

The authors provide genetic and cell-biological evidence indicating that MIG-10B interacts with ABI-1 to organize the actin cytoskeleton at presynaptic sites and that the C. elegans Synapsin ortholog snn-1 acts downstream from MIG-10B to instruct synaptic vesicle clustering at presynaptic sites, likely bridging vesicle interaction with the MIG-10B-organized actin cytoskeleton. The findings support a model in which MIG-10B localizes to presynaptic sites in response to Netrin, organizes the actin cytoskeleton through an ABI-1-mediated interaction with the WAVE complex, and instructs vesicle clustering at presynaptic sites through Synapsin.
Keywords: MIG-10/lamellipodin, synaptic vesicle clustering, thermotaxis, Netrin, ABI-1/ABI1, C. elegans
Abstract
The chemotrophic factor Netrin can simultaneously instruct different neurodevelopmental programs in individual neurons in vivo. How neurons correctly interpret the Netrin signal and undergo the appropriate neurodevelopmental response is not understood. Here we identify MIG-10 isoforms as critical determinants of individual cellular responses to Netrin. We determined that distinct MIG-10 isoforms, varying only in their N-terminal motifs, can localize to specific subcellular domains and are differentially required for discrete neurodevelopmental processes in vivo. We identified MIG-10B as an isoform uniquely capable of localizing to presynaptic regions and instructing synaptic vesicle clustering in response to Netrin. MIG-10B interacts with Abl-interacting protein-1 (ABI-1)/Abi1, a component of the WAVE complex, to organize the actin cytoskeleton at presynaptic sites and instruct vesicle clustering through SNN-1/Synapsin. We identified a motif in the MIG-10B N-terminal domain that is required for its function and localization to presynaptic sites. With this motif, we engineered a dominant-negative MIG-10B construct that disrupts vesicle clustering and animal thermotaxis behavior when expressed in a single neuron in vivo. Our findings indicate that the unique N-terminal domains confer distinct MIG-10 isoforms with unique capabilities to localize to distinct subcellular compartments, organize the actin cytoskeleton at these sites, and instruct distinct Netrin-dependent neurodevelopmental programs.
Netrin is a chemotrophic factor that can simultaneously instruct various neurodevelopmental outcomes in individual cells during development (Winberg et al. 1998; Livesey 1999; Colon-Ramos et al. 2007; Round and Stein 2007; Poon et al. 2008; Quinn and Wadsworth 2008; Manitt et al. 2009; Xu et al. 2010; Park et al. 2011). For example, during axon guidance, neurons differentially respond to Netrin signaling depending on the cell-specific expression of extracellular receptors, the cellular cytosolic cAMP levels, the extracellular matrix laminin composition near the growth cone, or serotonin signaling, among other factors (Hopker et al. 1999; Ming et al. 1999, 2001; Song and Poo 1999; Forcet et al. 2002; Graef et al. 2003; Ren et al. 2004; Wen et al. 2004; Bonnin et al. 2007). The mechanisms that modulate cell-specific responses to Netrin are important in instructing the diverse outgrowth responses observed in vivo.
Netrin is required for additional neurodevelopmental processes other than axon outgrowth; Netrin can also instruct cell migration, dendritic branching, axon arborization, or synapse formation (Hedgecock et al. 1990; Ishii et al. 1992; Colon-Ramos et al. 2007; Poon et al. 2008; Manitt et al. 2009; Xu et al. 2010; Park et al. 2011; Smith et al. 2012; Stavoe and Colon-Ramos 2012; Timofeev et al. 2012). The ability of a neuron to respond cell-specifically to Netrin in guidance or synapse formation is also critical for orchestrated circuit assembly in vivo (Salie et al. 2005; Colon-Ramos 2009; Sanes and Yamagata 2009; Shen and Cowan 2010; Williams et al. 2010). For example, connectivity between two interneurons in the nematode brain, AIY and RIA, is orchestrated by glia-derived UNC-6/Netrin. Netrin promotes guidance in the postsynaptic RIA neuron and presynaptic assembly in the AIY neuron. By instructing different neurodevelopmental outcomes in a cell-specific manner, Netrin orchestrates the correct formation of the AIY:RIA synapse at a glia-specified coordinate (Colon-Ramos et al. 2007). We do not yet understand how Netrin instructs distinct neurodevelopmental outcomes, such as axon arborization or synapse formation, in specific cells.
We recently determined that Netrin instructs AIY synaptic vesicle clustering through a Rac GTPase pathway downstream from the UNC-40/DCC receptor (Stavoe and Colon-Ramos 2012). Netrin localizes its receptor, UNC-40/DCC, to AIY presynaptic regions. UNC-40/DCC then interacts with CED-5/DOCK180, a Rac GEF. Through this interaction, CED-5 is localized to AIY presynaptic regions to activate CED-10/Rac1 (Stavoe and Colon-Ramos 2012). CED-10 can, in turn, interact with MIG-10/Lamellipodin, an adaptor molecule (Quinn et al. 2008). The CED-10 pathway is necessary for proper localization of MIG-10 to AIY presynaptic regions. Localization of this signaling complex to the Netrin-specified coordinate results in the organization of the actin cytoskeleton and synaptic vesicle clustering in presynaptic regions (Stavoe and Colon-Ramos 2012). Interestingly, this same signaling module acts in other neurons to instruct axon guidance, axon arborization, and cell migration (Gitai et al. 2003; Lundquist 2003; Adler et al. 2006; Chang et al. 2006; Quinn et al. 2006, 2008; Quinn and Wadsworth 2008; Manitt et al. 2009). It is not understood how these conserved signaling mechanisms result in distinct neurodevelopmental outcomes downstream from Netrin.
Here we examine the Caenorhabditis elegans AIY interneuron, the HSN (hermaphrodite-specific neuron) motor neuron, and the neurosecretory motor (NSM) neuron to understand how different neurodevelopmental outcomes are instructed downstream from Netrin in vivo. We determined that different MIG-10 isoforms are required cell-specifically to instruct a neuron's individual response to Netrin. We also determined that MIG-10 isoforms can localize to different subcellular compartments in response to Netrin. Specifically, we identified a MIG-10 isoform, MIG-10B, which has the unique ability to localize to presynaptic regions and is required to instruct synaptic vesicle clustering in response to Netrin. MIG-10B subcellular localization to presynaptic sites is mediated by a unique 21-amino-acid N-terminal motif and is dependent on both Netrin and active zone proteins SYD-1 and SYD-2. We determined that MIG-10B interacts with Abl-interacting protein-1 (ABI-1), a component of the WAVE complex, to organize the actin cytoskeleton in presynaptic regions. Our findings indicate that MIG-10B organization of the actin cytoskeleton through ABI-1 is crucial for synaptic vesicle clustering and that this interaction with synaptic vesicles is at least partially mediated by the synaptic vesicle-associated protein SNN-1/Synapsin. Underscoring the importance of the MIG-10B N-terminal helix in its localization, we demonstrated that we can disrupt MIG-10B localization through the cell-specific expression of its N-terminal motif. Cell-specific expression of the MIG-10B N-terminal motif acts as a dominant-negative disrupting actin organization and synaptic vesicle clustering in AIY. We provide a functional consequence for the reported phenotype, demonstrating that single-cell expression of this dominant-negative construct disrupts AIY-dependent thermotaxis behavior. Together, our findings demonstrate that MIG-10 isoforms, varying only in their N-terminal motifs, can localize to specific subcellular domains, organize the actin cytoskeleton at these domains, and instruct distinct neurodevelopmental outcomes downstream from Netrin.
Results
A specific MIG-10 isoform localizes to presynaptic regions in response to Netrin
In the AIY interneuron of C. elegans, MIG-10 is required downstream from Netrin to instruct proper synaptic vesicle clustering in presynaptic regions (Fig. 1A–F; Stavoe and Colon-Ramos 2012). To determine how MIG-10 regulates synaptic vesicle clustering in AIY, we examined its subcellular localization. The mig-10 gene has been shown to encode at least two protein isoforms, MIG-10A and MIG-10B (Fig. 1G; Manser et al. 1997; Quinn et al. 2006). The possible expression of a third protein isoform, MIG-10C, has also been suggested based on partial cDNA sequencing (http://www.wormbase.org/db/get?name=mig-10;class=Gene_name). To examine whether MIG-10C is also expressed in wild-type animals, we amplified and sequenced the entire coding region of MIG-10C from cDNA collected from a mixed stage population. Our data indicate that C. elegans expresses the mRNA for all three of the predicted MIG-10 isoforms (Supplemental Fig. S1A). Consistent with previous expression pattern reports for MIG-10A and MIG-10B, we also observed distinct expression patterns for all three isoforms in different tissues during the development of C. elegans (Supplemental Fig. S1B–J; Quinn et al. 2006).
Figure 1.

MIG-10B localizes to AIY presynaptic regions in response to Netrin. (A) Confocal micrograph of cytoplasmic GFP expressed cell-specifically in AIY, depicting the morphology of AIY. (B–F) Synaptic vesicle pattern in maximal projection confocal micrographs of GFP∷RAB-3 in AIY in wild-type (B), mig-10(ct41) (D), and mig-10(ok2499) (E) mutant animals and schematics (C,F). The schematic diagram of wild-type AIY (shown in C) indicates neurite zones. Zone 1 is asynaptic and projects anteriorly from the cell body. AIY forms synapses onto RIA, AIZ, and RIB in zone 2. Zone 3 is within the nerve ring and is the distal portion of the neurite after the neurite turns dorsally (Colon-Ramos et al. 2007). Bar: B, 5 μm (applies to A,B,D,E). (G) Diagram of the mig-10 gene, with exons as boxes and introns as lines. The first three schematics show the relative positions of the three MIG-10 isoforms—MIG-10A, MIG-10B, and MIG-10C—drawn to scale. Shared exons code for conserved domains. Cartoon diagram of the conserved protein domains in the top right corner. Ras association (RA), plekstrin homology (PH), proline-rich (Prl), and FPPP (FP) (Holt and Daly 2005). The fourth and fifth schematics include a diagram of the rescuing fosmids, drawn to scale and aligned with the genetic regions of mig-10 and the isoforms. Modified image adapted with permission from WormBase (http://www.wormbase.org). (H–J) Confocal micrographs showing the cell-specific subcellular localization of MIG-10A∷GFP (H), MIG-10B∷GFP (I), and MIG-10C∷GFP (J). Note that MIG-10A and MIG-10C signal is diffuse throughout the neurite, including asynaptic regions (bracket) (H,J), while MIG-10B is enriched in presynaptic regions (I, arrows). (K,L) Confocal micrographs showing MIG-10B∷GFP subcellular localization in AIY in unc-6(ev400) (K) and unc-40(e271) (L) mutant animals. (I) Note that MIG-10B∷GFP is less enriched in zone 2 in the two mutant backgrounds, as compared with wild-type animals. (M–O) Confocal micrographs of zones 2 and 3 of a transgenic animal expressing MIG-10B∷GFP (M) and mCh∷RAB-3 (N) in AIY. Merge is shown in O. Synaptic regions are expanded in these images to allow evaluation of partial colocalization (arrows). In all images, the asterisk (*) represents the location of the cell body, and the dashed box encloses zone 2. Bar: H, 5 μm (applies to H–L); M, 5 μm (applies to M–O).
We then examined the subcellular localization of these three MIG-10 isoforms in AIY by fusing the MIG-10 isoforms to GFP and expressing them cell-specifically in AIY. Interestingly, despite the large degree of identity between these isoforms, we observed differences in their subcellular localization in AIY. We observed that MIG-10A∷GFP and MIG-10C∷GFP display diffuse localization throughout the entire neurite of AIY, including the asynaptic regions and in a pattern that resembled that seen for GFP when cell-specifically expressed in AIY (Fig. 1A,H,J; Stavoe and Colon-Ramos 2012). Interestingly, we observed that MIG-10B selectively localized to presynaptic regions and was absent from the asynaptic regions of AIY (Fig. 1I).
The MIG-10B∷GFP localization pattern is reminiscent of that of GFP∷RAB-3 (Fig. 1B). Thus, we hypothesized that MIG-10B∷GFP and mCh∷RAB-3 colocalize in AIY presynaptic regions. Examination of a double-labeled strain expressing MIG-10B∷GFP and mCh∷RAB-3 showed that these two proteins partially colocalized in presynaptic regions (Fig. 1M–O). Consistent with these findings, we observed that MIG-10B can colocalize with F-actin structures in presynaptic regions (Stavoe and Colon-Ramos 2012; data not shown). Together, these data indicate that MIG-10B can partially colocalize with F-actin and synaptic vesicle clusters in AIY.
We previously determined that Netrin, UNC-40, CED-5, and CED-10 instruct AIY synaptic vesicle clustering upstream of MIG-10 (Stavoe and Colon-Ramos 2012). Therefore, we next examined whether UNC-6/Netrin and its receptor, UNC-40, are required for presynaptic MIG-10B localization by visualizing MIG-10B∷GFP in unc-6(ev400) and unc-40(e271) mutant backgrounds (Fig. 1K,L). In agreement with our previous findings, we observed that MIG-10B∷GFP localization to presynaptic regions is significantly decreased in unc-6(ev400), unc-40(e271), ced-5(n1812), and ced-10(n3246) mutants, suggesting that the UNC-6, UNC-40, and downstream pathway components are required for MIG-10B localization to presynaptic regions (Fig. 1K,L; Stavoe and Colon-Ramos 2012; data not shown). Together, our data indicate that MIG-10B specifically localizes to presynaptic regions in response to Netrin and the downstream Rac signaling components.
MIG-10B is sufficient for AIY synaptic vesicle clustering
MIG-10B, but not MIG-10A or MIG-10C, is enriched in AIY presynaptic regions. We hypothesized that MIG-10 localization to presynaptic regions is important for synaptic vesicle clustering downstream from Netrin. Therefore, we predicted that the MIG-10B isoform, but not the other isoforms, would rescue the AIY synaptic vesicle clustering defect in mig-10(ct41) mutant animals. To test our hypothesis, we first examined rescue in mig-10(ct41) mutant animals carrying a genomic fragment containing the promoter regions and all coding regions of mig-10a and mig-10b (fosmid WRM066dB08) (Fig. 2A). We observed that 80% of mig-10(ct41) mutant animals carrying this genomic fragment displayed wild-type presynaptic patterning in AIY zone 2 (n = 66) (Fig. 2B,E). These results indicate that MIG-10C is not necessary for synaptic vesicle clustering and that MIG-10A and MIG-10B are sufficient for AIY synaptic vesicle clustering.
Figure 2.

MIG-10B is sufficient and necessary to rescue synaptic vesicle clustering. (A) Diagram of the mig-10 gene, as in Figure 1G. The sixth, seventh, eighth, and ninth schematics correspond to the recombineered fosmids used to assay the necessity of MIG-10B. Red boxes represent the introduction of a cassette with a stop codon in lieu of the mig-10a unique exon [MIG-10A(−); MIG-10B(+)] or in lieu of the mig-10b unique exon [MIG-10A(+); MIG-10B(−)].Green boxes represent the introduction of GFP in-frame after the mig-10 coding region (“MIG-10:GFP”). For the “exon swap” recombineered fosmid, the shared exon A/C sequence (in blue) was introduced in place of the unique exon B. Image modified with permission from WormBase (http://www.wormbase.org). (B) Quantification of the percentage of animals displaying the AIY presynaptic defect. Rescuing fosmids (see A) were expressed in mig-10(ct41) mutants and scored for rescue of the AIY presynaptic defect. Note that only fosmid MIG-10A(+);MIG-10B(−) is not capable of rescuing the AIY presynaptic defect. (***) P < 0.0001 between indicated groups by Fisher's exact test. Error bars represent 95% confidence interval. (C,D) Distribution of synaptic vesicles in AIY in wild-type (C) and mig-10(ct41) mutants (D). (E–H) Distribution of synaptic vesicles in AIY in mig-10(ct41) mutants expressing rescuing constructs fosmid MIG-10A (+); MIG-10B (+) (E), fosmid MIG-10B (+) (F), fosmid MIG-10A(−); MIG-10B(+) (G), or fosmid MIG-10A(+); MIG-10B(−) (H). Note that the distribution of synaptic vesicles is disrupted in mig-10(ct41) mutants (shown in D) and is rescued in E–G, but not in H. (I,J) Distribution of synaptic vesicles in AIY in mig-10(ct41) mutants (I) and mig-10(ct41) mutants expressing rescuing construct MIG-10:GFP (J). Note that MIG-10:GFP (shown in J) rescues the mig-10(ct41) synaptic patterning defect. In all images, the asterisk (*) represents the location of the cell body, and the dashed box encloses zone 2. Bar: C, 5 μm (applies to C–J).
To determine whether just MIG-10B was sufficient for AIY synaptic vesicle clustering, we then examined whether a genomic fragment containing mig-10b, but not mig-10a, was capable of rescuing the AIY mig-10(ct41) presynaptic phenotype (fosmid WRM0613bB07) (Fig. 2A). We observed that 85% of mig-10(ct41) mutant animals carrying this genomic fragment displayed wild-type presynaptic patterning in AIY zone 2 (n = 68) (Fig. 2B,F). This rescue is not significantly different from that of the fosmid containing both mig-10a and mig-10b. Therefore, our data indicate that MIG-10B is sufficient for rescuing the mig-10(ct41) presynaptic defect in AIY. Together, these results demonstrate that MIG-10A and MIG-10C are not necessary for synaptic vesicle clustering and that MIG-10B is sufficient for AIY synaptic vesicle clustering.
MIG-10A and MIG-10B act redundantly in HSN to instruct cell migration and axon defasciculation
Previous studies conducted in HSN demonstrated that MIG-10 is required downstream from UNC-6 during cell migration and axon outgrowth (Supplemental Fig. S2A–C; Adler et al. 2006; Chang et al. 2006; Quinn et al. 2008). In these studies, both the MIG-10A and MIG-10B isoforms rescued the reported mig-10 defects when expressed cell-specifically in HSN, indicating that these isoforms act redundantly in the HSN motor neuron to regulate cell migration and axon guidance (Adler et al. 2006; Chang et al. 2006).
To better understand the isoform-specific requirement of MIG-10 during HSN development, we first conducted a detailed characterization of HSN development in mig-10(ct41) mutant animals. Consistent with previous reports, we observed that 80% of mig-10(ct41) mutant HSNL neurons fail to complete anterior migration (n = 30) (Supplemental Fig. S2E; Manser and Wood 1990). In wild-type animals, the HSN axon fasciculates with the ventral nerve cord (VNC) for a short distance, then defasciculates and grows anteriorly, and finally refasciculates with the VNC to grow anteriorly to the nerve ring (Supplemental Fig. S2A,B,D; Desai et al. 1988; Forrester and Garriga 1997; Adler et al. 2006; Asakura et al. 2007). These events allow HSN to form en passant synapses onto vulval muscles and two VC neurons (White et al. 1986; Shen and Bargmann 2003; Patel et al. 2006). We observed that mig-10(ct41) mutant animals exhibit a moderately penetrant HSN defasciculation defect (Supplemental Fig. S2C,D). In mig-10(ct41) mutant animals in which HSN does project to the correct presynaptic region, we observed normal clustering of synaptic vesicles at the presynaptic sites (data not shown).
We then examined whether the MIG-10A and MIG-10B isoforms can rescue these developmental defects in HSN. Consistent with previous studies, we observed that both isoforms were equally capable of rescuing cell migration and defasciculation in HSN (Supplemental Fig. S2D,E; Adler et al. 2006; Chang et al. 2006; Quinn et al. 2008). Thus, our data indicate that MIG-10A and MIG-10B can act redundantly in HSN cell migration and axon defasciculation.
We then examined the subcellular localization of these isoforms during HSN development. Consistent with previous reports, we observed that both MIG-10A and MIG-10B asymmetrically localized to the ventral side of HSN cell body during polarized axon outgrowth (Adler et al. 2006; Quinn et al. 2008; data not shown). Interestingly, we observed that the MIG-10A and MIG-10B isoforms displayed different localization patterns after these initial outgrowth events were completed. Upon conclusion of axon outgrowth, we observed that MIG-10A became dispersed throughout the HSN neurite (Supplemental Fig. S2F). Analysis of MIG-10C also revealed that, like MIG-10A, it became dispersed throughout the neurite after completion of HSN axon outgrowth (Supplemental Fig. S2H). Conversely, during this same developmental period, we observed that MIG-10B became enriched at the newly formed HSN presynaptic regions (Supplemental Fig. S2G; Shen and Bargmann 2003). MIG-10B localization to the presynaptic sites persisted to adulthood (data not shown). Although our studies did not uncover a role for MIG-10 in HSN presynaptic assembly, our cell-biological data in both HSN and AIY demonstrate that MIG-10B, but not MIG-10A or MIG-10C, can specifically localize to presynaptic regions. In addition, our data confirm previous studies that MIG-10A and MIG-10B act redundantly in HSN cell migration (Adler et al. 2006; Chang et al. 2006).
MIG-10B is necessary for rescue of AIY synaptic vesicle clustering
Data in HSN indicate that MIG-10A and MIG-10B can act redundantly in some cellular contexts to mediate Netrin-dependent neurodevelopmental events. To determine whether MIG-10A and MIG-10B also act redundantly in AIY synaptic vesicle clustering, we next examined the necessity of MIG-10B for rescue of the mig-10(ct41) synaptic vesicle defect in AIY. Unlike in HSN, we cannot use cell-specific promoters in AIY that are activated prior to synaptogenesis to achieve rescue of the mig-10(ct41) mutant phenotype (Stavoe and Colon-Ramos 2012). Instead, we used recombinogenic engineering (recombineering) to create transgenes in which we disrupted the expression of either MIG-10A [called MIG10A(−); MIG10B(+) for simplicity] or MIG-10B [called MIG10A(+); MIG10B(−)] (Fig. 2A). We first examined the functionality of these newly generated transgenes by assaying their capacity to rescue the mig-10(ct41) developmental defects in HSN. As expected, given the redundancy of MIG-10A and MIG-10B in HSN, both MIG10A(−); MIG10B(+) and MIG10A(+); MIG10B(−) fully rescued the mig-10(ct41) developmental defects in HSN, demonstrating that these transgenes were functional (Supplemental Fig. S2D,E). We then examined the capacity of these transgenes to rescue the mig-10(ct41) mutant presynaptic defect in AIY.
Consistent with our previous observation that MIG-10B is sufficient to instruct synaptic vesicle clustering in AIY, we observed that the MIG10A(−); MIG10B(+) transgene rescued the mig-10(ct41) presynaptic patterning defect (Fig. 2A,B,G). However, we observed that the MIG10A(+); MIG10B(−) transgene, which rescued the mig-10(ct41) developmental defects in HSN, did not rescue the mig-10(ct41) synaptic vesicle clustering defect in AIY (Fig. 2A,B,H). Together, these data indicate that MIG-10A is neither necessary nor sufficient to instruct AIY synaptic vesicle clustering. Furthermore, our data also suggest that MIG-10B is necessary and sufficient for proper AIY synaptic vesicle clustering in response to Netrin. Together, our data support the hypothesis that MIG-10B is uniquely capable of localizing to presynaptic regions and instructing synaptic vesicle clustering in response to Netrin.
MIG-10A and MIG-10C, but not MIG-10B, rescue an axon arborization defect in the NSM neuron
Based on the findings that MIG-10B rescues AIY synaptic vesicle clustering and HSN guidance defects, one might conclude that the MIG-10B isoform can perform all cellular functions of MIG-10. We set out to test this hypothesis by examining the mig-10(ct41) mutant phenotype in other C. elegans neurons. In particular, the axonal arbor structures of the NSM neuron displayed a drastic reduction in branch number in mig-10(ct41) mutants as compared with wild type (Fig. 3A–D). We first examined whether the MIG-10B(+) (WRM0613bB07) fosmid, which is sufficient to rescue the synaptic vesicle clustering in AIY, was also sufficient for rescue of the axon arbor defect in the NSM neuron of mig-10 mutants (Fig. 2A,B). We observed that while expression of the MIG-10A(+);MIG-10B(+) (WRM066dB08) fosmid resulted in rescue of the NSM axon branching defect (85.4% of transgenic animals displayed wild-type arborization, compared with 89.5% of wild-type animals and 18.2% of mig-10(ct41) mutant animals; n = 48, 57 and 44, respectively), expression of the MIG-10B(+) fosmid did not rescue the mig-10(ct41) branching phenotype (22.5% of animals displayed wild-type arborization; n = 40). To further examine the cell-specific requirement of these isoforms, we expressed MIG-10A∷GFP, MIG-10B∷GFP, or MIG-10C∷GFP in the NSM neuron of mig-10(ct41) mutant animals (Fig. 3C,E–G). Consistent with the fosmid rescue experiments, we observed that while cell-specific expression of MIG-10A or MIG-10C rescued the mutant phenotype (Fig. 3C,E,G), cell-specific expression of the MIG-10B isoform in the NSM did not rescue the phenotype (Fig. 3C,F). These data illustrate that the MIG-10B isoform cannot perform all of the cellular functions of MIG-10. Rather, these data support the hypothesis that specific MIG-10 isoforms are required for distinct neurodevelopmental programs in individual neurons.
Figure 3.

MIG-10A and MIG-10C rescue an axon arborization defect in the NSM neuron. (A) Schematic of the pharyngeal NSM neuron. Note the presence of axonal arbors extending from the ventral neurite in the pharyngeal isthmus. The bracket indicates the pharyngeal isthmus region in which axon arbors typically form. Modified image adapted with permission from Wormatlas (http://www.wormatlas.org). (B) Confocal micrograph of a wild-type animal expressing GFP under the control of the NSM-specific tph-1 promoter (Sze et al. 2002). (C) Quantification of the axon arborization defect in the NSM. Total primary branches extending off of the ventral NSM neurite were counted for each genotype. Cell-specific expression of MIG-10A∷GFP or MIG-10C∷GFP results in statistically significant rescue of the arborization defect with respect to mig-10(ct41) mutants, but NSM-specific expression of MIG-10B∷GFP does not result in rescue. Error bars represent SEM. (*) P < 0.05 between indicated groups and mig-10(ct41). Post-hoc analysis was performed using Tukey's HSD. (D–G) Confocal micrographs of mig-10(ct41) mutant (D) or mig-10(ct41) mutants expressing MIG-10 cDNA constructs under the control of the NSM-specific tph-1 promoter (Sze et al. 2002) (E–G). Note the presence of axon arbors in the bracketed region in wild-type (B) or rescued (E,G) animals, but note the absence in arbors in mig-10(ct41) mutant animals (D) or mig-10(ct41) mutant animals expressing MIG-10B (F). Bar: B, 5 μm (applies to B,D–G).
The MIG-10B unique exon is necessary for its subcellular localization to presynaptic regions
MIG-10A, MIG-10B, and MIG-10C isoforms share 629 amino acids of identity (>80% identity) and differ only in their N-terminal regions. The 629-amino-acid region of identity contains the structural domains known to be required for MIG-10 function: a Ras association domain that binds directly to CED-10, a Pleckstrin homology domain, and proline-rich domains (Fig. 1G; Chang et al. 2006; Quinn et al. 2008). The N-terminal region corresponds to an unstructured motif predicted to form an amphipathic α helix that varies in length and composition between MIG-10A, MIG-10C, and MIG-10B (Supplemental Fig. S3A,B).
Given that this N-terminal region is the only difference between MIG-10A/C and MIG-10B isoforms, we examined whether it conferred to MIG-10B the unique capacity to localize to presynaptic regions. We first cell-specifically expressed MIG-10B without the unique MIG-10B exon fused to GFP in AIY and assayed its subcellular localization. This construct contains the regions shared by all three MIG-10 isoforms. We observed that deletion of the unique N-terminal region of MIG-10B eliminated the capacity of the construct to localize and cluster specifically in AIY presynaptic regions (data not shown). These data indicate that the unique MIG-10B N terminus is necessary for MIG-10B clustering in presynaptic regions. Although the MIG-10B N terminus is necessary for its localization, we also determined that it was not sufficient to drive presynaptic localization (Supplemental Fig. S3C). Therefore, our results indicate that the MIG-10B N-terminal motif is necessary, but not sufficient, to instruct presynaptic enrichment of MIG-10B.
MIG-10B subcellular localization to presynaptic sites is necessary for AIY synaptic vesicle clustering
The localization of MIG-10B to presynaptic sites and its requirement downstream from Netrin for synaptic vesicle clustering suggest that MIG-10B subcellular localization is important for instructing AIY synaptic vesicle clustering in response to Netrin.
To test this hypothesis, we examined whether the MIG-10B N terminus, which is required for MIG-10B subcellular localization, was also required for its function in AIY. We used a fosmid containing only the MIG-10B genomic region (WRM0613bB07) and replaced the MIG-10B unique exon with the MIG-10A/C shared exon (Fig. 2A). This “exon swap” construct is different from the recombineered fosmid constructs described earlier. Instead of disrupting the expression of specific MIG-10 isoforms, the “exon swap” fosmid drives MIG-10A protein expression using mig-10b endogenous promoter elements and therefore in tissues that normally express MIG-10B, including AIY (Supplemental Fig. S1E–G). We reasoned that this fosmid would express a functional MIG-10A isoform, which would allow us to assess whether the isoform requirement in AIY is truly dependent on the MIG-10B unique N-terminal domain.
To examine this, we determined whether this transgene was capable of rescuing the mig-10(ct41) presynaptic defect in AIY. We observed that the transgene was not capable of rescuing the AIY presynaptic defect in mig-10(ct41) mutants (Fig. 4A,D). This genomic fragment, however, was capable of rescuing the HSN cell migration and defasciculation defects, suggesting that the construct is functional (Supplemental Fig. S2D,E). These data indicate that expression of functional MIG-10A in AIY is not sufficient to rescue the presynaptic defect, even when under the regulatory elements of MIG-10B. The data also demonstrate that the MIG-10B unique exon, which confers to this isoform the specific capacity to localize to presynaptic regions in response to Netrin, is required for AIY synaptic vesicle clustering. Together, our data strongly support a model in which MIG-10B localizes to presynaptic regions in response to Netrin, and this localization, which is dependent on its unique N-terminal domain, is critical for instructing synaptic vesicle clustering at the Netrin-specified coordinate.
Figure 4.

MIG-10B unique N-terminal helix is required for proper AIY synaptic vesicle clustering. (A) Quantification of the percentage of animals displaying the AIY presynaptic defect. Rescuing fosmids (see Fig. 2A) were expressed in mig-10(ct41) mutants and scored for rescue. (***) P < 0.0001 between indicated groups by Fisher's exact test. Error bars represent 95% confidence interval. (B–D) Distribution of synaptic vesicles in AIY in wild-type (B), mig-10(ct41) mutants (C), or mig-10(ct41) mutants expressing the “exon swap” construct (D). The “exon swap” construct contains the first MIG-10A exon in place of the unique MIG-10B exon, thus expressing MIG-10A under the MIG-10B endogenous promoter (see Fig. 2A). Note that the distribution of synaptic vesicles is disrupted in mig-10(ct41) mutants (shown in C) and is not rescued in D. In all images, the asterisk (*) represents the location of the cell body, and the dashed box encloses zone 2. Bar: B, 5 μm (applies to B–D).
SYD-1 and SYD-2/Liprin α are required for MIG-10B presynaptic localization
MIG-10B localizes to presynaptic sites in response to Netrin, and this subcellular localization is in part mediated through CED-10, which can directly interact with MIG-10 through the Ras association domain (Fig. 1G; Quinn et al. 2008; Stavoe and Colon-Ramos 2012). The Ras association domain is shared by all three isoforms of MIG-10, and CED-10 has also been shown to instruct the polarized localization of MIG-10A to the ventral side of the HSN growth cone in response to Netrin (Adler et al. 2006; Quinn et al. 2008). Therefore, this pathway, although necessary, cannot explain the unique capacity for MIG-10B, and not MIG-10A, to localize to presynaptic regions in response to Netrin.
We hypothesized the existence of a second necessary pathway, which would act in parallel to the Rac GTPase pathway to instruct MIG-10B subcellular localization specifically to presynaptic sites. To examine this hypothesis, we investigated MIG-10B∷GFP localization in syd-1(ju82) and syd-2(ju37) mutants animals. SYD-1 is a component of the active zone, is required for proper localization of presynaptic proteins, and is a regulator of SYD-2. SYD-2 is the nematode homolog of Liprin α, a scaffolding protein that organizes active zone formation (Zhen and Jin 1999; Dai et al. 2006; Patel et al. 2006). Interestingly, we observed that MIG-10B subcellular localization to presynaptic sites is dependent on SYD-1 and SYD-2, as MIG-10B∷GFP displays a significant decrease in enrichment in synaptic regions in syd-1(ju82) and syd-2(ju37) mutant animals (Fig. 5A–C; data not shown).
Figure 5.
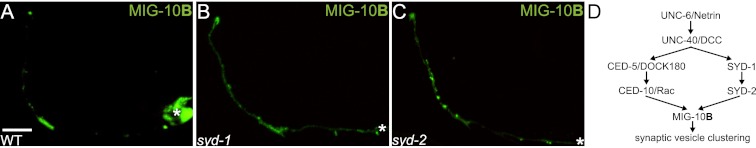
Active zone proteins SYD-1 and SYD-2 are necessary for MIG-10B synaptic localization. (A–C) Confocal micrographs showing MIG-10B∷GFP subcellular localization expressed cell-specifically in AIY in wild-type (A) and syd-1(ju82) (B) and syd-2(ju37) (C) mutant animals. Note that MIG-10B∷GFP is less enriched in zone 2 and is more diffuse in zone 3 in the two mutant backgrounds as compared with wild-type animals. (D) Schematic of the Rac GTPase and active zone protein pathways converging on MIG-10B to instruct MIG-10B localization and synaptic vesicle clustering. In all images, the asterisk (*) represents the location of the cell body. Bar: A, 5 μm (applies to A–C).
We previously demonstrated that mig-10 is not required for the subcellular localization of SYD-1∷GFP and SYD-2∷GFP (Stavoe and Colon-Ramos 2012). In light of those findings and our new observations, we hypothesized that SYD-1 and SYD-2/Liprin-α would act upstream of MIG-10B to instruct its subcellular localization. The Rac GTPase pathway also acts upstream of MIG-10B to instruct its subcellular localization. To better understand the relationship between SYD-1/SYD-2-instructed MIG-10B localization and Rac GTPase-instructed MIG-10B localization, we examined the subcellular localization of CED-5∷GFP in syd-1(ju82) and syd-2(ju37) mutants animals. Consistent with our previous findings, which demonstrated that SYD-1∷GFP and SYD-2∷GFP subcellular localization is not dependent on ced-5 (Stavoe and Colon-Ramos 2012), we also observed that CED-5∷GFP subcellular localization is not dependent on syd-1 or syd-2 (Stavoe and Colon-Ramos 2012; data not shown). Together, our findings suggest that the Rac GTPase pathway and the active zone assembly pathway are acting in parallel downstream from Netrin signaling. Our new observations now indicate that these two parallel pathways converge at the level of MIG-10B, as they are both required to instruct its subcellular localization to presynaptic sites (Fig. 5D).
MIG-10B interacts with ABI-1/ABI1 to organize the actin cytoskeleton at presynaptic sites and instructs synaptic vesicle clustering through synapsin
To better understand the role of MIG-10B in instructing vesicle clustering at the synapse, we next performed a LexA-based yeast two-hybrid analysis to identify proteins that interact with MIG-10B. Out of 73 positive hits, 32 corresponded to ABI-1. ABI-1 is an integral member of the WAVE complex and functions with the ARP2/3 complex to instruct cytoskeletal remodeling (Bompard and Caron 2004; Takenawa and Suetsugu 2007; Schmidt et al. 2009). ABI-1 was also recently shown to directly bind to and interact with the MIG-10A isoform. Furthermore, ABI-1 is required for excretory cell development and axon outgrowth, likely through an interaction with MIG-10 (Quinn and Xu 2012; MA McShea, KL Schmidt, ML Dubuke, CE Baldiga, M Sullender, A Reis, S Zhang, SM O'Toole, MC Jeffers, RM Warden, et al., in prep.). In addition, abi-1 is expressed in AIY (Schmidt et al. 2009). We showed previously that mig-10 is required for proper F-actin localization to and enrichment in AIY presynaptic regions (Fig. 6D,E; Stavoe and Colon-Ramos 2012). Thus, we hypothesized that the interaction between ABI-1 and MIG-10 was required for actin organization and synaptic vesicle clustering in AIY.
Figure 6.

MIG-10B and ABI-1 organize the actin cytoskeleton at presynaptic sites to instruct synaptic vesicle clustering through synapsin. (A–C) Confocal micrographs of GFP∷RAB-3 expressed in AIY in wild-type (A) and abi-1(tm494) (B) and snn-1(tm2557) (C) mutant animals. Note that the presynaptic pattern is disrupted in abi-1(tm494) and snn-1(tm2557) mutants. (D–F) Confocal micrographs of animals expressing the F-actin probe UtrCH∷GFP in AIY in wild-type (D) and mig-10(ct41) (E) and mig-10(ok2499) (F) mutant animals. Note that F-actin is no longer enriched in zone 2 in both mig-10 mutant alleles as compared with wild-type animals. (G–L) Confocal micrographs of a transgenic animal expressing SNN-1A∷GFP (G) and RAB-3∷mCh (H) in AIY. Merge is shown in I. Note that SNN-1A∷GFP and RAB-3∷mCh colocalize in AIY presynaptic regions and are enriched in zone 2 (dashed box). (J–L) In a mig-10(ct41) mutant animal, both SNN-1A∷GFP (J) and RAB-3∷mCh (K) are disrupted and no longer enriched in zone 2. Merge is shown in L. (M) Quantification of penetrance of AIY synaptic vesicle clustering defect in wild-type (n = 318), mig-10(ct41) (n = 141), and abi-1(tm494) (n = 75) mutant animals. Unlike mig-10(ct41) and snn-1(tm2557) alleles, abi-1(tm494) is not a null allele, which likely accounts for the reduced penetrance of presynaptic defect. (***) P < 0.0001 between indicated groups by Fisher's exact test. Error bars represent 95% confidence interval. (N) Transheterozygote analysis. Quantification of penetrance of AIY synaptic vesicle clustering defect in wild-type (n = 318), abi-1(tm494)/+ heterozygote (n = 28), mig-10(ct41)/+ heterozygote (n = 51), snn-1(tm2557)/+ heterozygote (n = 14), abi-1(tm494)/+;mig-10(ct41)/+ transheterozygote (n = 30), snn-1/+;mig-10(ct41)/+ transheterozygote (n = 40), and abi-1(tm494)/+;snn-1(tm2557)/+ transheterozygote (n = 35) animals. Note that transheterozygote animals display significantly higher penetrance of AIY presynaptic defects when compared with wild-type and heterozygote animals. (**) P < 0.01 between indicated groups; (***) P < 0.005 between indicated groups by Fisher's exact test. Error bars represent 95% confidence interval. In all images, the asterisk (*) represents the location of the cell body, and the dashed box encloses zone 2. Bar: A, 5 μm (applies to A–L).
To examine this, we first investigated the requirement of ABI-1 in synaptic vesicle clustering in AIY. Members of the WAVE and ARP2/3 complexes, including ABI-1, are required for viability in C. elegans, and null alleles result in early embryonic lethality (Soto et al. 2002; Withee et al. 2004). Therefore, we examined abi-1(tm494), a weak loss-of-function allele, for defects in AIY synaptic vesicle clustering. We observed that 40% of abi-1(tm494) mutants have reduced synaptic vesicle clustering in zone 2 (n = 75) (Fig. 6B,M). The disrupted synaptic vesicle pattern in abi-1(tm494) mutants is qualitatively similar to that of mig-10 mutants, suggesting that these genes may instruct AIY synaptic vesicle clustering through the same pathway.
To test for a possible genetic interaction between abi-1 and mig-10, we performed transheterozygote analyses. We hypothesized that if ABI-1 and MIG-10 genetically interact to instruct AIY synaptic vesicle clustering, transheterozygote animals would display AIY synaptic patterning defects. Consistent with ABI-1 and MIG-10 interacting in synaptic vesicle clustering, we observed that abi-1/+;mig-10/+ transheterozygote animals displayed synaptic vesicle clustering defects in AIY (Fig. 6N). Together, our data suggest that MIG-10 and ABI-1 interact to instruct AIY synaptic vesicle clustering.
To better understand the link between MIG-10B, the actin cytoskeleton, and vesicle clustering, we next examined whether mig-10 genetically interacts with snn-1/synapsin in synaptic vesicle clustering. Synapsin (SNN-1 in C. elegans) is a vesicle protein that links synaptic vesicles to the presynaptic cytoskeletal matrix by interacting with actin (De Camilli et al. 1983; Bahler and Greengard 1987; Rosahl et al. 1995). In vertebrates, loss of synapsin results in reduced number and density of synaptic vesicles in presynaptic terminals (Ferreira et al. 1995; Takei et al. 1995; Bloom et al. 2003). The only C. elegans synapsin homolog, SNN-1, is most similar to vertebrate synapsin II, which is required for hippocampal synapse formation and regulates the reserve pool of vesicles (Ferreira et al. 1995; Gitler et al. 2008). Thus, we hypothesized that SNN-1 could link synaptic vesicles to the MIG-10B/ABI-1-organized actin cytoskeleton at presynaptic sites. To examine this hypothesis, we first examined AIY synaptic vesicle clustering in snn-1(tm2557) mutant animals. We observed that snn-1(tm2557) mutant animals displayed a synaptic vesicle clustering defect (Fig. 6C). The AIY synaptic vesicle clustering phenotype of snn-1 mutants is qualitatively similar to that of mig-10 mutants, suggesting that these genes could act in the same pathway to instruct synaptic vesicle clustering in AIY.
We then examined whether mig-10, snn-1, and abi-1 genetically interacted by conducting transheterozygote experiments and assaying synaptic vesicle clustering in AIY. Similar to mig-10(ct41) and abi-1(tm494), snn-1(tm2557) is a recessive mutation, and heterozygote animals display predominantly wild-type phenotypes. However, mig-10/+;snn-1/+ and abi-1/+;snn-1/+ transheterozygote animals display synaptic vesicle clustering defects in AIY (Fig. 6N). These findings indicate that mig-10, snn-1, and abi-1 genetically interact to instruct synaptic vesicle clustering in AIY and are consistent with our hypothesis that SNN-1 links synaptic vesicles to the MIG-10B/ABI-1-organized actin cytoskeleton at presynaptic sites.
To further examine this hypothesis, we then examined GFP∷SNN-1 subcellular localization in AIY. Consistent with SNN-1 association with synaptic vesicles, we observed that GFP∷SNN-1 localizes to synaptic regions in AIY and colocalized with the vesicle-associated protein mCh∷RAB-3 (Fig. 6G–I). We hypothesized that if SNN-1 links synaptic vesicles to the MIG-10B/ABI-1-organized actin cytoskeleton, then SNN-1 would act downstream from MIG-10 in synaptic vesicle clustering. To test this hypothesis, we examined SNN-1 localization in mig-10(ct41) mutant animals and observed that SNN-1 enrichment at presynaptic sites is dependent on mig-10 (Fig. 6J–L). Also consistent with SNN-1 acting downstream from MIG-10 in synaptic vesicle clustering, we observed that MIG-10B localization to presynaptic sites is not significantly different in snn-1(tm2557) mutants as compared with wild-type animals (data not shown). Together, our data suggest that MIG-10B interacts with ABI-1 to organize the actin cytoskeleton, which then instructs synaptic vesicle clustering through SNN-1/Synapsin.
AIY-specific overexpression of the MIG-10B unique N terminus disrupts synaptic vesicle clustering and alters thermotactic behavior
Our findings suggest a model in which the MIG-10B N terminus mediates a key interaction necessary for the subcellular localization of MIG-10B. MIG-10B subcellular localization is required for organizing the actin cytoskeleton and vesicle clustering at presynaptic sites. Thus, we hypothesized that overexpression of the MIG-10B N terminus would interfere with endogenous MIG-10B localization and function, acting as a dominant negative. To examine this hypothesis, we expressed the MIG-10B N terminus in AIY and assayed full-length MIG-10B∷GFP subcellular localization. Indeed, we observed that overexpression of the MIG-10B N terminus disrupts the localization of the full-length MIG-10B∷GFP to presynaptic sites, suggesting that the MIG-10B N terminus interferes with endogenous MIG-10B localization (Fig. 7A,B).
Figure 7.

Overexpression of the MIG-10B N terminus disrupts AIY synaptic vesicle clustering and thermotactic behavior. (A) Confocal micrograph showing the subcellular localization of MIG-10B∷GFP in wild-type animals overexpressing MIG-10B N-terminal helix. Note that overexpression of the MIG-10B N-terminal helix affects MIG-10B∷GFP localization (cf. Fig. 1I). (B) Quantification of MIG-10B∷GFP distribution in AIY presynaptic zone 2 in wild-type animals (n = 49) and wild-type animals overexpressing the MIG-10B N-terminal helix (n = 45). (***) P < 0.0001 between indicated groups by unpaired t-test. Error bars represent SEM (C,D) F-actin organization in AIY in wild-type animals overexpressing MIG-10A in AIY (C) or the MIG-10B N-terminal helix in AIY (D). Note that overexpression of the MIG-10B N-terminal helix disrupts F-actin enrichment in zone 2. (E,F) Synaptic vesicle distribution in AIY in wild-type animals overexpressing the MIG-10A N terminus in AIY (E) or the MIG-10B N-terminal helix in AIY (F). Note that overexpression of the MIG-10B N-terminal helix disrupts synaptic vesicle clustering in AIY. (G) Quantification of the percentage of animals displaying a disrupted presynaptic pattern in AIY in wild-type animals (n = 67) and wild-type animals overexpressing MIG-10A (n = 108) or the MIG-10B N-terminal helix (n = 60). (***) P < 0.0001 between indicated groups by Fisher's exact test. Error bars represent 95% confidence interval. (H) Thermotaxis (TTX) index of wild-type animals (n = 5 independent behavioral assays) and wild-type animals overexpressing MIG-10A (n = 5 independent behavioral assays) or the MIG-10B N-terminal helix (n = 6 independent behavioral assays). In each assay, ∼20 animals were tracked for 1 h on a thermotaxis gradient, and their behavior was quantified (see the Materials and Methods). Note that overexpression of the MIG-10B N-terminal helix results in abnormal thermotaxis behavior. (*) P < 0.05 between the MIG-10B N-terminal helix and controls by Fischer individual and Tukey simultaneous confidence intervals. Error bars represent SEM.
We then examined whether disruption of MIG-10B localization also affected F-actin organization at presynaptic sites (Fig. 6D–F; Stavoe and Colon-Ramos 2012). We observed that F-actin enrichment in AIY zone 2 was disrupted in the presence of the MIG-10B dominant-negative construct but was normal when the MIG-10A construct was expressed in AIY (Fig. 7C,D). Thus, our data are consistent with our hypothesis that MIG-10B subcellular localization is necessary to instruct actin organization at AIY presynaptic sites.
We next investigated whether cell-specific expression of the dominant-negative MIG-10B N terminus also affected synaptic vesicle clustering in AIY. Consistent with our model, we observed that overexpression of the MIG-10B N terminus disrupted synaptic vesicle clusters in AIY (Fig. 7F,G). In addition, we determined that expression of the dominant-negative construct in mig-10(ct41) mutants did not enhance the synaptic defects observed in the dominant negative alone (Supplemental Fig. S3C–E). Importantly, we also observed that overexpression of the N terminus of MIG-10A, an isoform that does not localize to presynaptic sites and is not required for synaptic vesicle clustering, did not affect GFP∷RAB-3 localization to presynaptic regions (Fig. 7E,G). Our data are consistent with the MIG-10B N-terminal motif acting as a dominant negative, disrupting endogenous MIG-10B localization and function in actin organization and vesicle clustering at presynaptic sites.
Unlike mig-10(ct41) mutants, in which neurodevelopment is disrupted in multiple cellular contexts, AIY-specific expression of the MIG-10B dominant-negative fragment in wild-type animals affects only AIY. This provided an opportunity to examine the functional consequence of the AIY synaptic vesicle clustering defect in vivo and with single-cell resolution. The AIY interneuron is critical for thermotaxis behavior, and cell-specific ablation of AIY results in abnormal negative thermotaxis (movement down temperature gradients) (Mori and Ohshima 1995; Hobert et al. 1997). To examine the functional consequence of the observed defects, we expressed the dominant-negative MIG-10B N-terminal fragment cell-specifically in AIY and assayed the thermotaxis behavior as previously described (Ryu and Samuel 2002; Ramot et al. 2008). Interestingly, we observed that expression of the MIG-10B N terminus, but not of the MIG-10A N terminus, disrupted the thermotaxis behavior in a manner consistent with AIY loss of function (Fig. 7H; Mori and Ohshima 1995; Hobert et al. 1997). Animals expressing the MIG-10B N-terminal fragment also exhibited reduced positive thermotaxis up temperature gradients toward their cultivation temperature (Ramot et al. 2008; data not shown). These data are consistent with our model and demonstrate a functional consequence for the AIY synaptic vesicle clustering phenotype. To our knowledge, this is the first report of in vivo cell-specific disruption of synaptic vesicle clustering resulting in a specific behavioral perturbation in an animal.
Discussion
Chemotrophic factors such as Netrin instruct diverse neurodevelopmental outcomes (Hedgecock et al. 1990; Ishii et al. 1992; Gitai et al. 2003; Lundquist 2003; Adler et al. 2006; Chang et al. 2006; Quinn et al. 2006, 2008; Colon-Ramos et al. 2007; Poon et al. 2008; Quinn and Wadsworth 2008; Manitt et al. 2009; Xu et al. 2010; Park et al. 2011; Smith et al. 2012; Stavoe and Colon-Ramos 2012; Timofeev et al. 2012). Signaling modules, such as the Rac GTPase pathway and the adaptor protein MIG-10, are required downstream from Netrin for cell migration, axon guidance, axon arborization, and synaptic vesicle clustering (Adler et al. 2006; Chang et al. 2006; Quinn et al. 2006, 2008; Manitt et al. 2009; Stavoe and Colon-Ramos 2012). How does signaling through this common module result in different, cell-specific neurodevelopmental outcomes in response to Netrin? Our findings regarding the requirement of the MIG-10 isoforms downstream from Netrin provide insights into how similar signaling pathways result in distinct cellular responses in vivo.
The mig-10 gene produces three isoforms: MIG-10A, MIG-10B, and MIG-10C. These three isoforms share >80% identity, and these areas of identity include the known functional regions required for modulating the actin cytoskeleton. The three isoforms differ only in small motifs at their N termini. We found here that these motifs confer unique cell-biological properties to the isoforms. For example, we found that while both MIG-10A and MIG-10B respond to UNC-40 during cell migration and axon defasciculation (in HSN), only the MIG-10B isoform is capable of localizing to presynaptic sites in response to Netrin. In AIY, this presynaptic localization is required for accumulation of synaptic vesicles downstream from MIG-10B at the Netrin-specified coordinate. We predict that the other MIG-10 isoforms likely perform unique cell-biological functions as well. In support of this hypothesis, we found that the MIG-10A and MIG-10C isoforms are capable of instructing post-embryonic axon arborization in the NSM neuron, whereas the presynaptically localized MIG-10B isoform is dispensable for this function.
The subcellular localization conferred by the N-terminal motif is critical for the capacity of MIG-10B to regulate synaptic vesicle clustering in response to Netrin. This is perhaps best demonstrated by the inability of the MIG-10A isoform to rescue the mig-10(ct41) presynaptic patterning phenotype in AIY, even when expressed under the control of MIG-10B regulatory elements. Our finding that this same construct does rescue HSN guidance indicates that the MIG-10A construct retains the capacity to organize the actin cytoskeleton during cell migration and axon outgrowth. Nonetheless, this capacity, which resides in the shared region of these isoforms, is not sufficient for synaptic rescue. The fact that this construct cannot rescue mig-10(ct41) mutant AIY synaptic vesicle clustering defects indicates that MIG-10B subcellular localization, conferred in part by the unique N terminus, is essential for its role in organizing synaptic vesicle clustering in response to Netrin.
The N-terminal motif of MRL (MIG-10/RIAM/Lamellipodin) proteins, including that of MIG-10B, is predicted to form an amphipathic α helix. A recent study demonstrated that this motif mediates the interaction of RIAM (a MIG-10 vertebrate ortholog) with Talin (Lee et al. 2009). This interaction is critical for the recruitment of Talin and the subsequent activation of integrins. Although we did not find evidence for a role of Talin in synaptic vesicle clustering in AIY (data not shown), these studies indicate that the N-terminal domain is structurally conserved throughout evolution and that it can mediate important MRL protein functions.
We do not yet know the identity of the element that interacts with the MIG-10B N terminus to drive its subcellular localization to presynaptic sites. We do know, however, that MIG-10B synaptic localization is dependent on both Netrin and active zone proteins SYD-1 and SYD-2. While our data do not indicate whether this localization dependence is mediated by the MIG-10B N terminus or whether MIG-10B directly interacts with active zone proteins, our cell-biological data indicate the existence of a genetic interaction necessary for MIG-10B subcellular localization. Together with previous findings, our study suggests that the Rac GTPase pathway and the active zone assembly pathway converge to instruct MIG-10B subcellular localization at presynaptic sites. However, it should be noted that the mig-10(ct41);syd-1(ju82) phenotype is enhanced as compared with the single-mutant phenotypes (Stavoe and Colon-Ramos 2012). Therefore, SYD-1 and SYD-2 are likely required for other MIG-10-independent roles in synaptic vesicle clustering. Together, our findings support a model in which MIG-10B interacts with a synaptic element via its N-terminal motif, and this interaction is critical for MIG-10B function and its unique subcellular localization in response to Netrin.
Consistent with this model, we found that we can disrupt MIG-10 function in AIY by cell-specifically expressing just the N terminus of the MIG-10B isoform. We hypothesize that this construct impedes full-length MIG-10B interaction with a partner crucial for its presynaptic localization. Consistent with this hypothesis, we observed a significant disruption of full-length MIG-10B localization in AIY neurons overexpressing the MIG-10B N terminus. This AIY-specific disruption of MIG-10B localization results in mislocalization of RAB-3 and F-actin, reminiscent of the mig-10(ct41) mutant phenotype (Fig. 7D,F,G). Furthermore, we demonstrated that this synaptic vesicle disruption is functionally significant, as these otherwise wild-type animals display perturbed thermotaxis behavior. Importantly, we did not detect any cell-biological or functional consequence from overexpression of the MIG-10A N terminus. We show that disruption of synaptic vesicle clustering, with single-cell resolution and in vivo, results in a specific behavioral perturbation in the animal. Our findings underscore the importance of the MIG-10B isoform in instructing F-actin accumulation and synaptic vesicle clustering downstream from Netrin and provide a functional consequence for the synaptic vesicle clustering defect in AIY.
To elucidate how MIG-10B instructs AIY synaptic vesicle clustering, we performed a yeast two-hybrid screen with full-length MIG-10B. The major interaction partner from the screen was ABI-1/Abi1, a component of the WAVE complex. We show that ABI-1 genetically interacts with MIG-10B to direct actin organization at presynaptic sites and synaptic vesicle clustering through SNN-1/Synapsin. Our findings underscore the role of MIG-10B in organizing the actin cytoskeleton at presynaptic sites in response to Netrin signaling and the importance of the actin cytoskeleton in synaptic vesicle clustering. It is important to note that the MIG-10 pathway is likely only one of the modules downstream from Netrin that direct AIY presynaptic assembly, and MIG-10 does so by instructing vesicle clustering at the synapse.
ABI-1 was also recently identified as a MIG-10A interactor, indicating that the interaction between MIG-10B and ABI-1 is not isoform-specific. ABI-1 functions with MIG-10 during guided cell migration, cellular morphogenesis, and asymmetric axon outgrowth during guidance (Quinn and Xu 2012; MA McShea, KL Schmidt, ML Dubuke, CE Baldiga, M Sullender, A Reis, S Zhang, SM O'Toole, MC Jeffers, RM Warden, et al., in prep.). All of these functions are likely mediated by ABI-1 interaction with the ARP2/3 complex and actin polymerization. Together, these discoveries indicate that different MIG-10 isoforms can modulate actin organization through an interaction with ABI-1. These findings are consistent with the conserved capacity of MRL proteins to modulate cytoskeletal dynamics in both vertebrates and invertebrates. Therefore, our study supports a model in which the N-terminal motif of each MIG-10 isoform drives its subcellular localization in response to Netrin. This subcellular localization of MIG-10 then instructs actin reorganization in distinct cellular compartments and results in different neurodevelopmental outcomes in response to Netrin.
Material and methods
Strains and genetics
Worms were raised on NGM plates at 20°C using OP50 Escherichia coli as a food source. N2 Bristol was used as the wild-type reference strain. The following mutant strains were obtained through the Caenorhabditis Genetics Center: ced-10(n3246)IV, unc-40(e271)I, unc-6(ev400)X, mig-10(ct41)III, and ced-5(n1812)IV. The mig-10(ct41) lesion corresponds to an early stop codon in the second mig-10 shared exon (in MIG-10B, Q101* glutamine-to-amber stop codon) and affects all three mig-10 isoforms (Manser et al. 1997).
Molecular biology and transgenic lines
See the Supplemental Material for strain and cloning information. Other than fosmids, all AIY expression constructs allowed for AIY-specific expression using the ttx-3 promoter.
Recombineering was performed as per Dolphin and Hope (2006), Sarov et al. (2006), Zhang et al. (2008), and Tursun et al. (2009). Recombineering strains were obtained from the Biological Resource Branch at NCI (http://web.ncifcrf.gov/research/brb/recombineeringInformation.aspx) or provided by the Alfred Fisher laboratory (University of Pittsburgh) or the Oliver Hobert laboratory (Columbia University Medical Center).
Fluorescence microscopy and confocal imaging
Images of fluorescently tagged fusion proteins (eGFP, GFP, and mCherry with excitation wavelengths of 488 and 561 nm) were captured at room temperature in live C. elegans using a 60× CFI Plan Apo VC, NA 1.4, oil objective on an UltraView VoX spinning-disc confocal microscope on a NikonTi-E stand (PerkinElmer) with a Hammamatsu C9100-50 camera. Images were acquired and processed with Volocity software (Improvision by Perkin Elmer). All images are maximal projections (“Extended Focus” in Volocity). Final processing of images, including rotations and cropping, was conducted with Adobe Photoshop CS4 (Adobe Systems Inc.). All quantifications were performed on maximal projections of raw data. Worms were immobilized using 10 mM levamisole (Sigma) and oriented with anterior to the left and dorsal up.
Quantification
To quantify the enrichment of MIG-10B∷GFP in AIY, we first identified extrachromosomal transgenic lines that displayed stable expression of MIG-10B∷GFP (for example, olaEx324). To ensure consistency across experiments, one extrachromosomal line for each construct was crossed into the specified mutant backgrounds. Quantification was completed by using the line-scan function of ImageJ and tracing the AIY process. Quantifications were performed blindly, without knowledge of the genotype. The fluorescence intensities in zones 2 and 3 were determined. To measure the fluorescence intensity in zone 2, zone 2 was morphologically defined in adult animals as the region of the AIY process that encompasses the dorsal turn as the process exits the anterior VNC and enters the nerve ring (∼5 μm) (boxed in all figures). This anatomical definition of zone 2 was informed by the reconstructions of electron microscopy (EM) micrographs in White et al. (1986). To determine the average fluorescence intensity in zone 3, zone 3 was morphologically defined as the region of the AIY neurite immediately dorsal to the zone 2 region extending to the end of the dorsal midline. Total fluorescence intensity of the zone 2 region was divided by total fluorescence intensity of both zones 2 and 3, yielding MIG-10B∷GFP enrichment in zone 2.
Quantification of rescue of AIY synaptic vesicle clustering in mig-10(ct41) mutants was performed by scoring the GFP∷RAB-3 localization pattern in AIY zone 2. AIY presynaptic defect was defined as a lack of GFP∷RAB-3 enrichment in zone 2.
Quantification of cell migration of HSNL was achieved by collecting confocal micrographs of mutants and wild-type L4 animals and measuring the distance from the center of vulva to the near edge of the HSNL cell body. In wild-type animals, >90% of the HSNL cell bodies are within 20 μm of the vulva (data not shown). Therefore, distances from the vulva >20 μm were scored as abnormal HSN cell migration. Defasciculation was quantified by measuring the distance from the top-most segment of the HSNL axon to the VNC. Significance was determined using a two-tailed unpaired t-test.
Quantification of the axon arborization defect in the NSM was performed by collecting confocal micrographs of mutant and wild-type adult neurons and manually counting the number of primary branches extending from the ventral NSM neurite.
Secondary structure prediction
The secondary structure of the N-terminal unique exons of MIG-10 isoforms was predicted using PBIL analysis algorithms DSC and MLRC and PHD programs (Rost et al. 1994; Geourjon and Deleage 1995; Deleage et al. 1997). Helical wheel analysis was performed as described (Lee et al. 2009).
Yeast two-hybrid assay
The yeast two-hyrbid screen of MIG-10B full-length cDNA bait was performed by Dualsystems Biotech AG. To build the bait construct for screening, the full-length MIG-10B cDNA was originally amplified from a mixed-stage population and cloned into the vector pLexA-DIR (Dualsystems Biotech AG). The bait construct was transformed into the strain NMY32 [MATa his3Δ200 trp-1-901 leu2-3,112 (lexAop)8-ADE2 LYS2∷(lexAop)4-HIS3 URA∷(lexAop)8-lacZ GAL4] using standard procedures (Gietz and Woods 2001). Correct expression of MIG-10B bait was verified by Western blotting of cell extracts using a mouse monoclonal antibody directed against the LexA domain (Dualsystems Biotech AG). The absence of self-activation was verified by cotransformation of the bait with a control prey and selection on minimal medium lacking the amino acids tryptophan, leucine, and histidine (selective medium). For the yeast two-hybrid screen, the MIG-10B bait was cotransformed with a C. elegans cDNA library into NMY32.We screened 3,100,000 transformants, yielding 96 transformants that grew on selective medium. Positive transformants were tested for β-galactosidase activity using a PXG β-galactosidase assay (Dualsystems Biotech AG). Seventy-three of the 96 initial positive transformants showed β-galactosidase activity and were considered to be true positives. Library plasmids were isolated from true positive clones, and their identity was determined by sequencing.
Thermotaxis assay
C. elegans strains were grown and maintained as described by Sulston and Hodgkin (1988). For the thermotaxis assays, the tested strains were grown in a Tritech research incubator at 25°C.
A linear temperature gradient assay ranging from 18°C to 22°C was established on agar surfaces by placing bioassay dishes (Fisher 241 × 241 × 20 mm) containing 200 mL of thermotaxis medium (3 g of NaCL, 17 g L−1 Bacto agar, 25 mM KPO4) in the middle of temperature-controlled rectangular aluminum plates. The temperatures at the ends of the aluminum plates were adjusted using thermoelectric controllers (TECs; 5 cm2, 226 W). The TECs were thermostatically controlled using a circuit consisting of TEC temperature controllers (Ferrotec FTC100) with the support of a bidirectional power control and H-bridge amplifiers (Ferrotec FTX700). The temperature on the surface was measured using RTD probes (McMaster-Carr, #6568T46) that regulate the TECs. A 5-MP monochrome CMOS camera (BCE-B050-U, Mightex Systems) equipped with a megapixel lens with a 2/3-in format and an 8-mm focal length and F1.4 F-stop (M0814-MP, Computar) was used to image the worms. Worms were illuminated using superbright red-light-emitting diodes arranged over a square (the diode frame being 16.25 × 16.25 in, with the diodes themselves being 14.75 × 14.75) to produce a dark-type illumination. Video images were captured at 2 frames per second (fps) using the included Mightex software (Mightex Cam Demo version 1.2.1) (Ryu and Samuel 2002).
Fifteen to 25 young adult worms were washed in 10 mL of NGM buffer at room temperature and transferred in a 2-μL droplet along the center of the agar surfaces once the linear gradients were established. After waiting 5 min to ensure that the worms had escaped the droplets, video images of the plate were captured for 1 h at 2 fps.
After the assays were completed, the plates were subdivided in eight equal-sized regions, and the final positions of the animals were scored. The thermotaxis index was calculated using the previously described formula
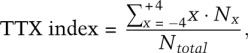 |
where Nx is the number of animals in each region x (x = −4 ∼ +4), and Ntotal is the total number of animals on a test plate (Sugi et al. 2011).
Statistical analyses
Statistical significance for categorical data was determined using Fischer's exact test. Error bars for categorical data were calculated using 95% confidence intervals. For Figure 7B, statistical significance for continuous data was determined by Fischer's exact test because there were only two conditions. Error bars for continuous data were calculated using standard errors of the mean (SEM). For Figure 3A, statistical significance for continuous data was determined using one-way ANOVA with post hoc analysis by Fischer individual and Tukey simultaneous confidence intervals using Minitab16 software.
Acknowledgments
We thank the Caenorhabditis Genetic Center for strains. Recombineering reagents and advice were kindly provided by NCI BRB Preclinical Repository, O. Hobert, B. Tursun, C. Dolphin, A. Fisher, and Y. Zhang. We thank Z. Altun (http://www.wormatlas.org) for diagrams used in the figures. In particular, we thank K. Shen for helpful discussions, generous sharing of advice, and reagents. We also thank C. Gao, G. Chatterjee, and N. Cook for technical assistance, and M. Hammarlund, S. Margolis, C. Smith, M. Hurwitz, and members of the Colón-Ramos laboratory for thoughtful comments on the manuscript. This work was funded by the following grants to D.A.C.-R.: R00 NS057931, a fellowship from the Klingenstein Foundation and the Alfred P. Sloan Foundation, and a March of Dimes Research Grant. A.K.H.S. was supported by Cellular and Molecular Biology Training Grant T32-GM007223; J.C.N. was supported by Interdepartmental Neuroscience Program Training Grant T32-NS41228; L.A.M.V. was supported by a diversity supplement to R00 NS057931. A.D.T.S. and M.K. were supported by the NSF and NIH Pioneer Award. A.K.H.S. and D.A.C.R. designed the cell-biological and genetic experiments and analyzed the data. A.K.H.S. performed the experiments. J.C.N. and A.K.H.S. generated cloning and recombineering constructs and generated transgenic lines. L.A.M.V., A.K.H.S., D.A.C.R., M.K., and A.D.T.S. designed, performed, and analyzed behavior experiments. J.C.N. performed the NSM experiments. A.K.H.S., J.C.N., and D.A.C.R. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.193409.112.
References
- Adler CE, Fetter RD, Bargmann CI 2006. UNC-6/Netrin induces neuronal asymmetry and defines the site of axon formation. Nat Neurosci 9: 511–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura T, Ogura K, Goshima Y 2007. UNC-6 expression by the vulval precursor cells of Caenorhabditis elegans is required for the complex axon guidance of the HSN neurons. Dev Biol 304: 800–810 [DOI] [PubMed] [Google Scholar]
- Bahler M, Greengard P 1987. Synapsin I bundles F-actin in a phosphorylation-dependent manner. Nature 326: 704–707 [DOI] [PubMed] [Google Scholar]
- Bloom O, Evergren E, Tomilin N, Kjaerulff O, Low P, Brodin L, Pieribone VA, Greengard P, Shupliakov O 2003. Colocalization of synapsin and actin during synaptic vesicle recycling. J Cell Biol 161: 737–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bompard G, Caron E 2004. Regulation of WASP/WAVE proteins: Making a long story short. J Cell Biol 166: 957–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin A, Torii M, Wang L, Rakic P, Levitt P 2007. Serotonin modulates the response of embryonic thalamocortical axons to netrin-1. Nat Neurosci 10: 588–597 [DOI] [PubMed] [Google Scholar]
- Chang C, Adler CE, Krause M, Clark SG, Gertler FB, Tessier-Lavigne M, Bargmann CI 2006. MIG-10/lamellipodin and AGE-1/PI3K promote axon guidance and outgrowth in response to slit and netrin. Curr Biol 16: 854–862 [DOI] [PubMed] [Google Scholar]
- Colon-Ramos DA 2009. Synapse formation in developing neural circuits. Curr Top Dev Biol 87: 53–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colon-Ramos DA, Margeta MA, Shen K 2007. Glia promote local synaptogenesis through UNC-6 (netrin) signaling in C. elegans. Science 318: 103–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Taru H, Deken SL, Grill B, Ackley B, Nonet ML, Jin YS 2006. SYD-2 Liprin-α organizes presynaptic active zone formation through ELKS. Nat Neurosci 9: 1479–1487 [DOI] [PubMed] [Google Scholar]
- De Camilli P, Harris SM Jr, Huttner WB, Greengard P 1983. Synapsin I (Protein I), a nerve terminal-specific phosphoprotein. II. Its specific association with synaptic vesicles demonstrated by immunocytochemistry in agarose-embedded synaptosomes. J Cell Biol 96: 1355–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleage G, Blanchet C, Geourjon C 1997. Protein structure prediction. Implications for the biologist. Biochimie 79: 681–686 [DOI] [PubMed] [Google Scholar]
- Desai C, Garriga G, Mcintire SL, Horvitz HR 1988. A genetic pathway for the development of the Caenorhabditis-elegans HSN motor neurons. Nature 336: 638–646 [DOI] [PubMed] [Google Scholar]
- Dolphin CT, Hope IA 2006. Caenorhabditis elegans reporter fusion genes generated by seamless modification of large genomic DNA clones. Nucleic Acids Res 34: e72 doi: 10.1093/nar/gkl1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Han HQ, Greengard P, Kosik KS 1995. Suppression of synapsin II inhibits the formation and maintenance of synapses in hippocampal culture. Proc Natl Acad Sci 92: 9225–9229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcet C, Stein E, Pays L, Corset V, Llambi F, Tessier-Lavigne M, Mehlen P 2002. Netrin-1-mediated axon outgrowth requires deleted in colorectal cancer-dependent MAPK activation. Nature 417: 443–447 [DOI] [PubMed] [Google Scholar]
- Forrester WC, Garriga G 1997. Genes necessary for C-elegans cell and growth cone migrations. Development 124: 1831–1843 [DOI] [PubMed] [Google Scholar]
- Geourjon C, Deleage G 1995. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci 11: 681–684 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods RA 2001. Genetic transformation of yeast. BioTechniques 30: 816–831 [DOI] [PubMed] [Google Scholar]
- Gitai Z, Yu TW, Lundquist EA, Tessier-Lavigne M, Bargmann CI 2003. The netrin receptor UNC-40/DCC stimulates axon attraction and outgrowth through enabled and, in parallel, Rac and UNC-115/AbLIM. Neuron 37: 53–65 [DOI] [PubMed] [Google Scholar]
- Gitler D, Cheng Q, Greengard P, Augustine GJ 2008. Synapsin IIa controls the reserve pool of glutamatergic synaptic vesicles. J Neurosci 28: 10835–10843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR 2003. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell 113: 657–670 [DOI] [PubMed] [Google Scholar]
- Hedgecock EM, Culotti JG, Hall DH 1990. The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron 4: 61–85 [DOI] [PubMed] [Google Scholar]
- Hobert O, Mori I, Yamashita Y, Honda H, Ohshima Y, Liu Y, Ruvkun G 1997. Regulation of interneuron function in the C. elegans thermoregulatory pathway by the ttx-3 LIM homeobox gene. Neuron 19: 345–357 [DOI] [PubMed] [Google Scholar]
- Holt LJ, Daly RJ 2005. Adapter protein connections: The MRL and Grb7 protein families. Growth Factors 23: 193–201 [DOI] [PubMed] [Google Scholar]
- Hopker VH, Shewan D, Tessier-Lavigne M, Poo M, Holt C 1999. Growth-cone attraction to netrin-1 is converted to repulsion by laminin-1. Nature 401: 69–73 [DOI] [PubMed] [Google Scholar]
- Ishii N, Wadsworth WG, Stern BD, Culotti JG, Hedgecock EM 1992. Unc-6, a laminin-related protein, guides cell and pioneer axon migrations in C-Elegans. Neuron 9: 873–881 [DOI] [PubMed] [Google Scholar]
- Lee HS, Lim CJ, Puzon-McLaughlin W, Shattil SJ, Ginsberg MH 2009. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J Biol Chem 284: 5119–5127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey FJ 1999. Netrins and netrin receptors. Cell Mol Life Sci 56: 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist EA 2003. Rac proteins and the control of axon development. Curr Opin Neurobiol 13: 384–390 [DOI] [PubMed] [Google Scholar]
- Manitt C, Nikolakopoulou AM, Almario DR, Nguyen SA, Cohen-Cory S 2009. Netrin participates in the development of retinotectal synaptic connectivity by modulating axon arborization and synapse formation in the developing brain. J Neurosci 29: 11065–11077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser J, Wood WB 1990. Mutations affecting embryonic cell migrations in Caenorhabditis elegans. Dev Genet 11: 49–64 [DOI] [PubMed] [Google Scholar]
- Manser J, Roonprapunt C, Margolis B 1997. C. elegans cell migration gene mig-10 shares similarities with a family of SH2 domain proteins and acts cell nonautonomously in excretory canal development. Dev Biol 184: 150–164 [DOI] [PubMed] [Google Scholar]
- Ming G, Song H, Berninger B, Inagaki N, Tessier-Lavigne M, Poo M 1999. Phospholipase C-γ and phosphoinositide 3-kinase mediate cytoplasmic signaling in nerve growth cone guidance. Neuron 23: 139–148 [DOI] [PubMed] [Google Scholar]
- Ming G, Henley J, Tessier-Lavigne M, Song H, Poo M 2001. Electrical activity modulates growth cone guidance by diffusible factors. Neuron 29: 441–452 [DOI] [PubMed] [Google Scholar]
- Mori I, Ohshima Y 1995. Neural regulation of thermotaxis in Caenorhabditis elegans. Nature 376: 344–348 [DOI] [PubMed] [Google Scholar]
- Park J, Knezevich PL, Wung W, O'Hanlon SN, Goyal A, Benedetti KL, Barsi-Rhyne BJ, Raman M, Mock N, Bremer M, et al. 2011. A conserved juxtacrine signal regulates synaptic partner recognition in Caenorhabditis elegans. Neural Dev 6: 28 doi: 10.1186/1749-8104-6-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MR, Lehrman EK, Poon VY, Crump JG, Zhen M, Bargmann CI, Shen K 2006. Hierarchical assembly of presynaptic components in defined C. elegans synapses. Nat Neurosci 9: 1488–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon VY, Klassen MP, Shen K 2008. UNC-6/netrin and its receptor UNC-5 locally exclude presynaptic components from dendrites. Nature 455: 669–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn CC, Wadsworth WG 2008. Axon guidance: Asymmetric signaling orients polarized outgrowth. Trends Cell Biol 18: 597–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn CC, Xu Y 2012. MIG-10 functions with ABI-1 to mediate the UNC-6 and SLT-1 axon guidance signaling pathways. PLoS Genet (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn CC, Pfeil DS, Chen E, Stovall EL, Harden MV, Gavin MK, Forrester WC, Ryder EF, Soto MC, Wadsworth WG 2006. UNC-6/netrin and SLT-1/slit guidance cues orient axon outgrowth mediated by MIG-10/RIAM/lamellipodin. Curr Biol 16: 845–853 [DOI] [PubMed] [Google Scholar]
- Quinn CC, Pfeil DS, Wadsworth WG 2008. CED-10/Rac1 mediates axon guidance by regulating the asymmetric distribution of MIG-10/lamellipodin. Curr Biol 18: 808–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramot D, MacInnis BL, Lee HC, Goodman MB 2008. Thermotaxis is a robust mechanism for thermoregulation in Caenorhabditis elegans nematodes. J Neurosci 28: 12546–12557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren XR, Ming GL, Xie Y, Hong Y, Sun DM, Zhao ZQ, Feng Z, Wang Q, Shim S, Chen ZF, et al. 2004. Focal adhesion kinase in netrin-1 signaling. Nat Neurosci 7: 1204–1212 [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Sudhof TC 1995. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375: 488–493 [DOI] [PubMed] [Google Scholar]
- Rost B, Sander C, Schneider R 1994. PHD—an automatic mail server for protein secondary structure prediction. Comput Appl Biosci 10: 53–60 [DOI] [PubMed] [Google Scholar]
- Round J, Stein E 2007. Netrin signaling leading to directed growth cone steering. Curr Opin Neurobiol 17: 15–21 [DOI] [PubMed] [Google Scholar]
- Ryu WS, Samuel ADT 2002. Thermotaxis in Caenorhabditis elegans analyzed by measuring responses to defined thermal stimuli. J Neurosci 22: 5727–5733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salie R, Niederkofler V, Arber S 2005. Patterning molecules; multitasking in the nervous system. Neuron 45: 189–192 [DOI] [PubMed] [Google Scholar]
- Sanes JR, Yamagata M 2009. Many paths to synaptic specificity. Annu Rev Cell Dev Biol 25: 161–195 [DOI] [PubMed] [Google Scholar]
- Sarov M, Schneider S, Pozniakovski A, Roguev A, Ernst S, Zhang Y, Hyman AA, Stewart AF 2006. A recombineering pipeline for functional genomics applied to Caenorhabditis elegans. Nat Methods 3: 839–844 [DOI] [PubMed] [Google Scholar]
- Schmidt KL, Marcus-Gueret N, Adeleye A, Webber J, Baillie D, Stringham EG 2009. The cell migration molecule UNC-53/NAV2 is linked to the ARP2/3 complex by ABI-1. Development 136: 563–574 [DOI] [PubMed] [Google Scholar]
- Shen K, Bargmann CI 2003. The immunoglobulin superfamily protein SYG-1 determines the location of specific synapses in C. elegans. Cell 112: 619–630 [DOI] [PubMed] [Google Scholar]
- Shen K, Cowan CW 2010. Guidance molecules in synapse formation and plasticity. Cold Spring Harb Perspect Biol 2: a001842 doi: 10.1101/cshperspect.a001842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Watson JD, VanHoven MK, Colon-Ramos DA, Miller DM 3rd 2012. Netrin (UNC-6) mediates dendritic self-avoidance. Nat Neurosci 15: 731–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HJ, Poo MM 1999. Signal transduction underlying growth cone guidance by diffusible factors. Curr Opin Neurobiol 9: 355–363 [DOI] [PubMed] [Google Scholar]
- Soto MC, Qadota H, Kasuya K, Inoue M, Tsuboi D, Mello CC, Kaibuchi K 2002. The GEX-2 and GEX-3 proteins are required for tissue morphogenesis and cell migrations in C. elegans. Genes Dev 16: 620–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavoe AK, Colon-Ramos DA 2012. Netrin instructs synaptic vesicle clustering through Rac GTPase, MIG-10, and the actin cytoskeleton. J Cell Biol 197: 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugi T, Nishida Y, Mori I 2011. Regulation of behavioral plasticity by systemic temperature signaling in Caenorhabditis elegans. Nat Neurosci 14: 984–992 [DOI] [PubMed] [Google Scholar]
- Sulston J, Hodgkin J 1988. Methods. In The nematode Caenorhabditis elegans (ed. WB Wood), pp. 587–606. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Sze JY, Zhang SY, Li J, Ruvkun G 2002. The C-elegans POU-domain transcription factor UNC-86 regulates the tph-1 tryptophan hydroxylase gene and neurite outgrowth in specific serotonergic neurons. Development 129: 3901–3911 [DOI] [PubMed] [Google Scholar]
- Takei Y, Harada A, Takeda S, Kobayashi K, Terada S, Noda T, Takahashi T, Hirokawa N 1995. Synapsin I deficiency results in the structural change in the presynaptic terminals in the murine nervous system. J Cell Biol 131: 1789–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenawa T, Suetsugu S 2007. The WASP–WAVE protein network: Connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol 8: 37–48 [DOI] [PubMed] [Google Scholar]
- Timofeev K, Joly W, Hadjieconomou D, Salecker I 2012. Localized netrins act as positional cues to control layer-specific targeting of photoreceptor axons in Drosophila. Neuron 75: 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tursun B, Cochella L, Carrera I, Hobert O 2009. A toolkit and robust pipeline for the generation of fosmid-based reporter genes in C. elegans. PLoS ONE 4: e4625 doi: 10.1371/journal.pone.0004625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Guirland C, Ming GL, Zheng JQ 2004. A CaMKII/calcineurin switch controls the direction of Ca(2+)-dependent growth cone guidance. Neuron 43: 835–846 [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S 1986. The structure of the nervous system of the nematode Caenorhanditis elegans. Philos Trans R Soc Lond 314: 1–340 [DOI] [PubMed] [Google Scholar]
- Williams ME, de Wit J, Ghosh A 2010. Molecular mechanisms of synaptic specificity in developing neural circuits. Neuron 68: 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winberg ML, Mitchell KJ, Goodman CS 1998. Genetic analysis of the mechanisms controlling target selection: Complementary and combinatorial functions of netrins, semaphorins, and IgCAMs. Cell 93: 581–591 [DOI] [PubMed] [Google Scholar]
- Withee J, Galligan B, Hawkins N, Garriga G 2004. Caenorhabditis elegans WASP and Ena/VASP proteins play compensatory roles in morphogenesis and neuronal cell migration. Genetics 167: 1165–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Goldman JS, Rymar VV, Forget C, Lo PS, Bull SJ, Vereker E, Barker PA, Trudeau LE, Sadikot AF, et al. 2010. Critical roles for the netrin receptor deleted in colorectal cancer in dopaminergic neuronal precursor migration, axon guidance, and axon arborization. Neuroscience 169: 932–949 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Nash L, Fisher AL 2008. A simplified, robust, and streamlined procedure for the production of C. elegans transgenes via recombineering. BMC Dev Biol 8: 119 doi: 10.1186/1471-213X-8-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen M, Jin Y 1999. The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature 401: 371–375 [DOI] [PubMed] [Google Scholar]