

Abstract
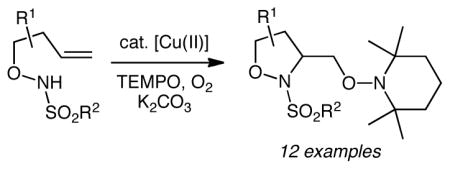
Isoxazolidines are useful in organic synthesis, drug discovery and chemical biology endeavors. A new stereoselective synthesis of methyleneoxy-substituted isoxazolidines is disclosed. The method involves copper-catalyzed aminooxygenation/cyclization of N-sulfonyl-O-butenyl hydroxylamines in the presence of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl radical (TEMPO) and O2, and provides substituted isoxazolidines in excellent yields and diastereoselectivities. We also demonstrate selective mono N-O reduction followed by oxidation of the remaining N-O bond to reveal a 2-amino-γ-lactone. Reduction of the γ-lactone reveals the corresponding aminodiol.
Isoxazolidines have demonstrated a range of biological activities including antibiotic,1 gene expression regulation2 and cancer cell cytotoxicity.3 While [3+2] dipolar cycloaddition reactions are most frequently used to synthesize isoxazolidines,4,5 the intramolecular addition of hydroxylamines onto alkenes and allenes has emerged as an important alternative strategy.6,7 These two synthetic strategies are largely complementary: the former is usually an entirely intermolecular process while the latter is intramolecular in the first step of the addition to the alkene. Because of these different aspects, different stereocontrolling elements are involved in each. Herein we report a copper-catalyzed addition of a nitrogen and oxygen across the alkene, resulting in methyleneoxy-functionalized isoxazolidines.
Our lab has recently disclosed that copper(II) salts can be used to efficiently promote and catalyze intramolecular alkene aminooxygenation.8 This reaction has been used to synthesize methyleneoxy functionalized indolines, pyrrolidines and cyclic ureas. The present contribution broadens the scope of the copper-catalyzed alkene aminooxygenation reaction to the synthesis of isoxazolidines. The resulting methyleneoxy functionalized isoxazolidines might serve directly as pharmacological leads or alternatively N-O bond reduction/oxidation could lead to synthetically useful 2-amino-γ-lactones and 1,3-aminoalcohols (vide infra).
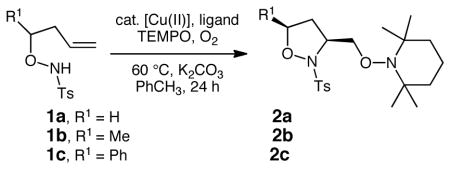
The N-sulfonyl-O-butenyl hydroxyl amine substrates 1 were synthesized via aldehyde allylation, subsequent Mitsunobu reaction with N-hydroxyphthalimide, N-deprotection and N-sulfonylation, as previously reported.6,7 Substrates 1a-c were then subjected to various copper(II)-catalyzed aminooxygenation conditions to determine the optimal protocol (Table 1). We found the reaction of N-tosyl-O-butenyl hydroxyl amine 1a to be very efficient at 60 °C when Cu(OTf)2 was used with the 2,2-isopropylidinebis(oxazoline) ligand 3 or when Cu(2-ethylhexanoate)2 [Cu(eh)2] was used as the catalyst (Table 1, entries 3 and 4). [We generally prefer Cu(2-ethylhexanoate)2 in our copper-carboxylate promoted and catalyzed reactions due to its high solubility in non-polar organic solvents.]8b,d Substrate 1a is significantly more reactive than the γ-pentenyl sulfonamide substrates we had previously examined in this aminooxygenation reaction8 and 1a actually suffered substantial decomposition at 110 and 120 °C, the temperatures previously examined substrates reacted best at.8 The oxygen source in this aminooxygenation reaction is (2,2,6,6-tetramethylpiperidin-1-yl)oxyl radical (TEMPO) and the stoichiometric oxidant is O2 (1 atm, balloon). We initially used 3 equiv of TEMPO based on our previously reported aminooxygenation reactions,8 but we found midway during these studies that 1.2 equiv of TEMPO is sufficient for excellent conversion. We also found that a 10% O2 in N2 atmosphere (1 atm, balloon) is sufficient for good conversion (Table 1, entry 6), thereby enabling a safer process.
Table 1.
Optimization of the aminooxygenationa
| |||||
|---|---|---|---|---|---|
| entry | R1 | CuX2 | ligand | yield (%) | cis/trans |
| 1b | H | Cu(OTf)2 | -- | 60c | -- |
| 2b | H | Cu(OTf)2 | Bipy | 55c | -- |
| 3 | H | Cu(OTf)2 | 3 | >95c | -- |
| 4b | H | Cu(eh)2 | -- | 83 | -- |
| 5 | H | Cu(eh)2 | -- | 81 | -- |
| 6d | H | Cu(eh)2 | -- | 70 | -- |
| 7e | H | Cu(eh)2 | -- | 65 (90c) | -- |
| 8b | Me | Cu(OTf)2 | 3 | 95c | 3:1 |
| 9b | Me | Cu(eh)2 | -- | 75 | 10:1 |
| 10b | Ph | Cu(OTf)2 | 3 | 95c | 8:1 |
| 11b | Ph | Cu(eh)2 | -- | 83 | 10:1 |
| 12 | Ph | Cu(eh)2 | -- | 82 | 10:1 |
Reaction conditions: 1 (0.165 mmol), K2CO3 (0.165 mmol) and TEMPO (0.198 mmol, 1.2 equiv) was added to a solution of CuX2 (0.033 mmol, 20 mol%) [Cu(OTf)2 and ligands (25 mol%) were complexed at 60 °C for 2 h prior to substrate addition] and the reaction mixture was heated at 60 °C under O2 (1 atm, balloon) for 24 h. Yields are of product isolated after flash chromatography on SiO2 unless otherwise noted.
3 equiv of TEMPO was used in this reaction.
% Conversion based on crude 1H NMR analysis.
10% O2 in N2 (1 atm, balloon) was used.
15 mol% Cu(eh)2 was used.
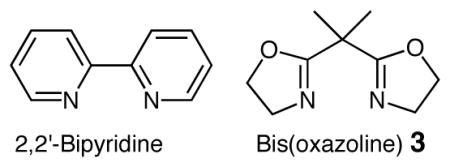
Substrates with α-substituents (1b, R1 = Me and 1c, R1 = Ph) provided moderate 3,5-cis isoxazolidine diastereoselectivity with the Cu(OTf)2•3 catalyst and higher diastereoselectivity with Cu(eh)2 as catalyst (Table 1, entries 8–12). The relative configuration of the major diastereomers were assigned by nOe.
A series of N-sulfonyl-O-butenylamines 1 were subjected to the optimal conditions (Table 1, entries 4 and 5) to establish the substrate scope (Scheme 1). A number of functional groups (aryl-Br, aryl-Cl, thiophene, Ns, SES, allyl) were stable to the reaction conditions. α-Substituted hydroxylamines provided the corresponding isoxazolidines 2d-j and 2l with good to excellent 3,5-cis diastereoselectivity while a β-phenyl substrate provided the 3,4-trans diastereomer 2k selectively (stereochemistry assigned by nOe). Substrates with internal alkenes and ones that could potentially form 6-membered rings were not examined in this limited study.
Scheme 1. Isoxazolidine Scope.

R3 = 2,2,6,6-tetramethylpiperidinyl
The proposed reaction mechanism is outlined in Scheme 2.8 Cis-aminocupration via the seven-membered chair-like transition state A gives the unstable organocopper(II) intermediate. For the diastereoselective reactions, placement of the substituent in a less sterically demanding pseudo-equatorial position leads to the major diasteromer. Homolysis of the carbon-copper bond gives Cu(I) and the carbon radical intermediate then undergoes rapid trapping with TEMPO radical to provide isoxazolidine 2. Reoxidation of Cu(I) to Cu(II) by TEMPO/O2 completes the catalytic cycle.
Scheme 2.

Proposed Reaction Mechanism
While isoxazolidines are useful in their own right, they can also be easily converted to 1,3-amino alcohols.9 The ring N-O bond of isoxazolidine 2c was selectively cleaved via hydrogenation, catalyzed by 20 mol% Pearlman’s catalyst [20% (by weight) Pd(OH)2 on carbon] to provide the acyclic 1,3-amino alcohol 4 in 70% yield along with 10% recovered 2c (Scheme 3).9c We initially anticipated that oxidation of amino alcohol 4 with m-CPBA8a,b would provide the corresponding lactol. Instead, γ-lactone 5 was obtained exclusively. Lactone 5 could be reduced to the corresponding 1,4-diol 6 with NaBH4. Such γ-lactones and 3-amino-1,4-diols are important intermediates in the synthesis of HPA-12, a CERT antagonist that can be used as a treatment for sphingolipid misregulation which in turn can cause autoimmune disorders.10
Scheme 3.

N-O bond reduction and oxidation
Summary and Conclusions
A very concise, efficient and diastereoselective method for the synthesis of variously functionalized isoxazolidines has been presented. Such compounds may prove useful in drug discovery and organic synthesis endeavors. This method could provide access to disubstituted chiral isoxazolidines if the synthetic route to the hydroxylamine substrate involved an enantioselective step,6f,6g e.g. an enantioselective aldehyde allylation reaction.11 Sequential N-O reduction and N-O oxidation reactions as shown in Scheme 3 can provide unnatural amino acid synthons such as γ-lactone 5 and structures related to CERT antagonists such as aminodiol 6.
Experimental Section
General Experimental Information
Reagents were used as supplied unless otherwise noted. m-CPBA was 70–75% composition by weight. 1H NMR spectra were recorded in CDCl3, C6D6 or CD3OD (referenced at 7.26 ppm, residual CHCl3, 7.16 ppm, residual C6H6 and 3.34 ppm, residual CH3OH) at 300, 400 or 500 MHz. 13C NMR spectra were recorded in CDCl3 (77.0 ppm as internal reference) at 75.5 MHz. Characteristic peaks in the IR spectra are reported in wave numbers, cm−1. Melting points are reported as uncorrected. Reactions were run in oven-dried glassware under Argon unless otherwise noted. Solvents were dried by filtration through alumina.
N-(but-3-enyloxy)-4-methylbenzenesulfonamide (1a) (Representative Procedure)6,7
A 0 °C solution of 3-butene-1-ol (500.0 mg, 6.9 mmol), triphenylphosphine (2.2 g, 8.3 mmol) and N-hydroxyphthalimide (1.3 g, 8.3 mmol) in THF (56 mL) was treated with diethylazodicarboxylate (3.6 mL, 8.3 mmol, 40 wt% in toluene), added over 30 min. The reaction was warmed to rt and stirred for 4 h, then hydrazine monohydrate (0.77 mL, 15.8 mmol) was added drop-wise. After 2 h, the mixture was filtered through Celite and concentrated in vacuo. The resulting hydroxylamine was dissolved in CH2Cl2 (69 mL), treated with pyridine (1.7 mL, 20.8 mmol) and p-TsCl (1.6 g, 8.3 mmol) and stirred for 16 h at rt. The mixture was quenched with water and extracted with CH2Cl2 (2×30 mL). The organics were washed with 1M HCl (20 mL), brine (20 mL), dried over Na2SO4 and concentrated in vacuo. Flash chromatography on SiO2 (10–30% EtOAc in hexanes) gave N-tosyl hydroxylamine 1a (600 mg, 30% yield, yellow oil). 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J=8.0 Hz, 2 H), 7.35 (d, J=8.0 Hz, 2 H), 5.78-5.74 (m, 1 H), 5.13-5.05 (m, 2 H), 4.05 (t, J=12.0 Hz, 2 H), 2.46 (s, 3 H), 2.46-2.34 (m, 2 H); 13C NMR (75.5 MHz, CDCl3) δ 144.7, 134.1, 133.4, 129.5, 128.5, 116.9, 76.1, 32.4, 21.5; IR (thin film) 3222, 2946, 1642, 1598, 1335, 1164 cm−1; HRMS (ESI) calcd for [M+Na]+ C11H15O3N1Na1S1: 264.0666, found: 264.0665.
(±)-4-methyl-N-(pent-4-en-2-yloxy)benzenesulfonamide (1b)
Pent-4-en-2-ol was converted to 1b (326 mg, 22% yield, clear oil). 1H NMR (300 MHz, CDCl3) δ 7.80 (d, J=8.1 Hz, 2H), 7.32 (d, J=8.1 Hz, 2H), 6.99 (s, 1H), 5.80-5.68 (m, 1H), 5.08-5.03 (m, 2H), 4.21-4.15 (m, 1H), 2.43 (s, 3H), 2.39-3.30 (m, 1H), 2.25-2.18 (m, 1H), 1.18 (d, J=9.5 Hz, 3H); 13C NMR (75.5 MHz, CDCl3) δ 144.7, 133.9, 133.6, 129.5, 128.5, 117.4, 81.8, 39.1, 21.6, 18.2; IR (thin film) 3222, 1314, 1167, 1091 cm−1; HRMS (EI) calcd for [M]+ C12H17O3N1S1: 255.09152, found: 255.0924.
(±)-4-methyl-N-(1-phenylbut-3-enyloxy)benzenesulfonamide (1c)7
1-Phenyl-but-3-en-1-ol12 was converted to 1c (481.2 mg, 42% yield, white solid). m.p. = 105 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J=8.4 Hz, 2 H), 7.36-7.28 (m, 5H), 7.28-7.25 (m, 2H), 6.67 (s, 1H), 5.80-5.71 (m, 1H), 5.10-5.01 (m, 3H), 2.68-2.60 (m, 1H), 2.49 (s, 3H), 2.48-2.45 (m, 1H); 13C NMR (75.5 MHz, CDCl3) δ 144.8, 139.6, 133.8, 133.7, 129.6, 128.7, 128.4, 128.2, 127.1, 117.6, 88.0, 39.6, 21.6; IR (Thin Film) 3224, 1642, 1598, 1339, 1166 cm−1; HRMS (ESI) calcd for [M+Na]+ C17H19O3N1S1: 340.0978, found: 340.0975.
(±)-N-(1-(4-chlorophenyl)but-3-enyloxy)-4-methylbenzenesulfonamide (1d)
1-(4-Chlorophenyl)-but-3-en-1-ol12 was converted to 1d (1.5 g, 81% yield, white solid). mp = 107–110 °C; 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J=8.0 Hz, 2 H), 7.33-7.28 (m, 4H), 7.19 (d, J=8.5 Hz, 2H), 6.77 (s, 1H), 5.76-5.68 (m, 1H), 5.08-4.99 (m, 3H), 2.65-2.58 (m, 1H), 2.45 (s, 3H), 2.45-2.41 (m, 1H); 13C NMR (75.5 MHz, CDCl3) δ 144.9, 138.1, 133.9, 133.5, 133.2, 129.6, 128.6, 128.6, 128.5, 117.9, 87.2, 39.5, 21.6; IR (thin film) 3189, 1643, 1592, 1333, 1158, 913 cm−1; HRMS (ESI) calcd for [M+Na]+ C17H18O3N1S1Cl1Na1: 374.0588, found: 374.0586.
(±)-N-(1-(4-chlorophenyl)but-3-enyloxy)-2-(trimethylsilyl)ethanesulfonamide (1e)
1-(4-Chlorophenyl)-but-3-en-1-ol12 was converted to 1e (104 mg, 26% yield, white solid) as above except with 2-trimethylsilylethanesulfonyl chloride. mp = 92–94 °C; 1H-NMR (400 MHz, CDCl3) δ 7.35 (d, J= 8.4 Hz, 2H), 7.30 (d, J= 8.8 Hz, 2 H), 6.72 (s, 1H), 5.83-5.76 (m, 1H), 5.12-5.06 (m, 2H), 5.00 (dd, J=6.4 Hz, J= 7.6 Hz, 1 H), 3.15-3.08 (m, 2H), 2.68-2.64 (m, 1H), 2.49-2.46 (m, 1H), 1.05-0.89 (m, 2H), 0.07 (s, 9H); 13C NMR (75.5 MHz, CDCl3) δ 137.9, 134.1, 133.2, 128.6, 128.5, 117.9, 86.9, 45.5, 39.5, 9.7, 0.0, IR (thin film) 3223, 2923, 2852, 1597, 1338, 1167, 1091, 919, 813, 705; HRMS (ESI) calcd for [M+Na]+ C15H24Cl1NO3S1Si1Na1: 384.0827, found: 384.0832.
(±)-N-(1-(4-chlorophenyl)but-3-enyloxy)-4-nitrobenzenesulfonamide (1f)
1-(4-Chlorophenyl)-but-3-en-1-ol12 was converted to 1f (126 mg, 30% yield, yellow solid) as above except with 4-nitrobenzenesulfonyl chloride. mp = 127–129 °C; 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J= 8.8 Hz, 2H), 8.11 (d, J=8.8 Hz, 2H), 7.32 (d, J= 8.4 Hz, 1H), 7.20 (d, J=8 Hz, 2H), 6.95 (s, 1H), 5.78-5.69 (m, 1H), 5.14-5.04 (m, 3H), 2.66-2.58 (m, 1H), 2.49-2.42 (m, 1H); 13C NMR (75.5 MHz, CDCl3), δ 150.8, 142.2, 137.5, 134.5, 133.1, 130.0, 128.8, 128.5, 124.2, 118.3, 88.9, 39.4; IR (thin film) 3224, 1606, 1531, 1348, 1312, 1173, 1089, 1013, 854, 744 cm−1; HRMS (EI) calcd for [M-C3H5]+ C13H10N2O5Cl1S1: 340.9977, found: 340.9993.
(±)-4-nitro-N-(pent-4-en-2-yloxy)benzenesulfonamide (1g)
Pent-4-en-2-ol was converted to sulfonamide 1g (1.20 g, 64% yield, white solid) as above except with 4-nitrobenzenesulfonyl chloride. mp = 72–75 °C; 1H NMR (500 MHz, CDCl3) δ 8.38 (d, J=8.5 Hz, 2H), 8.12 (d, J=9.0 Hz, 2H), 7.14 (s, 1H), 5.79-5.74 (m, 1H), 5.10-5.06 (m, 2H), 4.27-4.23 (m, 1H), 2.36-2.32 (m, 1H), 2.28-2.24 (m, 1H), 1.21 (d, J=6.5 Hz, 3 H); 13C NMR (75.5 MHz, CDCl3) δ 150.7, 142.3, 133.7, 129.9, 124.1, 117.8, 82.6, 39.1, 30.9, 18.2; IR (thin film) 3212, 1608, 1535, 1445, 1348, 1167 cm−1; HRMS (EI) calcd for [M]+ C11H14O5N2S1: 286.0625, found: 286.0618.
(±)-4-methyl-N-(1-(thiophen-2-yl)but-3-enyloxy)benzenesulfonamide (1h)7
1-(Thiophen-2-yl)but-3-en-1-ol12 was converted to 1h (90 mg, 44% yield, yellow solid). mp = 80–82 °C; 1H NMR (400 MHz, CDCl3), δ 7.72 (d, J= 8.4 Hz, 2H), 7.24 (d, J= 8.0 Hz, 2H), 7.19 (t, J= 5.2 Hz, 1H), 6.98 (d, J= 8.4 Hz, 1H), 6.88 (dd, J= 3.4 Hz, 5.0 Hz, 1H), 6.75 (s, 1H), 5.76-5.66 (m, 1H), 5.18 (t, J= 7.0 Hz, 1H), 5.07-5.01 (m, 2H), 2.70-2.63 (m, 1H), 2.54-2.46 (m, 1H), 2.36 (s, 3H); 13C NMR (75.5 MHz, CDCl3) δ 144.8, 142.2, 133.6, 133.2, 129.6, 128.6, 127.1, 126.6, 125.8, 118.0, 83.2, 39.6, 29.6, 21.6; IR(thin film) 3223, 2923, 1339, 1167, 1091, 920, 813, 705 cm−1; HRMS (ESI) calcd for [M+Na]+ C15H17O3N1Na1S2: 346.0542, found: 346.0526.
(±)-N-(1-(4-bromophenyl)but-3-enyloxy)-4-methylbenzenesulfonamide (1i)
1-(4-Bromophenyl)-but-3-en-1-ol12 was converted to 1i (1.2 g, 70% yield, white solid). mp = 115–118 °C; 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J=8.5 Hz, 2H), 7.45 (d, J=8.0 Hz, 2H), 7.32 (d, J=8.0 Hz, 2H), 7.13 (d, J=8.5 Hz, 2H), 6.75 (s, 1H), 5.76-5.69 (m, 1H), 5.08-5.04 (m, 2H), 5.00 (t, J=5.6 Hz, 1H), 2.62-2.58 (m, 1H), 2.45 (s, 3H), 2.45-2.39 (m, 1H); 13C NMR (75.5 MHz, CDCl3) δ 144.9, 138.6, 133.5, 133.2, 131.6, 129.6, 128.9, 128.6, 122.2, 118.0, 87.3, 39.4, 21.7; IR (thin film) 3213, 1597, 1166, 1091, 732 cm−1; HRMS (ESI) calcd for [M+Na]+ C17H18O3N1S1Br1Na1: 420.0063, 418.0088, found: 420.0051; 418.0109.
(±)-N-(hepta-1,6-dien-4-yloxy)-4-methylbenzenesulfonamide (1j)
Hepta-1,6-dien-4-ol13 was converted to 1j (162 mg, 26% yield, white solid). mp = 52–54 °C; 1H NMR (400 MHz, CDCl3), 7.81 (d, J= 8.0 Hz, 2H), 7.32 (d, J= 8.0 Hz, 2H), 6.84 (s, 1H), 5.84-5.73 (m, 1H), 5.10-5.05 (m, 4H), 4.20-4.16 (t, J = 6.0 Hz, 1H), 2.44 (s, 3H), 2.35-2.32 (m, 4H); 13C NMR (75.5 MHz, CDCl3) δ 144.8, 133.9, 133.6, 129.6, 128.4, 117.6, 85.0, 41.0, 36.6, 21.7; IR (thin film), 3220, 2929, 1642, 1334, 1168, 1091, 1019, 918, 814, 731 cm−1; HRMS (ESI) calcd. for [M+Na]+ C14H19O3N1Na1S1: 304.0987, found: 304.0978.
(±)-4-methyl-N-(2-phenylbut-3-enyloxy)benzenesulfonamide (1k)
2-Phenyl-but-3-en-1-ol14 was converted to 1k (120 mg, 56% yield, clear oil). 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J= 8.0 Hz, 2H), 7.37-7.19 (m, 7H), 6.93 (s, 1 H), 6.02-5.90 (m, 1 H), 5.16-5.07 (m, 2H), 4.31 (d, J= 10.0 Hz, 2H), 3.76-3.68 (m, 1H), 2.42 (s, 3H); 13C NMR (75.5 MHz, CDCl3) δ 144.7, 140.3, 137.8, 133.3, 129.5, 128.5, 128.4, 128.0, 126.8, 116.5, 79.7, 48.2, 21.6; IR (thin film) 3223, 2960, 1453, 1336, 1167, 1091, 918, 757 cm−1; HRMS (ESI) calcd for [M+Na]+ C17H19O3N1Na1S1: 340.0969, found: 340.0978.
(±)-4-Methyl-N-(3-methyl-1-phenylbut-3-enyloxy)benzenesulfonamide (1l)
3-Methyl-1-phenylbut-3-en-1-ol15 was converted to 1l (105 mg, 51% yield, yellow solid). mp = 83–85 °C; 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J= 8.0 Hz, 2H), 7.33-7.27 (m, 7H), 6.62 (s, 1H), 5.15 (dd, J = 5.6 Hz, 8.0 Hz, 1H), 4.83 (s, 1H), 4.73 (s, 1H), 2.59 (dd, J = 8.4 Hz, 14.4 Hz, 1H), 2.41 (s, 3 H), 2.35 (dd, J= 5.2 Hz, 14.4 Hz, 1H), 1.81 (s, 3H); 13C NMR (75.5 MHz, CDCl3) δ 144.7, 141.3, 140.0, 133.7, 129.6, 128.7, 127.1, 113.5, 88.2, 44.1, 23.4, 21.7; IR (thin film), 3223, 2923, 1597, 1454, 1339, 1167, 812, 727 cm−1; HRMS (ESI) calcd for [M+Na]+ C18H21O3N1Na1S1: 354.1134, found: 354.1134.
(±)-2,2,6,6-tetramethyl-1-((2-tosylisoxazolidin-3-yl)methoxy)piperidine (2a) (Representative Procedure)
N-tosyl hydroxylamine 1a (40 mg, 0.165 mmol), copper(II) 2-ethylhexanoate (11.5 mg, 0.033 mmol), TEMPO (30.9 mg, 0.198 mmol) and K2CO3 (22.8 mg, 0.165 mmol) were placed in a 100 mL flask (14/20 neck) and dissolved in toluene (1.65 mL). O2 (1atm) was introduced via balloon attached to a short vacuum hose, that in turn was attached to an adapter (14/20). The solution was heated to 60 °C and stirred for 24 h, then was diluted with EtOAc, filtered through Celite and concentrated. Flash chromatography on SiO2 (10–30% EtOAc:hexanes) afforded 51.6 mg of isoxazolidine 2a as a white solid in 79% yield. mp = 101–102 °C; 1H NMR (500 MHz, C6D6) δ 7.98 (d, J=8.0 Hz, 2H), 6.72 (d, J=7.5 Hz, 2 H), 4.58-4.56 (m, 1H), 4.07-4.04 (m, 1H), 4.00 (dd, J= 7.5 Hz, 15.7 Hz, 1H), 3.85 (dd, J= 6.5 Hz, 9.0 Hz, 1H), 3.55-3.51 (m, 1H), 1.97-1.94 (m, 1H), 1.79 (s, 3H), 1.69-1.66 (m, 1H), 1.42-1.40 (m, 4H), 1.28-1.15 (m, 12H); 13C NMR (75.5 MHz, CDCl3) δ 144.8, 133.1, 129.6, 129.1, 77.7, 69.8, 59.9, 57.6, 39.5, 33.0, 31.9, 30.8, 21.6, 20.0, 16.9; IR (thin film) 1361, 1329, 1162, 1088 cm−1; HRMS (ESI) calcd for [M+H]+ C20H33O4N2S1: 397.2156, found: 397.2159.
(±)-2,2,6,6-tetramethyl-1-(((3S,5S)-5-methyl-2-tosylisoxazolidin-3-yl)methoxy)piperidine (2b)
1b was converted to (48 mg, 75% yield, white solid, d.r. = 10:1) 2b as above (except 3 equiv of TEMPO was used). The diastereomers were separated by HPLC (5% EtOAc in hexanes). Major diastereomer: mp = 111–113 °C; 1H NMR (400 MHz, C6D6) δ 8.01 (d, J=8.4 Hz, 2H), 6.73 (d, J=8.8 Hz, 2H), 4.65-4.61 (m, 1H), 4.51-4.46 (m, 1H), 4.20 (dd, J= 6.4 Hz, 9.2 Hz, 1H), 3.96 (dd, J= 6.4 Hz, 9.0 Hz, 1H), 2.12 (ddd, J= 6.0 Hz, 8.0 Hz, 11.1 Hz, 1H), 1.81 (s, 3H), 1.48-1.18 (m, 5H), 1.14-1.13 (m, 12H), 0.91 (d, J= 6.0 Hz, 3H); 13C NMR (75.5 MHz, CDCl3) δ 144.7, 133.4, 129.6, 129.1, 78.3, 77.8, 59.9, 58.4, 39.5. 30.9, 21.6, 18.1, 17.0; IR (film) 1598, 1359, 1333, 1166, 813 cm−1; HRMS (ESI) calcd for [M+H] + C21H35O4N2S1: 411.2312 found: 411.2310.
(±)-2,2,6,6-tetramethyl-1-(((3S,5R)-5-phenyl-2-tosylisoxazolidin-3-yl)methoxy)piperidine (2c)
1c was converted to 2c (49.4 mg, 83% yield, 10:1 dr). The diastereomers were separated by HPLC (5% EtOAc in hexanes). Major diastereomer: white solid, mp = 131–133 °C; 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J=8.4 Hz, 2H), 7.28-7.21 (m, 7H), 5.16 (dd, J=6.4 Hz, 10.0 Hz, 1H), 4.50-4.46 (m, 1H), 4.11-4.08 (dd, J= 6.0 Hz, 9.2 Hz, 1H), 3.88 (dd, J= 6.6 Hz, 9.4 Hz, 1H), 2.76 (ddd, J= 6.4 Hz, 8.2 Hz, 12.4 Hz, 1H), 2.38 (s, 3H), 2.13 (ddd, J= 7.2 Hz, 10.2 Hz, 12.8 Hz, 1H), 1.51-1.39 (m, 4H), 1.15-1.09 (m, 12H); 13C NMR (75.5 MHz, CDCl3) δ 144.8, 137.1, 133.3, 129.6, 129.3, 128.63, 128.57, 126.9, 83.1, 78.2, 60.1, 58.7, 40.5, 39.6, 33.9, 21.7, 20.1, 17.1; IR (thin film) 1598, 1360, 1333, 1164, 813 cm−1; HRMS (ESI) calcd for [M+H]+ C26H37O4N2S1: 473.2469, found: 473.2474.
(±)-1-(((3S,5R)-5-(4-chlorophenyl)-2-tosylisoxazolidin-3-yl)methoxy)-2,2,6,6-tetramethylpiperidine (2d)
1d was converted to 2d (50.8 mg, 64% yield, white solid, >20:1 d.r.) using the conditions for 2b. mp = 128–129 °C; 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J=8.4 Hz, 2H), 7.32-7.21 (m, 6H), 5.21 (dd, J= 6.6 Hz, 9.8 Hz, 1H), 4.55-4.48 (m, 1 H), 4.04 (dd, J= 6.0 Hz, 9.2 Hz, 1H), 3.89 (dd, J= 6.6 Hz, 8.4 Hz, 1H), 2.79 (ddd, J= 6.6 Hz, 8.2 Hz, 12.6 Hz, 1H), 2.43 (s, 3H), 2.12 (ddd, J= 7.2 Hz, 10.0 Hz, 12.2 Hz, 1H), 1.55-1.41(m, 4H), 1.28-1.07 (m, 12H); 13C NMR (75.5 MHz, CDCl3) δ 144.9, 135.7, 134.4, 133.1, 129.2, 128.7, 128.2, 82.3, 78.0, 60.1, 58.6, 40.4, 39.6, 33.0, 30.9, 21.7, 20.1, 17.0; IR (thin film) 1596, 1492, 1360, 1166, 817 cm−1; HRMS (ESI) calcd for [M+H]+ C26H36O4N2S1Cl1: 507.2085, found: 507.2088.
(±)-1-(((3S,5R)-5-(4-chlorophenyl)-2-(2-(trimethylsilyl)ethylsulfonyl)isoxazolidin-3-yl)methoxy)-2,2,6,6-tetramethylpiperidine (2e)
1e was converted to 2e (35.2 mg, 62% yield, white solid) in >20:1 d.r. mp = 88–90 °C; 1H NMR (400 MHz, CDCl3) δ 7.35 (s, 4H), 5.47 (dd, J= 7.2 Hz, 1H), 4.59-4.55 (m, 1H), 4.00 (dd, J= 6.4 Hz, 9.2 Hz, 1H), 3.88 (dd, J= 6.4 Hz, 8.8 Hz, 1H), 3.29-3.13 (m, 2H), 2.85 (ddd, J= 7.2 Hz, 12.0 Hz, 17.8 Hz, 1H), 2.15 (ddd, J= 6.8 Hz, 9.8 Hz, 12.5 Hz, 1H), 1.54-1.44 (m, 4H), 1.19-1.09 (m, 12H), 0.03 (s, 9H); 13C NMR (75.5 MHz, CDCl3) δ 135.8, 134.4, 128.8, 128.3, 82.9, 77.8, 60.1, 56.9, 47.1, 40.0, 39.6, 33.0, 20.1, 16.9, 9.5, 0.0; IR (thin film) 2930, 1599, 1493, 1469, 1333, 1251, 1172, 1148, 1091, 1056, 1015, 860, 830, 736, 701 cm−1; HRMS (ESI) calcd for [M+H]+ C24H42O4N2Cl1S1Si1: 517.2318, found: 517.2335.
(±)-1-(((3S,5R)-5-(4-chlorophenyl)-2-(4-nitrophenylsulfonyl)isoxazolidin-3-yl)methoxy)-2,2,6,6-tetramethylpiperidine (2f)
1f was converted (at 70 °C) to 2f (36.6 mg, 65% yield, white solid, >20:1 d.r.). mp = 158–160 °C; 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J= 8.8 Hz, 2H), 8.18 (d, J= 8.8 Hz, 2H), 7.31 (d, J= 8.4 Hz, 2H), 7.25 (d, J= 8.8 Hz, 2H), 5.41 (dd, J= 6.8 Hz, 1H), 4.69-4.67 (m, 1 H), 4.04 (dd, J= 6.4 Hz, 9.2 Hz, 1H), 3.94 (dd, J= 5.4 Hz, 9.4 Hz, 1H), 2.88 (ddd, J= 8.4 Hz, 12.6 Hz, 17.1 Hz, 1H), 2.16 (ddd, J= 7.2 Hz, 10.0 Hz, 12.6 Hz, 1H), 1.46-1.43 (m, 4H), 1.25-1.14 (m, 12H); 13C NMR (75.5 MHz, CDCl3) δ 150.7, 142.5, 135.1, 134.7, 130.3, 128.9, 128.2, 124.1, 83.0, 77.6, 60.1, 58.3, 39.9, 39.6, 39.9, 32.9, 20.1, 17.0; IR (thin film), 2973, 2930, 1607, 1533, 1493, 1469, 1348, 1310, 1261, 1167, 1132, 1091, 1048, 1014, 955, 910, 854, 739, 622, 620 cm−1; HRMS (ESI) calcd for [M+H]+ C25H33O6N3Cl1S1: 538.1773, found: 538.1759.
(±)-2,2,6,6-tetramethyl-1-(((3S,5S)-5-methyl-2-(4-nitrophenylsulfonyl)isoxazolidin-3- yl)methoxy)piperidine (2g)
1g was converted to 2g (49 mg, 69% yield, white solid, >20:1 d.r.) using the above conditions except at 70 °C with 3 equiv TEMPO. mp = 146–147 °C; 1H NMR (500 MHz, CDCl3) δ 8.37 (d, J=8.5 Hz, 2H), 8.17 (d, J=8.5 Hz, 2H), 4.52-4.45 (m, 2H), 3.95 (dd, J= 6.7 Hz, 9.2 Hz, 1H), 3.83 (dd, J= 5.7 Hz, 9.2 Hz, 1H), 2.60 (ddd, J= 6.5 Hz, 8.0 Hz, 12.2 Hz, 1H), 1.72 (ddd, J= 7.0 Hz, 10.0 Hz, 12.2 Hz, 1H), 1.52-1.43 (m, 4H), 1.25 (s, 3H), 1.22-1.10 (m, 12H); 13C NMR (75.5 MHz, CDCl3) δ 150.6, 142.8, 130.2, 124.0, 78.4, 77.8, 60.0, 58.2, 39.5, 39.0, 33.0, 32.8, 30.9, 18.0, 16.9; IR (thin film) 1607, 1534, 1349, 1263, 1172 cm−1; HRMS (ESI) calcd for [M+H]+ C20H31O6N3S1: 441.1928, found: 441.1932.
(±)-2,2,6,6-tetramethyl-1-(((3S,5R)-5-(thiophen-2-yl)-2-tosylisoxazolidin-3-yl)methoxy)piperidine (2h)
1h was converted to 2h (43.6 mg, 76% yield, white solid, 5.5:1 d.r.). Major diastereomer: mp = 95–97 °C; 1H NMR (300 MHz, CDCl3) δ 7.87 (d, J= 8.1 Hz, 2 H), 7.33 (d, J= 8.1 Hz, 2H), 7.27 (d, J= 7.5 Hz, 1 H), 7.06 (d, J= 3.0 Hz, 1H), 6.95 (t, J= 5.4 Hz, 1H), 5.57 (dd, J= 6.3 Hz, 7.8 Hz, 1H), 4.60-4.56 (m, 1H), 4.08 (dd, J= 6.3 Hz, 8.7 Hz, 1H), 3.93 (dd, J= 6.8 Hz, 9.0 Hz, 1H), 2.92-2.82 (m, 1H), 2.43 (s, 1H), 2.28 (ddd, J= 7.0 Hz, 10.0 Hz, 12.4 Hz, 1H), 1.52-1.38 (m, 4H), 1.25-1.11 (m, 12H); 13C NMR (75.5 MHz, CDCl3) δ 144.8, 139.4, 133.2, 129.6, 129.2, 127.2, 126.7, 126.5, 78.8, 77.9, 60.0, 58.5, 40.3, 39.6, 33.1, 32.9, 21.6, 20.1, 17.0; IR (thin film) 2927, 1451, 1359, 1332, 1162, 1092, 1048, 812, 705, 671; HRMS (ESI) calcd for [M+H]+ C24H35O4N2S2: 479.2026, found: 479.2033.
(±)-1-(((3S,5R)-5-(4-bromophenyl)-2-tosylisoxazolidin-3-yl)methoxy)-2,2,6,6-tetramethylpiperidine (2i)
1i was converted to 2i (51.1 mg, 92% yield, white solid, >20:1 d.r.) using the method for 2b. mp = 137–138 °C; 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.0, 2H), 7.18 (d, J = 8.4 Hz, 2H), 5.21 (dd, J= 6.4 Hz, 9.4 Hz, 1H), 4.55-4.52 (m, 1H), 4.06 (dd, J= 6.0 Hz, 8.8 Hz, 1H), 3.91 (dd, J= 6.6 Hz, 9.0 Hz, 1H), 2.81 (ddd, J= 6.8 Hz, 8.0 Hz, 12.5 Hz, 1H), 2.43 (s, 3H) 2.13 (ddd, J= 7.2 Hz, 9.8 Hz, 11.7 Hz, 1H), 1.45-1.43 (m, 4H), 1.18-1.09 (m, 12H); 13C NMR (75.5 MHz, CDCl3) δ 144.9, 136.3, 133.1, 131.7, 129.6, 129.2, 128.5, 122.5, 82.4, 77.9, 60.1, 58.6, 40.4, 39.6, 33.1, 30.1, 21.7, 20.1, 17.1; IR (thin film) 1596, 1360, 1334, 1163, 671 cm−1; HRMS (ESI) calcd for [M+H]+ C26H36O4N2S1Br1: 551.1574, found: 551.1583.
(±)1-(((3R,5R)-5-allyl-2-tosylisoxazolidin-3-yl)methoxy)-2,2,6,6-tetramethylpiperidine (2j)
1j was converted to 2j (42.5 mg, 65% yield, colorless oil, 15:1 d.r.). The diastereomers were separated by HPLC (10% EtOAc in hexanes). Major diastereomer: 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.1, Hz, 2H), 5.77-5.63 (m, 1H), 5.08-5.05 (m, 2H), 4.42-4.25 (m, 2H), 3.97 (dd, J = 8.0 Hz, 12.2 Hz, 1H), 3.80 (dd, J = 8.0 Hz, 12.2 Hz, 1H), 2.48 (ddd, J = 6.4 Hz, 8.4 Hz, 12.2 Hz, 1H), 2.45 (s, 3H), 2.39-2.33 (m, 1H), 2.28-2.20 (m, 1H), 1.80 (ddd, J= 6.4 Hz, 9.4 Hz, 12.3 Hz, 1H), 1.45-1.43 (m, 4H), 1.16-1.09 (m, 12H); 13C NMR (75.5 MHz, CDCl3) δ 144.7, 133.4, 133.0, 129.6, 129.1, 117.8, 80.8, 78.2, 60.0, 58.1, 39.6, 37.2, 33.1, 32.8, 21.7, 20.1, 17.0; IR (thin film), 2928, 1451, 1334, 1359, 1164, 1056, 918, 813, 671 cm−1; HRMS (ESI) calcd. for [M+H]+ C23H37O4N2S1: 437.2455, found: 437.2469.
(±)-2,2,6,6-tetramethyl-1-(((3S,4R)-4-phenyl-2-tosylisoxazolidin-3-yl)methoxy)piperidine (2k)
1k was converted to 2k (48.8 mg, 82% yield), clear oil, 10:1 d.r. The diastereomers were separated by HPLC (5% EtOAc in hexanes). Major diastereomer: 1H NMR (400 MHz, C6D6) δ 8.05 (d, J= 8.4 Hz, 2H), 7.31 (d, J= 7.2 Hz, 2H), 7.08 (t, J= 7.2 Hz, 2H), 6.99 (t, J= 5.2 Hz, 1H), 6.74 (d, J= 7.6 Hz, 2H), 4.78-4.73 (m, 1H), 4.39-4.36 (m, 1H), 4.32 (dd, J= 6.0 Hz, 10.0 Hz, 1H), 4.24 (dd, J= 6.4 Hz, 10.2 Hz, 1H), 3.84 (t, J= 8.0 Hz, 1H), 3.57-3.50 (m, 1H), 1.81 (s, 3H), 1.44-1.42 (m, 4H), 1.26-1.03 (m, 12H); 13C NMR (75.5 MHz, CDCl3) δ 144.9, 136.4, 133.2, 129.7, 129.3, 128.8, 128.0, 127.6, 65.1, 59.9, 52.3, 39.6, 32.9, 21.7, 20.0, 17.0; IR (thin film) 2919, 1458, 1360, 1338, 1164, 1092, 1052, 908, 732 cm−1; HRMS (ESI) calcd. for [M+H]+ C26H37O4N2S1: 473.2484, found: 473.2469.
(±)-2,2,6,6-tetramethyl-1-(((3S,5S)-3-methyl-5-phenyl-2-tosylisoxazolidin-3-yl)methoxy)piperidine (2l)
1l was converted to 2l (49.5 mg, 85% yield, white solid, 12:1 d.r.) using the conditions for 2f. Major diastereomer: mp = 120–122 °C; 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J= 8.4 Hz, 2H), 7.37-7.23 (m, 7H), 5.54 (t, J= 8.4 Hz, 1H), 3.88 (ABq, ΔνAB = 88.6 Hz, JAB = 8.6 Hz, 2H), (dd, J= 8.0 Hz, 12.2 Hz, 1H), 2.42 (dd, J= 8.0 Hz, 12.2 Hz, 1H), 2.37 (s, 3H), 1.93 (s, 3H), 1.48-1.42 (m, 4H), 1.20-1.16 (m, 12H); 13C NMR (75.5 MHz, CDCl3) δ 144.1, 137.7, 135.9, 129.2, 128.7, 128.4, 128.3, 126.6, 81.5, 70.5, 60.2, 48.5, 39.7, 21.5, 21.3, 20.2, 16.9; IR (thin film) 2926, 1597, 1452, 1331, 1160, 1091, 812, 756, HRMS (ESI) calcd for [M+H]+ C27H39O4N2S1: 487.2623, found: 487.2623.
(±)-N-((2S,4R)-4-hydroxy-4-phenyl-1-(2,2,6,6-tetramethylpiperidin-1-yloxy)butan-2-yl)-4-methylbenzenesulfonamide (4)
A solution of isoxazolidine 2c (40 mgs, 0.08 mmols) and Pearlman’s catalyst [Pd(OH)2 on carbon, wet. Degussa type, 20% (wt.)] (6 mg, 0.017 mmols, 10 mol%) in EtOH (8 mL) was shaken under 3 atm H2 pressure (hydrogenator) for 18 h.9c Additional catalyst (6 mg, 10 mol%) was added and the mixture was shaken for 4 h. The mixture was diluted with EtOAc, filtered through Celite and concentrated under reduced pressure. Flash chromatography (10–60% EtOAc in hexanes) afforded 28 mg (70%) of amino-alcohol 4 (colorless oil). 2c (4 mg, 10%) was recovered. 1H NMR (CDCl3, 400 MHz) δ 7.73 (d, J=8.0 Hz, 2H), 7.32-7.23 (m, 7H), 5.08 (d, J= 8.0 Hz, 1H), 4.68 (t, J= 6.4 Hz, 1H), 3.71 (dd, J= 4.4 Hz, 8.8 Hz, 1H), 3.61 (dd, J= 5.6 Hz, 9.2 Hz, 1H), 3.50-3.46 (m, 1H), 2.39 (s, 3H), 1.98 (t, J= 8.0 Hz, 2H), 1.42-1.35 (m, 4H), 1.03-0.95 (m, 12H); 13C NMR (CDCl3, 75.5 MHz) δ 144.1, 143.4, 137.4, 129.6, 128.5, 127.7, 127.2, 125.8, 77.5, 72.1, 59.9, 51.8, 41.7, 39.6, 32.9, 21.4, 20.1, 16.9; IR (thin film) 3484 (br), 3279, 2928, 1598, 1494, 1453, 1374, 1330, 1159, 1093, 1047, 972, 910, 814, 732, 700, 665 cm−1; HRMS (ESI) calcd for [M+H]+ C26H39N2O4S1: 475.2625; found: 475.2636.
(±)-4-methyl-N-((3S,5R)-2-oxo-5-phenyl-tetrahydrofuran-3-yl)benzenesulfonamide (5)
Sulfonamide 4 (15 mg, 0.032 mmol) in CH2Cl2 (0.3 mL) was treated with m-CPBA (14.3 mg, 0.083 mmol of 77% pure material, 2 equiv), added over 5 min. After being stirred for 5 h, the mixture was quenched with Na2SO3 (aq) and extracted with CH2Cl2 (3 × 10 mL). The organic extracts were washed with brine, dried over Na2SO4 and concentrated in vacuo. Flash chromatography on SiO2 (20–80% EtOAc in hexanes) afforded lactone 5 (6.8 mg, 64% yield) as a waxy solid. 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J= 8.0 Hz, 2H), 7.41-7.21 (m, 7H), 5.69 (d, J= 8.0 Hz, 1H), 5.14 (d, J= 3.2 Hz, 1H), 4.02-3.96 (m, 1H), 2.83 (dd, J= 2.0 Hz, 8.6 Hz, 1H), 2.80 (dd, J= 2.0 Hz, 8.0 Hz, 1H) 2.42 (s, 3H); 13C NMR (75.5 MHz, CDCl3) 173.9, 144.3, 138.1, 135.8, 129.9, 129.0, 128.6, 127.2, 124.7, 78.6, 50.7, 37.6, 21.6; IR (thin film) 3264, 2924, 1786, 1597, 1496, 1452, 1333, 1254, 1161, 1092, 1009, 916 cm−1; HRMS (ESI) calcd. for [M+Na]+ C17H17O4N1Na1S1: 354.0757, found: 354.0771.
N-((2S,4R)-1,4-dihydroxy-4-phenylbutan-2-yl)-4-methylbenzenesulfonamide (6)
Crude 5 [obtained from 4 (58.5 mg, 0.12 mmol) as above] in EtOH (5 mL) and NaBH4 (22.7 mg, 0.6 mmol) were stirred at rt. After 6 h, the mixture was quenched with 5% K2CO3 (aq) (8 mL) and EtOH was removed in vacuo. The mixture was extracted with Et2O (4×20 mL) and the organic extracts were dried over Na2SO4 and concentrated. Flash chromatography on SiO2 (30–90% EtOAc in hexanes) provided amino-diol 6 (22.8 mg) in 56% yield (white solid). mp 125–127 °C; 1H NMR (400 MHz, CD3OD) δ 7.76 (d, J= 8.0 Hz, 2H), 7.39 (d, J= 8.0 Hz, 2H), 7.31-7.23 (m, 3H), 7.18 (d, J= 8.4 Hz, 2H) 4.60 (t, J= 6.8 Hz, 1H), 3.52 (dd, J = 4.0 Hz, 11.2 Hz, 1H), 3.41 (dd, J= 5.6 Hz, 11.2 Hz, 1H), 3.24-3.22 (m, 1H), 2.48 (s, 1H), 1.98-1.95 (m, 1H), 1.81-1.75 (m, 1H); 13C NMR (75.5 MHz, CD3OD) δ 145.7, 144.6, 139.9, 130.7, 129.2, 128.3, 128.1, 127.3, 72.0, 64.9, 54.2, 41.8, 21.5; IR (thin film), 3461, 3288, 2924, 2853, 1454, 1323, 1156, 1093, 1051, 815 cm−1; HRMS ESI calcd. for [M+Na]+ C17H21O4N1Na1S1: 358.1091, found: 358.1084.
Supplementary Material
Acknowledgments
The National Institutes of Health (GM 078383) is gratefully acknowledged for support of this research.
Footnotes
Supporting information: [NMR spectra for all new compounds, NMR spectra supporting stereochemical assignments]. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Nora GP, Miller MJ, Mollmann U. Bioorg Med Chem Lett. 2006;16:3966–3970. doi: 10.1016/j.bmcl.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 2.(a) Lee LW, Taylor CEC, Desaulniers JP, Zhang M, Hojfeldt JW, Pan Q, Mapp AK. Bioorg Med Chem Lett. 2009;19:6233–6236. doi: 10.1016/j.bmcl.2009.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Loh B, Vozzolo L, Mok BJ, Lee CC, Fitzmaurice RJ, Caddick S, Fassati A. Chem Biol Drug Des. 2010;75:461–474. doi: 10.1111/j.1747-0285.2010.00956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rescifina A, Chiacchio U, Corsaro A, Piperno A, Romeo R. Eur J Med Chem. 2011;46:129–136. doi: 10.1016/j.ejmech.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 4.Reviews: Martin JN, Jones RCF. In: Synthetic Application of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products. Padwa A, Pearson WH, editors. Chapter 1. Wiley and Sons; Hoboken, NJ: 2003. p. 1.Gothelf KV, Jorgensen KA. Chem Rev. 1998;98:863–909. doi: 10.1021/cr970324e.Stanley LM, Sibi MP. Chem Rev. 2008;108:2887–2902. doi: 10.1021/cr078371m.
- 5.Selected catalytic enantioselective [3+2] reactions: Evans DA, Song HJ, Fandrick KR. Org Lett. 2006;8:3351–3354. doi: 10.1021/ol061223i.Kano T, Hashimoto T, Maruoka K. J Am Chem Soc. 2005;127:11926–11927. doi: 10.1021/ja0523284.Kanemasa S, Oderaotoshi Y, Tanaka J, Wada E. J Am Chem Soc. 1998;120:12355–12356.Kobayashi S, Kawamura M. J Am Chem Soc. 1998;120:5840–5841.Suga H, Nakajima T, Itho K, Kakehi A. Org Lett. 2005;7:1431–1434. doi: 10.1021/ol050397h.Jen WS, Wiener JJM, MacMillan DWC. J Am Chem Soc. 2000;122:9874–9875.Karlsson S, Hogberg HE. Eur J Org Chem. 2003:2782–2791.Desimoni G, Faita G, Toscanini M, Boiocchi M. Chem Eur J. 2009;15:9674–9677. doi: 10.1002/chem.200900908.Barroso S, Blay G, Munoz MC, Pedro JR. Org Lett. 2011;13:402–405. doi: 10.1021/ol102716e.Chatterjee I, Frohlich R, Studer A. Angew Chem Int Ed. 2011;50:11257–11260. doi: 10.1002/anie.201105515.
- 6.Metal-catalyzed methods: Bates RW, Sa-Ei K. Org Lett. 2002;4:4225–4227. doi: 10.1021/ol026701d.Dongol KG, Tay BY, Xiang K. Synth Comm. 2006;36:1247–1257.Dongo KG, Tay BY. Tetrahedron Lett. 2006;47:927–930.Hay MB, Wolfe JP. Angew Chem Int Ed. 2007;46:6492–6494. doi: 10.1002/anie.200701386.Lemen GS, Giampietro NC, Hay MB, Wolfe JP. J Org Chem. 2009;74:2533–2540. doi: 10.1021/jo8027399.Bates R, Nemeth J, Snell R. Synthesis. 2008:1033–1038.Bates RW, Lu Y. Org Lett. 2010;12:3938–3941. doi: 10.1021/ol1016492.LeLonde RL, Wang ZJ, Mba M, Lackner AD, Toste FD. Angew Chem Int Ed. 2010;49:598–601. doi: 10.1002/anie.200905000.
- 7.A metal-free cyclization: Moriyama K, Izumisawa Y, Togo H. J Org Chem. 2011;76:7249–7255. doi: 10.1021/jo201113r.
- 8.Fuller PH, Kim JW, Chemler SR. J Am Chem Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m.Paderes MC, Chemler SR. Org Lett. 2009;11:1915–1918. doi: 10.1021/ol9003492.Paderes MC, Chemler SR. Eur J Org Chem. 2011:3679–3684. doi: 10.1002/ejoc.201100444.Paderes MC, Belding L, Fanovic B, Dudding T, Keister JB, Chemler SR. Chem Eur J. 2012;18:1711–1726. doi: 10.1002/chem.201101703.Sequeira FC, Bovino MT, Chipre AJ, Chemler SR. Synthesis. 2012;44:1481–1484. doi: 10.1055/s-0031-1289762.(f) The enantioselective desymmetrization of 1j with [Cu(R,R)-Ph-box]OTf2 as catalyst provided 2j in 17% ee.
- 9.N-O bond reduction: Tufariello JJ, Lee GE. J Am Chem Soc. 1980;102:373–374.Cicchi S, Bonanni M, Cardona F, Revuelta J, Goti A. Org Lett. 2003;5:1773–1776. doi: 10.1021/ol034434l.and references therein Degiorgis F, Lombardo M, Trombini C. Synthesis. 1997:1243–1245.
- 10.Duris A, Wiesenganger T, Moravcikova D, Baran P, Kozisek J, Daich A, Berkes D. Org Lett. 2011;13:1642–1645. doi: 10.1021/ol2001057. [DOI] [PubMed] [Google Scholar]
- 11.Yus M, Gonzalez-Gomez JC, Foubelo F. Chem Rev. 2011;111:7774–7854. doi: 10.1021/cr1004474. [DOI] [PubMed] [Google Scholar]
- 12.Taillier C, Hameury T, Bellosta V, Cossy J. Tetrahedron. 2007;63:4472–4490. [Google Scholar]
- 13.Liu K, Aricho JW, Taylor RE. J Org Chem. 2010;75:3953–3957. doi: 10.1021/jo100094d. [DOI] [PubMed] [Google Scholar]
- 14.Lin M, Lin L, Chuang T. Synlett. 2011:1871–1874. [Google Scholar]
- 15.Dussault P, Shchepin R, Xu C. Org Lett. 2010;12:4772–4775. doi: 10.1021/ol1018757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.