to the resolution of 3 parameters - KI, β and γ.
Table 1 summarizes the results of hetero-inhibition studies in which the effects of a wide range of forskolin analogues on [3H]-cytochalasin B binding were measured. Figure 5 and Table 1 summarize the results of similar studies in which the effects of a number of cytochalasin analogues on [3H]-cytochalasin B binding were measured.
Figure 5.

Modulation of [3H]-cytochalasin B binding to red cell membranes by cytochalasins. Ordinate: The ratio [3H]-CB binding in the presence of other cytochalasins to [3H]-CB binding in the absence of cytochalasins. Abscissa: [cytochalasin], μM (note the log scale). Results are shown as mean ± SEM of at least 3 separate measurements made in triplicate. CB binding was measured at 50 nM [3H]-CB. Results are shown for modulation by CB (○), CC (●) and CH (△). Curves drawn through the points were computed by nonlinear regression using equation 6. The results are: CB, Ki = KL = 98 ± 8.5 nM and α = β = γ = 6.284 ± 3.849, R2 = 0.997. CC, KI = 1.591 ± 431 μM, β = ∞, γ = 0.433 ± 0.003, R2 = 0.986. CH, KI = 98 ± 3 nM, β = 249 ± 45, γ = 0.449 ± 0.019, R2 = 0.94.
DISCUSSION
The human type 1 glucose transporter (GLUT1) is inhibited by diverse natural compounds. Methylxanthines and ATP act as mixed-type inhibitors of sugar exit reducing Vmax and increasing Km(app) (28–30). Cytochalasins and forskolins increase Km(app) for exchange transport and reduce Vmax for net entry (see here and (14, 15, 24). Tyrosine kinase inhibitors are competitive inhibitors of sugar import (31) and the androgens and catechins reduce Vmax for exit and increase Km(app) for uptake (32). These observations are consistent with the hypothesis that cytochalasins and forskolins bind at or close to the sugar export site and that the tyrosine kinase inhibitors, androgens and catechins bind at or close to the sugar import site (32). Methylxanthines and ATP appear to modulate transport by acting at a site(s) distinct from import and exit sites.
Recent studies have revealed an unexpected complexity in transport inhibition by import and export site ligands. When presented at very low concentrations, the exofacial site inhibitor maltose or the endofacial site inhibitors cytochalasin B (CB) or forskolin (FSK) modestly stimulate sugar import in human red blood cells (21, 22). As inhibitor concentration is increased, sugar import stimulation is replaced by transport inhibition. The simplest explanation for this behavior is that the sugar transporter contains 2 binding sites for exofacial inhibitors of sugar transport and two sites for endofacial inhibitors of transport. When one exofacial or endofacial site is occupied by inhibitor, the transporter is converted into a high affinity, high capacity sugar transporter that catalyzes greater net transport (22). As the concentration of inhibitor is increased further, the second exofacial or endofacial site is occupied by inhibitor and transport is fully inhibited. The goals of the present study were to better understand the kinetics of equilibrium ligand binding at the GLUT1 export site and to explore the ligand-features that determine ligand binding affinity and cooperativity.
At 4 °C, where transport and ligand binding measurements were made, modulations of [3H]-CB binding to GLUT1 by cytochalasin and forskolin analogs fall into 1 of 5 categories. Type 1) Simple, or pseudo-Michaelis inhibition in which CB binding is inhibited; Type 2) Schizo-modulation in which CB binding is enhanced at low [inhibitor] but is inhibited at higher [inhibitor]; Type 3) Biphasic inhibition in which high and low affinity components of inhibition are observed; Type 4) Binding stimulation in which only enhanced binding is observed, and Type 5) No modulation of binding. Similar ligand binding behavior has been observed with purified GLUT1 and with red cell sugar transport using a limited selection of endofacial site ligands (21). Type 2, 3 and 4 modulations of CB binding to GLUT1 are incompatible with the classic or alternating conformer model for carrier-mediated transport (10).
This conclusion is predicated on the simplifying assumption that ligand-binding sites correspond to substrate-binding sites. For example, it is assumed that the endofacial CB binding site corresponds to (or its occupancy is mutually exclusive with) the endofacial sugar-binding site. Similarly, it is assumed that the exofacial maltose-binding site corresponds to (or its occupancy is mutually exclusive with) the exofacial sugar-binding site. How might we alter our conclusions if a simple carrier also contains non catalytic but modulatory exo- and endofacial ligand-binding sites whose occupancy affects the affinity of substrate binding at catalytic sites? This has been considered in previous studies and rejected as a satisfactory explanation of extracellular maltose biphasic modulation of 3-O-methylglucose import or maltose modulation of CB binding to GLUT1 (10, 22). Moreover, if multiple ligand binding sites are presented by the endofacial conformation of GLUT1, the stoichiometry of ligand binding should be greater than 1 mol CB or FSK per mol GLUT1 and this has never been reported. However, we still do not know whether exofacial substrate and ligand binding sites are physically congruous or mutually exclusive or whether endofacial glucose, CB and FSK binding sites are physically congruous or mutually exclusive.
The simplest explanation for these behaviors is that the sugar transporter contains 2 binding sites for endofacial transport ligands. If binding of a single ligand species is studied, interactions between binding sites are homo-cooperative. If binding of one ligand (e.g. [3H]-CB or [3H]-FSK) is measured in the presence of a second ligand, homo- and hetero-cooperativity may be observed. When measuring CB binding in the presence of FSK, binding of the first CB molecule to unoccupied GLUT1 is described by the dissociation constant KL and binding of the second CB molecule is described by αKL where α is a dimensionless constant greater than zero. Binding of the first FSK molecule to unoccupied transporter is described by KI and binding of the second FSK molecule is described by βKI (β is a dimensionless constant greater than zero). When heterocomplexes are formed, binding of the first molecule (CB or FSK) is described by KL or KI and binding of the second molecule (FSK or CB) is described by γ KL or γ KI where γ is a dimensionless constant greater than zero. This model explains enhanced CB binding in the presence of at least 11 of the 20 “antagonists” tested in the present study. The following rules are observed for each type of binding modulation: Type 1 - β > 1, γ > 0.5, βKI < 7,200; Type 2 - β < 23,000, γ < 0.5, 9,000 < 1βKI < 52,000; Type 3 - 1,000 < β < 8,000, γ > 0.5, 150,000 < βKI < 790,000; Type 4 - β = ∞, γ < 0.5.
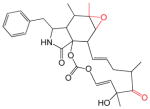
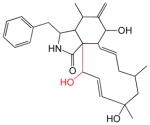
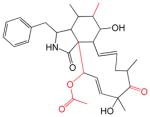
The cytochalasins (Table 2) share a common rigid bicyclic isoindoline core fused to a macrocycle. CB, CE, CH and CJ show the greatest affinity for site 1 (Kd(app) ranges from 12 to 100 nM). Dehydration of the macrocycle hydroxyl group to form cytochalasin A (CA) results in a 60-fold loss of affinity for GLUT1 (ΔΔG° = 2.28 kcal/mol). This hydroxyl group may serve as a hydrogen bond donor. Shortening the macrocycle by three carbon atoms (CJ) increases affinity by 8-fold (ΔΔG° = −1.2 kcal/mol), but introduction of an acetyl group to the macrocycle ketone produces a compensatory 10-fold reduction in binding affinity (compare CJ and CH). Addition of a carbonyl at position 5 of the shortened macrocycle further reduces binding affinity by 20-fold (compare CD and CH). CB and CA show the highest affinities for the second binding site (βKI = 600 and 900 nM respectively). The remaining cytochalasins display very large β parameters indicating that they cannot simultaneously occupy endofacial sites 1 and 2. However, when site 1 is occupied by CB, all cytochalasins have access to site 2 (γKI = 8 – 1,100 nM; CC and CD share the lowest affinities for this site).
Table 2.
The Cytochalasins
| Cytochalasin B (CB) | Cytochalasin E (CE) | ||
| KL = 0.1 μM β = 6.3 γ = 6.3 |

|
KL = 0.1 μM β = 7917 γ = 0.56 |
|
| Cytochalasin J (CJ) | Cytochalasin A (CA) | ||
| KL = 0.1 μM β = 6.3 γ = 6.3 |
|
KL = 0.82 μM β = 1.11 γ = 0.38 |

|
| Cytochalasin C (CC) | Cytochalasin H (CH) | ||
| KL = 1.6 μM β = ∞ γ = 0.43 |
|
KL = 0.1 μM β = 249 γ = 0.45 |

|
| Cytochalasin D (CD) | |||
| KL = 2.0 μM β = 101 γ = 0.56 |

|
||
The structures of the cytochalasins. KL, β and γ are also shown (taken from Table 1) to facilitate comparison. Groups modified from the parent molecule (CB) are highlighted in red.
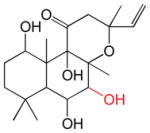
The forskolins (FSK; Table 3) are a family of labdane diterpenes known primarily for their activation of adenylyl cyclase (33). Schizo-modulation of CB binding to GLUT1 is most common among the forskolins in which positions 1 and 9 are deoxygenated or where the forskolins contain small substitutions or adducts (acetylation, fluoridation) at positions 1, 6, 7 and 9. Only one forskolin derivative produces biphasic inhibition and this contains a fluorophenylacetate group at position 7. Large changes to FSK at position 7 (deacetylation or introduction of a (2-(3-(4-fluorophenyl)-1-hydroxypropylamino)ethylamino)methanediol group) produce binding stimulation indicating that these adducts do not preclude CB binding but do prevent a second, identically modified forskolin derivative from binding. Acetylation of position 6 in combination with deacetylation of position 7 or introduction of an intermediate group at position 7 (fluorophenylpropanoic acid) appear to prevent forskolin binding altogether.
Table 3.
The forskolins
| FSK | 1DeO-FSK | 7DeA-FSK | 1A-FSK | ||||
| KL = 2800nM β = 2.4 γ = 0.9 |

|
KL = 1nM β = 22000 γ = 0.44 |

|
KL = 2nM β = ∞ γ = 0.45 |
|
KL = 3 nM β = 11923; γ = 0.44 |
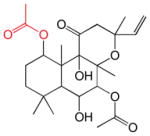
|
| 1,6DiA-FSK | 7FPPNEA-FSK | 14,15DiH-FSK | 7FPA-FSK | ||||
| KL = 11 nM β = 1500 γ = 0.49 |

|
KL = 25 nM β = 40400 γ = 0.43 |
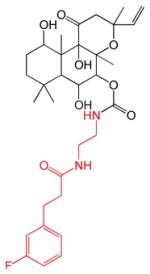
|
KL = 30 nM β = 243 γ = 0.6 |
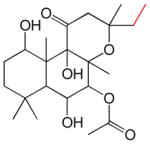
|
KL = 140 nM β = 1120 γ = 0.51 |

|
| 9DeO-FSK | 7FA-FSK | 6A-FSK | |||||
| KL = 430 nM β = 120 γ = 0.46 |

|
KL = 630 nM β = 45 γ = 0.48 |

|
KL = 950 nM β = 10000 γ = 0.42 |

|
||
| 7FPP-FSK | 6A,7DeA-FSK | ||||||
| KL = NM β = NA γ = NA |

|
KL = NA β = NA γ = NA |

|
||||
The structures of the forskolins. KL, β and γ are also shown (taken from Table 1) to facilitate comparison. Groups modified from the parent molecule (FSK) are highlighted in red.
Cytochalasins and forskolins show similar hetero-cooperativity parameters (γ = 0.49 ± 0.06) for binding to site 2 when site 1 is occupied by CB. This positive hetero-cooperativity means that forskolin or cytochalasin occupancy of site 1 increases the affinity of site 2 for CB by two-fold and vice versa. The exception appears to be forskolin, which displays a hetero-cooperativity parameter (γ) for binding to the transporter-CB complex of 0.93 suggesting that CB and FSK binding sites are only weakly interacting. Only 3 of 20 GLUT1 endofacial site ligands tested in the present study produce pseudo-Michaelis inhibition of CB binding. Twelve compounds enhance CB binding when applied at low concentrations. Three compounds display high and low affinity inhibition of CB binding. Only two compounds have no effect on CB binding.
Previous studies have examined cytochalasin B and forskolin inhibition of sugar transport and CB binding in human red cells (14, 25, 34, 35) and inhibition by cytochalasin B (36) or forskolin (37, 38) of energized D-galactose transport by the GalP sugar-H+ symport protein of Escherichia coli. The present study differs from previous studies in that our analysis of inhibition of ligand binding to GLUT1 allows for two interacting ligand binding sites. In their study of cytochalasin inhibition of [3H]-CB binding to red cell membranes (34). Rampal and coworkers found that only CA and CB inhibit CB binding with high affinity. The remaining cytochalasins (CC through CH) were extremely low affinity inhibitors (Ki(app) > 100 μM). While this result appears to contradict our observations, βKI for occupation of site 2 by cytochalasin analogs in our studies corresponds to Ki(app) observed by Rampal et al. (34). For example, CE inhibition of CB binding is characterized by β, γ and KI parameters of 7,900, 0.56 and 0.1 μM respectively. If one were to analyze our data assuming assuming a single endofacial binding site, these parameters would translate into Ki(app) > 1000 μM. Rampal et al. (34) report that Ki(app) ≥ 100 μM. As with our previous studies showing that low concentrations of “inhibitors” can produce a paradoxical stimulation of transport (21, 22), the key to the present observations is the use of inhibitor concentrations that span Kd(app) by ± 2 or more log units.
GLUT1 three-dimensional structure has been modeled using the crystal structure of E. coli GlpT as a homology template and human glucose-6-phosphate translocase as an “evolutionary template” to correct for missing residue assignments (16). The resulting structure has been docked with a variety of GLUT1 ligands (cytochalasin B, forskolin, glucose, galactose, mannose, quecertin and phloretin) to reveal potential substrate binding sites (16, 39). Salas-Burgos et al. propose that GLUT1 presents exofacial and endofacial binding sites for forskolin, phloretin and glucose but only an endofacial site for CB. An exofacial phloretin and glucose binding site is consistent with the observed competition between substrate and phloretin during sugar uptake. However, forskolin is a noncompetitive inhibitor of sugar uptake (see here and (14)), which is incompatible with docking analyses suggesting that exofacial glucose and forskolin compete for binding. Forskolin is proposed to make H-bond acceptor contacts with the GLUT1 endofacial site via hydroxyl or carbonyl groups at forskolin positions 6, 9 and 11. Contrary to this suggestion, elimination of the hydroxyl group at position 9 enhances ligand binding affinity. CB is proposed to dock with GLUT1 at a single site comprising portions of GLUT1 cytoplasmic loops 2, 6 and 10 (16). Biochemical data suggest that CB interacts with GLUT1 transmembrane helices 10 and 11 and loop 10 (12, 40, 41) although the absence of a photo-induced covalent interaction between CB and loops 2 and 6 does not eliminate binding interactions with these domains. Deviations between experimental and modeled behavior are not unexpected. The use of homology-based threading to model LacY on the known GlpT structure or to model GlpT on the known LacY structure results in broadly correct transporter architecture but mis-aligns active site residues (42).
Two explanations could account for multiple, interacting endofacial ligand binding sites. 1) Both sites exist on a single GLUT1 molecule. 2) Only one site is present within each GLUT1 molecule but GLUT1-GLUT1 interactions give rise to binding cooperativity. The first possibility is ruled out by GLUT1 CB binding stoichiometry which ranges from 0.5 – 1 mol CB per mol GLUT1 (4, 43, 44). The second possibility is supported by demonstrations that GLUT1 forms noncovalent dimers and tetramers in detergent micelles and lipid bilayers ((44–46) but see (47) for a counter view). Strong, negative cooperativity could be explained if a ligand, complexed within the endofacial binding site, extends its molecular envelope into the endofacial binding pocket of an adjacent subunit thereby inhibiting ligand binding at the second site (negative cooperativity; Figure 6A). Positive or weak negative cooperativity would be explained if endofacial ligand binding is an induced fit phenomenon which in turn promotes a similar or compensatory conformational change in the endofacial binding site of an adjacent GLUT1 molecule (Figure 6B). Resolution of these questions must await determination of the structure of the GLUT1-CB complex.
Figure 6.

Panel A shows a GLUT1 tetramer (see (46)) viewed from the interstitium. Two subunits (blue) are shown in the e2 conformation each presenting an import site and two subunits (a and b) are shown in the e1 conformation (their sugar binding sites are not visible because they are exposed to the cytoplasm). Panel B shows the e1 subunits a and b viewed normal to the bilayer and each sectioned directly through its catalytic center. If the GLUT1 region that binds cytochalasin B interacts with the same domain on the adjacent GLUT1 molecule (subunit b is rotated 180° relative to subunit a about the central axis of the tetramer), this would allow for the molecular envelope of a cytochalasin bound to subunit a to overlap with that of cytochalasin bound to subunit b. This, in turn, could account for strong negative cooperativity as observed, for example, with CA. Holman and Rees (12) suggest that the CB binding site lies close to TMs 10 and 11 which would require that TMs 9 and 12 most likely form the oligomerization interface. Panel C hypothesizes that ligand interaction with subunit a promotes a conformational change in both subunits which, in this instance enhances the affinity of subunit b for ligand. However, the ligand-induced conformational change could also reduce binding affinity at subunit b and thereby explain negative cooperativity.
The significance of GLUT1 cooperativity to GLUT1 function at physiologic temperature (37°C) remains to be established. It has been noted that purified, reduced (noncooperative) GLUT1 (8) is some 10 – 20-fold less active than red cell-resident GLUT1 (1). This could be explained if reduced, purified transporter behaves as a simple carrier (45). In order that multiple rounds of sugar uptake may proceed via a simple carrier, each transport cycle (which includes substrate binding at the exofacial surface, substrate translocation and substrate release at the endofacial surface) must include a conformational change that restores the exofacial substrate- binding site. Regeneration of the exofacial site is called relaxation (1) and, at 10°C, is 100-fold slower than substrate translocation (8). Red cell-resident (tetrameric) GLUT1, on the other hand, couples substrate translocation through one GLUT1 molecule to relaxation of a neighboring subunit and thereby avoids rate-limiting relaxation. The cost is significant - transport requires twice as much cooperative GLUT1 than noncooperative GLUT1 - but the catalytic advantage is even greater. Studies with reduced GLUT1 at 37°C will be necessary to determine whether insights obtained at lower temperatures are applicable to physiologic temperatures. GLUT1 cooperativity has not been studied systematically in other tissues but the hallmarks of cooperativity (GLUT1 sensitivity to reductant, interaction with quaternary-structure-sensitive polyclonal antibodies, formation of oligomers, dominant negative phenotypic behavior of heterologously expressed GLUT1 mutants) have been observed in cultured cells (45, 48–51) suggesting that GLUT1 may behave as a cooperative complex in most tissues where it is expressed.
Supplementary Material
Acknowledgments
This work was supported by NIH grants DK 36081 and DK 44888 (A.C.) ANA acknowledges a studentship from the BBSRC and support from GlaxoSmithKline plc. PJFH and RBH thank the BBSRC and the EU (EDICT consortium grant 201924) for research funding.
The abbreviations used are
- GLUT1
human erythrocyte glucose transport protein
- CA
cytochalasin A
- CB
cytochalasin B
- CC
cytochalasin C
- CD
cytochalasin D
- CE
cytochalasin E
- CH
cytochalasin H
- CJ
cytochalasin J
- FSK
forskolin
- 1DeO-FSK
1-deoxy-forskolin
- 7DeA-FSK
7-deacetyl-forskolin
- 1A-FSK
1-acetyl-forskolin
- 1,6DiA-FSK
1,60diacetyl-forskolin
- 7FPPNEA-FSK
14,15DiH-FSK, 14,15-dihydro-forskolin
- 7FPA-FSK
7-fluorophenylacetyl-forskolin
- 9DeO-FSK
9-deoxy-forskolin
- 7FA-FSK
7-fluoroacetyl-forskolin
- 6A-FSK
6-acetyl-forskolin
- 7FPP-FSK
7-fluorophenylpropionate-forskolin
- 6A,7DeA-FSK
6-acetyl, 7-deacetyl-forskolin
- EGTA
EDTA, ethylenediaminetetraacetic acid
- 3MG
3-O-methylglucose
Footnotes
The single exception is CA where •KI = 600
The details of the ligand binding model (differential equations describing Scheme 1) are available in the Supplementary Material, which may be accessed free of charge online at http://pubs.acs.org.
BIBLIOGRAPHY
- 1.Stein WD. Transport and diffusion across cell membranes. 1986:231–305. [Google Scholar]
- 2.Saier MH, Jr, Beatty JT, Goffeau A, Harley KT, Heijne WH, Huang SC, Jack DL, Jahn PS, Lew K, Liu J, Pao SS, Paulsen IT, Tseng TT, Virk PS. The major facilitator superfamily. J Mol Microbiol Biotechnol. 1999;1:257–279. [PubMed] [Google Scholar]
- 3.Kasahara M, Hinkle PC. Reconstitution and purification of the D-glucose transporter from human erythrocytes. J Biol Chem. 1977;252:7384–7390. [PubMed] [Google Scholar]
- 4.Zoccoli MA, Baldwin SA, Lienhard GE. The monosaccharide transport system of the human erythrocyte. Solubilization and characterization on the basis of cytochalasin B binding. J Biol Chem. 1978;253:6923–6930. [PubMed] [Google Scholar]
- 5.Takata K, Hirano H, Kasahara M. Transport of glucose across the blood-tissue barriers. Int Rev Cytol S. 1997;172:1–53. doi: 10.1016/s0074-7696(08)62357-8. [DOI] [PubMed] [Google Scholar]
- 6.Pascual JM, Wang D, Lecumberri B, Yang H, Mao X, Yang R, De Vivo DC. GLUT1 deficiency and other glucose transporter diseases. Eur J Endocrinol. 2004;150:627–633. doi: 10.1530/eje.0.1500627. [DOI] [PubMed] [Google Scholar]
- 7.Hruz PW, Mueckler MM. Structural analysis of the GLUT1 facilitative glucose transporter (review) Mol Membr Biol. 2001;18:183–193. doi: 10.1080/09687680110072140. [DOI] [PubMed] [Google Scholar]
- 8.Appleman JR, Lienhard GE. Kinetics of the purified glucose transporter. Direct measurement of the rates of interconversion of transporter conformers. Biochemistry. 1989;28:8221–8227. doi: 10.1021/bi00446a038. [DOI] [PubMed] [Google Scholar]
- 9.Cloherty EK, Heard KS, Carruthers A. Human erythrocyte sugar transport is incompatible with available carrier models. Biochemistry. 1996;35:10411–10421. doi: 10.1021/bi953077m. [DOI] [PubMed] [Google Scholar]
- 10.Sultzman LA, Carruthers A. Stop-flow analysis of cooperative interactions between GLUT1 sugar import and export sites. Biochemistry. 1999;38:6640–6650. doi: 10.1021/bi990130o. [DOI] [PubMed] [Google Scholar]
- 11.Mueckler M, Makepeace C. Analysis of transmembrane segment 8 of the GLUT1 glucose transporter by cysteine-scanning mutagenesis and substituted cysteine accessibility. J Biol Chem. 2004;279:10494–10499. doi: 10.1074/jbc.M310786200. [DOI] [PubMed] [Google Scholar]
- 12.Holman GD, Rees WD. Photolabelling of the hexose transporter at external and internal sites: fragmentation patterns and evidence for a conformational change. Biochim Biophys Acta. 1987;897:395–405. doi: 10.1016/0005-2736(87)90437-8. [DOI] [PubMed] [Google Scholar]
- 13.Bloch R. Inhibition of sugar transport in the human erythrocyte by cytochalasin B. Biochemistry. 1973;12:4799–4801. doi: 10.1021/bi00747a036. [DOI] [PubMed] [Google Scholar]
- 14.Sergeant S, Kim HD. Inhibition of 3-O-methylglucose transport in human erythrocytes by forskolin. J Biol Chem. 1985;260:14677–14682. [PubMed] [Google Scholar]
- 15.Carruthers A, Helgerson AL. Inhibitions of sugar transport produced by ligands binding at opposite sides of the membrane. Evidence for simultaneous occupation of the carrier by maltose and cytochalasin B. Biochemistry. 1991;30:3907–3915. doi: 10.1021/bi00230a015. [DOI] [PubMed] [Google Scholar]
- 16.Salas-Burgos A, Iserovich P, Zuniga F, Vera JC, Fischbarg J. Predicting the three-dimensional structure of the human facilitative glucose transporter glut1 by a novel evolutionary homology strategy: insights on the molecular mechanism of substrate migration, and binding sites for glucose and inhibitory molecules. Biophys J. 2004;87:2990–2999. doi: 10.1529/biophysj.104.047886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heard KS, Fidyk N, Carruthers A. ATP-dependent substrate occlusion by the human erythrocyte sugar transporter. Biochemistry. 2000;39:3005–3014. doi: 10.1021/bi991931u. [DOI] [PubMed] [Google Scholar]
- 18.Leitch JM, Carruthers A. ATP-dependent sugar transport complexity in human erythrocytes. American journal of physiology Cell physiology. 2007;292:C974–86. doi: 10.1152/ajpcell.00335.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blodgett DM, Carruthers A. Quench-Flow Analysis Reveals Multiple Phases of GluT1-Mediated Sugar Transport. Biochemistry. 2005;44:2650–2660. doi: 10.1021/bi048247m. [DOI] [PubMed] [Google Scholar]
- 20.Henderson PJ. A linear equation that describes the steady-state kinetics of enzymes and subcellular particles interacting with tightly bound inhibitors. Biochem J. 1972;127:321–333. doi: 10.1042/bj1270321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cloherty EK, Levine KB, Carruthers A. The red blood cell glucose transporter presents multiple, nucleotide-sensitive sugar exit sites. Biochemistry. 2001;40:15549–15561. doi: 10.1021/bi015586w. [DOI] [PubMed] [Google Scholar]
- 22.Hamill S, Cloherty EK, Carruthers A. The human erythrocyte sugar transporter presents two sugar import sites. Biochemistry. 1999;38:16974–16983. doi: 10.1021/bi9918792. [DOI] [PubMed] [Google Scholar]
- 23.Appleman JR, Lienhard GE. Rapid kinetics of the glucose transporter from human erythrocytes. Detection and measurement of a half-turnover of the purified transporter. J Biol Chem. 1985;260:4575–4578. [PubMed] [Google Scholar]
- 24.Basketter DA, Widdas WF. Asymmetry of the hexose transfer system in human erythrocytes. Comparison of the effects of cytochalasin B, phloretin and maltose as competitive inhibitors. J Physiol. 1978;278:389–401. doi: 10.1113/jphysiol.1978.sp012311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavis VR, Lee DP, Shenolikar S. Evidence that forskolin binds to the glucose transporter of human erythrocytes. J Biol Chem. 1987;262:14571–14575. [PubMed] [Google Scholar]
- 26.Gorga FR, Lienhard GE. Changes in the intrinsic fluorescence of the human erythrocyte monosaccharide transporter upon ligand binding. Biochemistry. 1982;21:1905–1908. doi: 10.1021/bi00537a031. [DOI] [PubMed] [Google Scholar]
- 27.Baldwin SA, Baldwin JM, Lienhard GE. Monosaccharide transporter of the human erythrocyte. Characterization of an improved preparation. Biochemistry. 1982;21:3836–3842. doi: 10.1021/bi00259a018. [DOI] [PubMed] [Google Scholar]
- 28.Carruthers A. Hexose transport across human erythrocyte membranes. In: Raess BU, Tunnicliff G, editors. The Red Cell Membrane. Humana Press; Clifton, N.J: 1989. pp. 249–279. [Google Scholar]
- 29.Challiss JR, Taylor LP, Holman GD. Sugar transport asymmetry in human erythrocytes--the effect of bulk haemoglobin removal and the addition of methylxanthines. Biochim Biophys Acta. 1980;602:155–166. doi: 10.1016/0005-2736(80)90298-9. [DOI] [PubMed] [Google Scholar]
- 30.Cloherty EK, Diamond DL, Heard KS, Carruthers A. Regulation of GLUT1-mediated sugar transport by an antiport/uniport switch mechanism. Biochemistry. 1996;35:13231–13239. doi: 10.1021/bi961208t. [DOI] [PubMed] [Google Scholar]
- 31.Vera JC, Reyes AM, Velasquez FV, Rivas CI, Zhang RH, Strobel P, Slebe JC, Nunez-Alarcon J, Golde DW. Direct inhibition of the hexose transporter GLUT1 by tyrosine kinase inhibitors. Biochemistry. 2001;40:777–90. doi: 10.1021/bi001660j. [DOI] [PubMed] [Google Scholar]
- 32.Naftalin RJ, Afzal I, Cunningham P, Halai M, Ross C, Salleh N, Milligan SR. Interactions of androgens, green tea catechins and the antiandrogen flutamide with the external glucose-binding site of the human erythrocyte glucose transporter GLUT1. Br J Pharmacol. 2003;140:487–499. doi: 10.1038/sj.bjp.0705460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morris DI, Robbins JD, Ruoho AE, Sutkowski EM, Seamon KB. Forskolin photoaffinity labels with specificity for adenylyl cyclase and the glucose transporter. Journal of Biological Chemistry. 1991;266:13377–13384. [PubMed] [Google Scholar]
- 34.Rampal AL, Pinkofsky HB, Jung CY. Structure of cytochalasins and cytochalasin B binding sites in human erythrocyte membranes. Biochemistry. 1980;19:679–683. doi: 10.1021/bi00545a011. [DOI] [PubMed] [Google Scholar]
- 35.Shanahan MF, Morris DP, Edwards BM. [3H]forskolin. Direct photoaffinity labeling of the erythrocyte D-glucose transporter. J Biol Chem. 1987;262:5978–5984. [PubMed] [Google Scholar]
- 36.Cairns MT, McDonald TP, Horne P, Henderson PJ, Baldwin SA. Cytochalasin B as a probe of protein structure and substrate recognition by the galactose/H+ transporter of Escherichia coli. Journal of Biological Chemistry. 1991;266:8176–8183. [PubMed] [Google Scholar]
- 37.Martin GE, Seamon KB, Brown FM, Shanahan MF, Roberts PE, Henderson PJ. Forskolin specifically inhibits the bacterial galactose-H+ transport protein, GalP. J Biol Chem. 1994;269:24870–24877. [PubMed] [Google Scholar]
- 38.Martin GE, Rutherford NG, Henderson PJ, Walmsley AR. Kinetics and thermodynamics of the binding of forskolin to the galactose-H+ transport protein, GalP, of Escherichia coli. Biochem J. 1995;308:261–268. doi: 10.1042/bj3080261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cunningham P, Afzal-Ahmed I, Naftalin RJ. Docking studies show that D-glucose and quercetin slide through the transporter GLUT1. J Biol Chem. 2006;281:5797–5803. doi: 10.1074/jbc.M509422200. [DOI] [PubMed] [Google Scholar]
- 40.Kasahara T, Kasahara M. Tryptophan 388 in putative transmembrane segment 10 of the rat glucose transporter Glut1 is essential for glucose transport. J Biol Chem. 1998;273:29113–29117. doi: 10.1074/jbc.273.44.29113. [DOI] [PubMed] [Google Scholar]
- 41.Garcia JC, Strube M, Leingang K, Keller K, Mueckler MM. Amino acid substitutions at tryptophan 388 and tryptophan 412 of the HepG2 (Glut1) glucose transporter inhibit transport activity and targeting to the plasma membrane in Xenopus oocytes. Journal of Biological Chemistry. 1992;267:7770–7776. [PubMed] [Google Scholar]
- 42.Lemieux MJ. Eukaryotic major facilitator superfamily transporter modeling based on the prokaryotic GlpT crystal structure (Review) Molecular membrane biology. 2007;24:333–341. doi: 10.1080/09687680701496507. [DOI] [PubMed] [Google Scholar]
- 43.Jung CY, Rampal AL. Cytochalasin B binding sites and the glucose transporter in human erythrocyte ghosts. J Biol Chem. 1977;252:5456–5463. [PubMed] [Google Scholar]
- 44.Hebert DN, Carruthers A. Cholate-solubilized erythrocyte glucose transporters exist as a mixture of homodimers and homotetramers. Biochemistry. 1991;30:4654–4658. doi: 10.1021/bi00233a003. [DOI] [PubMed] [Google Scholar]
- 45.Zottola RJ, Cloherty EK, Coderre PE, Hansen A, Hebert DN, Carruthers A. Glucose transporter function is controlled by transporter oligomeric structure. A single, intramolecular disulfide promotes GLUT1 tetramerization. Biochemistry. 1995;34:9734–9747. doi: 10.1021/bi00030a011. [DOI] [PubMed] [Google Scholar]
- 46.Graybill C, van Hoek AN, Desai D, Carruthers AM, Carruthers A. Ultrastructure of Human Erythrocyte GLUT1. Biochemistry. 2006;45:8096–8107. doi: 10.1021/bi060398x. [DOI] [PubMed] [Google Scholar]
- 47.Haneskog L, Andersson L, Brekkan E, Englund AK, Kameyama K, Liljas L, Greijer E, Fischbarg J, Lundahl P. Monomeric human red cell glucose transporter (Glut1) in non-ionic detergent solution and a semi-elliptical torus model for detergent binding to membrane proteins. Biochim Biophys Acta. 1996;1282:39–47. doi: 10.1016/0005-2736(96)00031-4. [DOI] [PubMed] [Google Scholar]
- 48.Levine KB, Robichaud TK, Hamill S, Sultzman LA, Carruthers A. Properties of the human erythrocyte glucose transport protein are determined by cellular context. Biochemistry. 2005;44:5606–5616. doi: 10.1021/bi0477541. [DOI] [PubMed] [Google Scholar]
- 49.Levine KB, Cloherty EK, Fidyk NJ, Carruthers A. Structural and physiologic determinants of human erythrocyte sugar transport regulation by adenosine triphosphate. Biochemistry. 1998;37:12221–12232. doi: 10.1021/bi980585y. [DOI] [PubMed] [Google Scholar]
- 50.Cloherty EK, Sultzman LA, Zottola RJ, Carruthers A. Net sugar transport is a multistep process. Evidence for cytosolic sugar binding sites in erythrocytes. Biochemistry. 1995;34:15395–15406. doi: 10.1021/bi00047a002. [DOI] [PubMed] [Google Scholar]
- 51.Pessino A, Hebert DN, Woon CW, Harrison SA, Clancy BM, Buxton JM, Carruthers A, Czech MP. Evidence that functional erythrocyte-type glucose transporters are oligomers. J Biol Chem. 1991;266:20213–20217. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.