

Abstract
A new silicon chip for protein microarray development, fabrication and validation is proposed. The chip is made of two areas with oxide layers of different thicknesses: an area with a 500 nm SiO2 layer dedicated to interferometric label-free detection and quantification of proteins and an area with 100 nm SiO2 providing enhanced fluorescence. The chip allows, within a single experiment performed on the same surface, label-free imaging of arrayed protein probes coupled with high sensitivity fluorescence detection of the molecular interaction counterparts. Such a combined chip is of high practical utility during assay development process to image arrays, check consistency and quality of the protein array, quantify the amount of immobilized probes and finally detect fluorescence of bioassays.
Keywords: Protein microarrays, Silicon, Label-free detection, Fluorescence detection, Allergens
1. Introduction
Multianalyte biosystems in array format have generated increased excitement in the last decade for their ability to simultaneously detect multiple analytes in a sample by an affinity-binding event at the surface interface (Seidel and Niessner, 2008). Protein chips can be used to discover new proteins, to analyze their binding behaviour or to study protein–protein complexes (Tomizaki et al., 2010; Yu et al., 2010; Wolf-Yadlin et al., 2009). They are becoming more and more appreciated for applications in medical diagnostics due to the fast and highly parallel analyses that can be performed with them (Hartmann et al., 2010). In protein microarrays, multiple proteins are immobilized on a surface and share the same superficial chemical environment regardless of their broad range of physicochemical properties and their different binding vocations. Protein microarray techniques, on the other hand, assume that the entire panel of arrayed proteins is reproducibly and quantitatively immobilized on the probing surface. In-depth investigation on the absolute amount of surface bound probes is indeed of utmost importance for protein chips, especially if quantitative data are required as in clinical applications such as antibody profiling for allergy diagnosis, responses to infectious agents, diabetes or autoimmune disorders. Label based methods to visualize printed slides were developed for DNA microarrays spot quality control prior to hybridization (Yue et al., 2001; Diehl et al., 2001; Hessner et al., 2003, 2004). Recently, a label-free imaging technique using photonic crystals for quantitative spot quality analysis was developed (George et al., 2010) and applied to DNA microarrays without altering standard microarray protocols. Though relevant, these works deal with DNA molecules, which are a class of chemicals with uniform physicochemical characteristics. Proteins, on the contrary, are different one from each other in their way of interacting with surfaces; for each protein, spotting conditions must be carefully optimized to keep solubility and structure, favour binding and obtain a good spot morphology.
In this work we propose a system comprising a silicon chip and a dual interferometric/fluorescence reading for protein microarray development, fabrication and validation. The system allows, within a single experiment on the same chip, label-free imaging of the arrayed protein probes coupled with the fluorescent detection of the molecular interaction counterparts.
Recently, a simple interferometric method has been described in which an optical phase shift resulting from the surface accumulation of biological mass at different binding sites is monitored to investigate molecular interactions (Ozkumur et al., 2008). In this technique a silicon substrate with a thermally grown oxide layer on top of a solid silicon support is used for probe molecules spotted in an array format. To quantify the optical phase shift, the reflectance spectrum of the surface is recorded onto a CCD camera by illuminating the substrate with a tunable laser and serially capturing the intensity image of the reflectance for different wavelengths. We have proven that this technique is quantitative by directly relating the measured optical thickness of the biolayers to the absolute amount of molecules on the surface (Özkumur et al., 2009). We have also recently reported on the use of silicon with a silicon dioxide top layer of appropriate thickness for fluorescently enhanced microarray substrate. Fluorescence is intensified on these slides due to the optical constructive interference between the incident and reflected light of the fluorescent radiation (Cretich et al., 2009). The silicon oxide surface of the chips for both label-free and fluorescent techniques was functionalized by adsorption of copoly(DMA–NAS–MAPS), a ter-copolymer based on N,N-dimethylacrylamide (DMA), N-acryloyloxysuccinimide (NAS) and 3-(trimethoxysilyl)propyl-methacrylate (MAPS) (Cretich et al., 2004) which does not alter the optical properties of the optimized SiO2 layers, thus allowing us to produce microarray supports characterized by a very low non specific background for interferometric detection (Ozkumur et al., 2010) and for high sensitivity protein assays in array format (Cretich et al., 2009, 2010).
Silicon technology and a proper modulation of the thermal oxide layer are at the basis of the above mentioned techniques. In this work we combined on the same silicon chip two areas with oxide layers optimized for label-free measurement and fluorescence enhancement: an area with a 500 nm SiO2 layer dedicated to interferometric label-free detection and quantification of proteins and an area with 100 nm SiO2 providing enhanced fluorescence (Daaboul et al., 2011; Cretich et al., 2009).
Such a chip is of high practical utility during the assay development process to image arrays of proteins, check consistency and quality of the spotted array, quantify the amount of immobilized probes and finally detect fluorescence of bioassays. The use of the chip does not alter the microarray protocol and provides a sensitivity down to the pg/mL range for antibody detection.
2. Materials and methods
2.1. Reagents
Tris, BSA, Tween 20, PBS tablets, SSC 20X, bovine milk casein, rabbit immunoglobulin G, carbonic anhydrase, bovine serum albumin, ovalbumin, β-lactoglobulin B, α-lactalbumin and lysozyme were purchased from Sigma (St. Louis, MO). Sheep anti-casein was purchased from Abnova (Taipei city, Taiwan), rabbit anti β-lactoglobulin from Bethyl Laboratories (Montgomery, TX), goat anti-α-lactalbumin from GeneTex Inc. (Irvine, CA) and rabbit anti-ovalbumin from AbCam (Cambridge, UK). Secondary antibodies (Cy3 labelled goat anti-mouse and anti-sheep IgG and mouse anti-goat IgG) were from Jackson ImmunoResearch (West Grove, PA).
2.2. Silicon chip microfabrication
The combination 500 nm and 100 nm SiO2 chips with bare silicon reference were fabricated using photolithography patterning processes and wet etching. Wafers of 500 nm thermally grown SiO2 on a silicon substrate were purchased from Silicon Valley Microelectronics (Santa Clara, CA). Acetone sonication for 10 min and oxygen plasma ashing at 300 sccm and 500 W for 10 min are used to remove organic residue on the surface. Hexamethyldisilazane (HMDS) and Shipley S1818 positive resist are spun onto the surface at 2 krpm for 30 s. The chip is exposed for 30 s at 15 mW and then developed for 45 s in the SUSS Mask Aligner MA6 and Micro-Dev resist developer, respectively, to form the bare silicon reference pattern. The exposed region of the wafer is etched 500 nm at 77 nm/min when submerged in buffered oxide etch (BOE) 6:1.
With the bare silicon reference exposed, the wafer is again patterned following the same cleaning, spinning, and developing steps. The developed pattern exposes the SiO2 which will be used for fluorescence enhancement while protecting the label-free measurement regions. Etching from 500 nm to 100 nm is performed with BOE 6:1 diluted 1:40 with DI to achieve an etch rate of 4 nm/min. Finally, the resist is striped to reveal a chip with label-free and enhanced fluorescence SiO2 regions.
2.3. Detection setup
The LED-based Interferometric Reflectance Imaging Sensor (IRIS) has been recently described elsewhere (Daaboul et al., 2011). Briefly, the principle of detection for the LED-IRIS is based on quantifying the shifts in the spectral reflectance signature to calculate the added biomass by sampling it at specific wavelengths and measuring the characteristic reflection intensities using a CCD camera (Retiga 2000R from QImaging). The sensor surface is sequentially illuminated using an ACULED VHL surface-mount LED package (Perkin-Elmer), which has four independently driven LEDs with peak emission wavelengths of 455 nm, 518, 598 nm, and 635 nm. The position of the LEDs’ emission spectra with respect to the reflectance curve is critical for allowing the accurate measurement of the shift in this curve due to a change in the thickness (biomass accumulation) of the top layer. After acquiring images of the substrate for each of the four wavelengths each pixel of the CCD represents an individual measurement of the reflective interference intensity at each wavelength, forming a 3-dimensional array of data (pixel, wavelength, intensity) for the entire sensor. The data points for each pixel are fitted to a curve derived using well-known formulations (Fresnel equations), which govern the behaviour of reflection from a semi-transparent bi-layered substrate (Ozkumur et al., 2008). To achieve repeatable measurements the intensity of the incident light must be monitored. Incident light is traditionally measured by using an external photodetector; however, to maintain the simplicity of the system without sacrificing sensivity an on-chip reference is utilized (Vedula et al., 2010). The reference is created by including a non-interfering region, such as a region etched to the bare silicon, within the field of view. By monitoring fluctuations in the average value of a user-defined subset for this region, typically more than 1000 pixels, fluctuations in incident light intensity are measured After fitting every pixel in the image, the surface topography of the sensor’s surface is presented in a grayscale image where brighter regions indicate greater thickness on the surface. To determine optical spot heights, the average value from pixels in an annular region outside of the spot (background) is subtracted from the average value of pixels inside the spot. By using previously determined calibration factors, this information can be converted to mass densities at each spot location on the chip to determine binding. Previous work have shown 1 nm of optical thickness correlates to an absorbed mass conversion factor of 1.21 ng/mm2 for BSA, 1.28 ng/mm2 for IgG, and 0.8 ng/mm2 for DNA (Özkumur et al., 2009).
2.4. Silicon coating by copoly(DMA–NAS–MAPS)
Silicon slides were immersed for 30 min in a solution of copoly(DMA–NAS–MAPS), at 1% (w/v) concentration in water solution of ammonium sulphate at 20% saturation level. Slides were washed with water and dried under vacuum at 80 °C for 15 min. Copoly(DMA–NAS–MAPS) has been chosen for the easiness and reproducibility of the coating procedure and for its ability to functionalize silicon without altering its optical properties.
2.5. Microarray experiments
In the study of the immobilization conditions, an array of rabbit immunoglobulin G, carbonic anhydrase, bovine serum albumin, ovalbumin, β-lactoglobulin B, α-lactalbumin and lysozyme at 1 mg/mL concentration was patterned using an SciFlexArrayer S5 spotter from Scienion. Every protein was spotted in three different buffers: PBS (pH 7.2), borate (borate/NaOH 50 mM pH 9) and acetate (6-EACA/acetate 20 mM pH 4.4).
Printed slides were placed in a humid chamber and incubated at room temperature overnight. The slides were then blocked by 50 mM ethanolamine in Tris/HCl 1 M pH 9 for 1 h, washed with water and dried by a stream of Nitrogen. Arrayed slides were then incubated in a humid chamber with 100 μL of the solution of the specific antibody in incubation buffer (Tris/HCl 0.05 M pH 7.6, NaCl 0.15 M, Tween 20 0.02%) with 1% (w/v) BSA, for 2 h at different concentrations of antibody (0, 2, 10, 25, and 50 ng/mL). For casein experiments, slides were incubated with policlonal anti-casein sheep antibody (1 ng/mL) for 2 h.
Slides were then washed with washing buffer (Tris/HCl 0.05 M pH 9, NaCl 0.25 M, Tween 20 0.05%) for 10 min, rinsed with water and then incubated with 100 μL of the solution of the specific labelled secondary antibody 0.001 mg/mL in incubation buffer for 1 h. Slides were then washed with PBS (10 min), rinsed with water and dried with a Nitrogen stream.
Scanning for fluorescence evaluation was performed by a ProScanArray scanner from Perkin Elmer (Boston, MA); silicon slides were analyzed using 80% and 90% Photomultiplier (PMT) and laser power. Fluorescence intensities of replicated spots were averaged.
2.6. Determination of the limit of detection (LOD)
In order to determine the minimum concentration of antibody that can be reliably detected (analytical sensitivity), the concentrations of antibody used were plotted versus the intensities of the corresponding detected fluorescence. We fitted the values with a linear regression and extrapolated the limit of detection (LOD) as the antibody concentration that exceed the mean fluorescence of the 0 ng/mL (blank) sample plus three standard deviations.
3. Results and discussion
3.1. Concept
In Fig. 1 the concept of the new silicon biochip, applied to allergen microarrays, is illustrated. The chip is a 15 mm × 15 mm square silicon slide divided into four quarters (Fig. 1a), three regions of the chip exhibit a silicon oxide layer of 100 nm whereas one region displays an oxide thickness of 500 nm. Each quarter of the chip is spotted with the same protein array within the same spotting session. After binding of proteins and blocking of unreacted sites, the quarter of the chip exhibiting the 500 nm oxide layer is imaged using the LED-based IRIS and the amount of immobilized protein for each condition is quantified (Özkumur et al., 2009); the morphology and consistency of spots are also verified by visual inspection of the images generated by the IRIS setup. After the interferometric imaging, the chip undergoes the conventional microarray experiment. In the protein microarray example shown here, an allergen array (see spotting scheme and experimental details in the supplementary information) is incubated first with serum samples containing allergen specific IgEs and then with fluorescent anti-IgE antibody. The microarray is finally analyzed by a laser induced fluorescence (LIF) scanner for quantification of fluorescent signals. Three sectors of the chip are dedicated to fluorescence detection to increase the number of replicates. Fig. 1b reports a composite image obtained, for the same allergen microarray, before the serum incubation (label free detection of immobilized allergens) and after incubations with an allergic patient’s serum and fluorescent secondary antibody. The fluorescent signals obtained can be associated, allergen by allergen, with the amount of immobilized protein in order to implement an internal quality control into the microarray. This is particularly important if we consider that the allergens are usually recombinant or purified proteins with high commercial value, often experiencing problems of solubility and stability and that the amount of fluorescence obtained for each allergen is diagnostic for the type of allergy and the severity of symptoms experienced by the patients. Nowadays, microarrays with up to 103 different arrayed allergens are commercially available and used in clinics, the proposed chip could therefore be a valuable tool to be used in the development, production and daily application of protein microarrays.
Fig. 1.
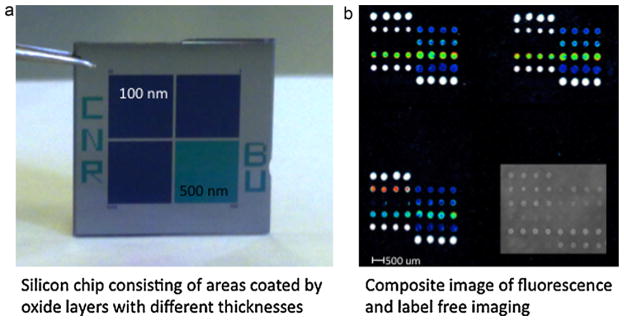
The silicon chip is a 15 mm × 15 mm square silicon slide divided into four quarters (a), three regions of the chip exhibit a silicon oxide layer of 100 nm whereas one region displays an oxide thickness of 500 nm. Each quarter of the chip is spotted with the same protein array within the same spotting session. The quarter of the chip exhibiting the 500 nm oxide layer is imaged by the LED-based Interferometric Reflectance Imaging Sensor (IRIS) the other three quarters are analyzed by a fluorescence scanner. Figure (b) reports a composite image obtained, for the same allergen microarray, before the serum incubation (label free detection of immobilized allergens) and after incubations with an allergic patient’s serum and fluorescent secondary antibody.
3.2. Optimization of probe solubilization conditions
As an example to demonstrate the practical utility of the silicon biochip as a tool for protein microarray development, we studied the solubilization and binding of casein from bovine milk, an important food allergen which solubility is pH and ionic strength dependent. The amount of casein immobilized on copoly(DMA–NAS–MAPS) coated Si/SiO2 slides using buffers with different pH and additives was studied. In this experiment, 1 mg/mL casein was added to PBS with 0.1% (w/v) SDS (1), PBS with 1% (w/v) SDS (2), PBS with 0.01% (v/v) Tween (3), borate pH 9 with 0.1% (w/v) SDS (4), borate pH 9 with 1% (w/v) SDS (5), borate pH 9 with 0.01% (v/v) Tween (6), saline-sodium citrate (SSC) buffer (7), phosphate/NaOH 50 mM pH 8.5 buffer (8) and NaOH 10 mM (9) and allowed to dissolve under stirring overnight. The casein solutions were then filtered and spotted (4 replicated spots for each conditions) on the silicon chip. After one night of incubation in a humid chamber, the surface was washed and the unreacted sites were blocked as reported in the experimental section. The surface was then imaged and the amount of immobilized protein quantified as described in Section 2. In Fig. 2a, the label free imaging of casein dissolved and spotted in different conditions (labelled from 1 to 9) is shown. The poor morphology of spots for conditions (2), (4), (5) and (6) can be observed; moreover we can notice that dissolution condition (3) led to absence of interferometric signal on the chip whereas condition (9) provided the highest signal with an acceptable spot morphology. The quantification of the amount of casein immobilized in the different conditions (expressed as ng/mm2) is reported in Fig. 2b, showing that dissolving and spotting casein in NaOH 10 mM, condition (9), provided more than 1 ng/mm2 immobilized protein on copoly(DMA–NAS–MAPS) coated Si/SiO2 slides. The array was then incubated with a polyclonal anti-casein antibody from sheep (1 ng/mL) followed by incubation with an anti-sheep IgG antibody labelled with Cy3 for fluorescence detection. The quantification of detected fluorescence signals is shown in Fig. 2c; as expected, condition (9) provided the highest fluorescence intensity confirming that, among the tested conditions, dissolution of casein in NaOH 10 mM yields the highest amount of protein available in spotting solution and an efficient immobilization environment.
Fig. 2.

(a) Label free image of casein from bovine milk dissolved and spotted in PBS with 0.1% (w/v) SDS (1), PBS with 1% (w/v) SDS (2), PBS with 0.01% (v/v) Tween (3), borate pH 9 with 0.1% (w/v) SDS (4), borate pH 9 with 1% (w/v) SDS (5), borate pH 9 with 0.01% (v/v) Tween (6), saline-sodium citrate (SSC) buffer (7), phosphate/NaOH 50 mM pH 8.5 buffer (8) and NaOH 10 mM (9). Dissolution condition (3) led to absence of interferometric signal on the chip whereas condition (9) provided the highest signal with an acceptable spot morphology. (b) Quantification of the amount of casein immobilized in the different conditions (expressed as ng/mm2). Dissolving and spotting casein in NaOH 10 mM, condition (9), provided more than 1 ng/mm2 immobilized protein on copoly(DMA–NAS–MAPS) coated Si/SiO2 slides. (c) Quantification of fluorescence after incubation with a polyclonal anti-casein antibody from sheep (1 ng/mL) followed by incubation with an anti-sheep IgG antibody labelled with Cy3 for fluorescence detection. As expected, condition (9), provided the highest fluorescence intensity.
3.3. Optimization of protein immobilization conditions
Due to a broad range of physicochemical characteristics, proteins differ in terms of ideal immobilization conditions (optimal pH to favour binding, use of detergent or additives to optimize spot morphology and to keep structure and/or activity). The absolute amount of immobilized protein is a crucial, though not sufficient, information during the development of an assay based on a protein array and also valuable information when checking the consistency of the derived data. The amount of protein, immobilized on copoly(DMA–NAS–MAPS) coated Si/SiO2 slides, using spotting buffers with different pH values was studied. In Fig. 3a a label free image of different proteins bound to the surface is shown. In these experiments rabbit immunoglobulin G, carbonic anhydrase, bovine serum albumin, ovalbumin, β-lactoglobulin B, α-lactalbumin and lysozyme were spotted at a concentration of 1 mg/mL in borate pH 9 buffer, PBS pH 7.2 and EACA–acetate pH 4.4 buffer. After one night of incubation in a humid chamber, the surface was washed and the unreacted sites were blocked as reported in the experimental section. The surface was then imaged and the amount of immobilized protein quantified. The amount of protein immobilized in each pH condition (expressed as ng/mm2) is shown in Fig. 3b and reported, with standard deviations of the measurements in Table 1. Reproducibility of protein binding within the same buffer condition is in line with microarray typical result variability. For each of the seven proteins used in this study, alkaline conditions (borate buffer pH 9) gave the highest yield of immobilization and reproducibility; this is probably due to the fact that in basic pH the reaction between the protein amino groups and the N-hydroxysuccinimide ester of the polymeric coating is favoured. In general, immobilization in neutral (pH 7.2) conditions provided higher yield and binding reproducibility than in acidic ones.
Fig. 3.

(a) Label-free imaging of the 500 nm SiO2 surface spotted with rabbit immunoglobulin G, carbonic anhydrase, bovine serum albumin, ovalbumin, β-lactoglobulin B, α-lactalbumin and lysozyme at the concentration of 1 mg/mL in a borate pH 9 buffer, PBS and EACA–acetate pH 4.4 buffer. The chip was imaged after overnight protein binding and washing. (b) Quantification of the amount of immobilized protein expressed as ng/mm2 of chip surface. For each of the spotted proteins, alkaline immobilization provides a higher yield of binding.
Table 1.
Quantification of the amount of immobilized proteins on copoly(DMA–NAS–MAPS) coated silicon chips determined by optical phase shift expressed in ng/mm2.
| pH 4.4 | pH 7.2 | pH 9 | |
|---|---|---|---|
| Rabbit immunoglobulin G | 2.80 ± 0.65 | 7.26 ± 1.11 | 11.12 ± 1.07 |
| Carbonic anhydrase | 4.02 ± 1.29 | 3.70 ± 0.99 | 8.25 ± 1.13 |
| Bovine serum albumin | 1.24 ± 0.61 | 1.94 ± 0.75 | 7.52 ± 0.58 |
| Ovoalbumin | 0.55 ± 0.02 | 2.39 ± 0.52 | 6.82 ± 0.65 |
| β lactoglobulin B | 0.21 ± 0.39 | 1.23 ± 0.15 | 3.99 ± 0.45 |
| α lactalbumin | 0.20 ± 0.14 | 1.95 ± 0.68 | 5.04 ± 0.43 |
| Lysozyme | 0.49 ± 0.46 | 4.20 ± 1.49 | 6.31 ± 0.60 |
As an example, for three out of the seven tested proteins (the three food allergens ovoalbumin, β-lactoglobulin B, α-lactalbumin) we have also investigated the analytical sensitivity for their specific antibodies through the determination of their limit of detection (LOD). The aim of this label free/fluorescence assay was to relate the activity of the antigen upon immobilization with its actual amount on the surface. The data reported here allow the estimation of the fraction of proteins that are recognized by their solution target. This fraction depends on how proteins are exposed and their structure maintained. The chips were incubated with different concentrations of anti-ovoalbumin, anti-β-lactoglobulin B and anti-α-lactalbumin policlonal antibodies followed by incubation with a fluorescent secondary antibody. A calibration curve reporting the fluorescent intensities obtained upon incubation with the specific antibodies in the ng/mL range was built for each protein. The minimum concentration of specific antibody tested was 2 ng/mL for anti-β-lactoglobulin B and anti-α-lactalbumin and 4 ng/mL for anti-ovoalbumin. For each protein, the specific antibody LOD was extrapolated from the fluorescent value corresponding to the blank sample plus three times the standard deviation (Wild, 2005). The calibration curves generated to determine LODs and the fluorescent values for the blank samples with the relative standard deviations are shown in Fig. 2 and Table 1 of the Supplementary Information. For all the specific antibodies tested we obtained very low LODs (in the pg/mL range) due to the fluorescence enhancement of the 100 nm silicon oxide layer. The values of LOD for each antibody, depending on the immobilization conditions of the capture protein, are reported in Table 2.
Table 2.
Limit of detection (LOD) determined for protein specific antibodies in pg/mL.
| Immobilized proteins | Immobilization conditions
|
||
|---|---|---|---|
| pH 4.4 | pH 7.2 | pH 9 | |
| β-Lactoglobulin B | 6 | 3 | 60 |
| α-Lactalbumin | 67 | 57 | 232 |
| Ovoalbumin | 25 | 37 | 28 |
Despite the higher amount of immobilized protein at pH 9, alkaline immobilization provides a LOD for β-lactoglobulin B and α-lactalbumin specific antibodies that is higher than the LODs provided by the corresponding immobilization in neutral and acidic buffers. This is due to the higher fluorescence of the blank samples (which is derived by aspecific interaction of the fluorescent secondary antibody) when proteins are immobilized in alkaline conditions. A comparison of the fluorescence values obtained for the blank samples are reported in Fig. 3 of the Supplementary Information. Indeed, in sandwich type microimmunoassay, specificity of the secondary antibody is of great importance in determining the actual LOD of an assay because it directly influences the signal provided by the blank samples.
4. Concluding remarks
Awareness of the real immobilization yield for different proteins, in distinct immobilization conditions, can help in developing consistent protein chips. The absolute amount of immobilized material is valuable information when developing protein microarrays; together with activity of the immobilized proteins and specificity of the secondary antibodies it will ultimately determine the sensitivity and reproducibility of protein microarrays. A correlation between interferometric measurements and fluorescence outcomes in model experiments as well as in a relevant clinical application such as allergy diagnosis is currently under way in our laboratories.
The new silicon biochip for dual label-free and fluorescence detection proposed here can be of high practical utility during the microarray assay development process. It allows, in a single experiment, performed on the same surface chemistry, to check spotting consistency, to optimize solubilization and binding conditions for the different proteins and to relate these data to the fluorescence of the final bioassay with a sensitivity in protein detection down to the pg/mL range. Without altering the standard microarray protocol, the new tool can be implemented in a routine use as an internal quality control for protein array production.
Supplementary Material
Acknowledgments
We thank Carlos A. Lopez and George G. Daaboul for helpful discussion.
Financial support from Regione Lombardia, project “FREE IMAGER” – ID 15900; Rif. SAL-39, SmartLighting ERC funded via the NSF under Cooperative Agreement EEC-0812056 and by NYS-TAR under contracts C080145 and C090145 and by the Wallace H. Coulter Foundation 2010 Coulter Translational Research Award.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bios.2011.03.016.
References
- Cretich M, Breda D, Damin F, Borghi M, Unlu SM, Burastero SE, Chiari M. Anal Bioanal Chem. 2010;398:1723–1733. doi: 10.1007/s00216-010-4077-x. [DOI] [PubMed] [Google Scholar]
- Cretich M, Di Carlo G, Longhi R, Gotti C, Spinella N, Coffa S, Galati C, Renna L, Chiari M. Anal Chem. 2009;81:5197–5203. doi: 10.1021/ac900658c. [DOI] [PubMed] [Google Scholar]
- Cretich M, Pirri G, Damin F, Solinas I, Chiari M. Anal Biochem. 2004;332:67–74. doi: 10.1016/j.ab.2004.05.041. [DOI] [PubMed] [Google Scholar]
- Daaboul GG, Vedula RS, Ahn S, Lopez CA, Reddington A, Ozkumur E, Unlu MS. Biosens Bioelectron. 2011;26:2221–2227. doi: 10.1016/j.bios.2010.09.038. [DOI] [PubMed] [Google Scholar]
- Diehl F, Grahlmann S, Beier M, Hoheisel JD. Nucleic Acids Res. 2001;29:e38. doi: 10.1093/nar/29.7.e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S, Block ID, Jones SI, Mathias PC, Chaudhery V, Vuttipittayamongkol P, Wu HY, Vodkin LO, Cunningham BT. Anal Chem. 2010;82:8551–8557. doi: 10.1021/ac101551c. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Roeraade J, Stoll D, Templin M, Joos TO. Anal Bioanal Chem. 2010;393:1407–1416. doi: 10.1007/s00216-008-2379-z. [DOI] [PubMed] [Google Scholar]
- Hessner MJ, Singh VK, Wang X, Khan S, Tschannen MR, Zahrt TC. BMC Genomics. 2004;5:12. doi: 10.1186/1471-2164-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessner MJ, Wang X, Hulse K, Meyer L, Wu Y, Nye S, Guo SW, Ghosh S. Nucleic Acids Res. 2003;31:e14. doi: 10.1093/nar/gng014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkumur E, Lopez CA, Yalcin A, Connor JH, Chiari M, Unlu SM. IEEE J Sel Top Quantum Electron. 2010;16:635–646. [Google Scholar]
- Ozkumur E, Needham JW, Bergstein DA, Gonzalez R, Cabodi M, Gershoni JM, Goldberg BB, Unlu MS. PNAS. 2008;105:7988–7992. doi: 10.1073/pnas.0711421105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özkumur E, Yalçin A, Cretich M, Lopez CA, Bergstein DA, Goldberg BB, Chiari M, Ünlü MS. Biosens Bioelectron. 2009;25:167–172. doi: 10.1016/j.bios.2009.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel M, Niessner R. Anal Bioanal Chem. 2008;391:1521–1544. doi: 10.1007/s00216-008-2039-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizaki K, Usui K, Mihara H. FEBS J. 2010;277 (9):1996–2005. doi: 10.1111/j.1742-4658.2010.07626.x. [DOI] [PubMed] [Google Scholar]
- Vedula R, Daaboul G, Reddington A, Ozkumur E, Bergstein DA, Unlu MS. J Mod Opt. 2010;57:1564–1569. [Google Scholar]
- Wild D, editor. The Immunoassay Handbook. Elsevier; 2005. [Google Scholar]
- Wolf-Yadlin A, Sevecka M, MacBeath G. Curr Opin Chem Biol. 2009;13:398–405. doi: 10.1016/j.cbpa.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XB, Schneiderhan-Marra N, Joos TO. Clin Chem. 2010;56:376–387. doi: 10.1373/clinchem.2009.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue H, Eastman PS, Wang BB, Minor J, Doctolero MH, Nuttall RL, Stack R, Becker JW, Montgomery JR, Vainer M. Nucleic Acids Res. 2001;29:e41. doi: 10.1093/nar/29.8.e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.