

Abstract
Decreased basal glutamate levels are observed in the rat nucleus accumbens (NA) core following cocaine self-administration. This disruption of glutamate homeostasis arises from a reduction in the export of glutamate via system xC− and is accompanied by a decrease in expression of xCT, the catalytic subunit of system xC−. A second hallmark of disrupted homeostasis is a decrease in expression and function of the major glutamate transporter, GLT-1. We have previously shown that chronic treatment with the antibiotic ceftriaxone restores xCT and GLT-1 expression following cocaine self-administration and attenuates both cue- and cocaine-primed reinstatement. Here we used a 3H-glutamate uptake assay and microdialysis to test the hypothesis that ceftriaxone restores the function of both GLT-1 and xCT (glutamate reuptake and export, respectively) in the NA core following cocaine self-administration. We also used electrophysiology to investigate the ability of ceftriaxone to normalize measures of synaptic plasticity following cocaine. We found that 5 d of ceftriaxone treatment following cocaine self-administration restores basal glutamate levels in the accumbens core, likely through an upregulation of system xC− function. We also found that ceftriaxone restores glutamate reuptake and attenuates the increase in synaptically released glutamate that accompanies cocaine-primed reinstatement. Ceftriaxone also reversed the cocaine-induced synaptic potentiation in the accumbens core, evidenced by normalized spontaneous EPSC amplitude and frequency and evoked EPSC amplitude. These data indicate that ceftriaxone normalizes multiple aspects of glutamate homeostasis following cocaine self-administration and thus holds the potential to reduce relapse in human cocaine addicts.
Introduction
Disrupted glutamate homeostasis is observed in the nucleus accumbens (NA) core following cocaine administration. The hallmarks of this homeostatic dysregulation are decreased basal levels of glutamate (Baker et al., 2003; Moussawi et al., 2011), enhanced synaptically released glutamate during cocaine-primed reinstatement (McFarland et al., 2003), and decreased sodium-dependent glutamate uptake accompanied by decreased expression of GLT-1 (Knackstedt et al., 2010). GLT-1 is the major glutamate transporter, accounting for ∼94% of total CNS glutamate uptake (Tanaka et al., 1997). System xC−, which exchanges extracellular cystine for intracellular glutamate in a sodium-independent manner, accounts for the majority of basal extracellular glutamate in the NA core (Baker et al., 2002). Cocaine produces deficits in NA core system xC− function, as well as decreased expression of its catalytic subunit, xCT (Knackstedt et al., 2010).
The β-lactam family of antibiotics increases the expression and function of GLT-1 in vitro and in vivo, with ceftriaxone possessing the greatest potency (Rothstein et al., 2005). GLT-1 upregulation is attributable to an increase in gene transcription through the nuclear factor-κβ signaling cascade (Lee et al., 2008). Ceftriaxone is also a transcriptional regulator of xCT, Nrf2, and thereby increases system xC− activity (Lewerenz et al., 2009). Accordingly, we have shown that ceftriaxone normalizes the expression of xCT and GLT-1 in the NA core following cocaine self-administration and extinction (Knackstedt et al., 2010). Furthermore, ceftriaxone attenuates cue- (Sari et al., 2009; Knackstedt et al., 2010) and cocaine- (Knackstedt et al., 2010) primed reinstatement, an effect that persists weeks after cessation of ceftriaxone treatment (Sondheimer and Knackstedt, 2011).
Here we investigated whether ceftriaxone can restore compromised glutamate transport and basal glutamate levels following cocaine self-administration. We also investigated the ability of ceftriaxone to reverse cocaine-induced alterations in synaptic plasticity by measuring spontaneous and evoked EPSCs in the NA core of animals with a history of cocaine self-administration.
Materials and Methods
Subjects.
Male Sprague Dawley rats (Charles River Laboratories) weighing 250–275 g were housed individually in a temperature-controlled vivarium on a 12 h/12 h reverse light cycle. All experiments were conducted during the dark phase. Animals were provided with ad libitum water and 20 g of food/day. All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. A total of 86 rats were used for this study.
Drugs.
Cocaine hydrochloride was generously donated by the National Institute on Drug Abuse and was dissolved in 0.9% physiological saline. For the purposes of self-administration, a 4 mg/ml solution was prepared and subjects received 0.25 mg/infusion. Cocaine-primed reinstatement testing was conducted using the dose of 10 mg/kg cocaine (i.p.). Ceftriaxone (Nova Plus) was dissolved in saline and administered in a dose of 200 mg/kg.
Surgery.
Rats were anesthetized with ketamine HCl (87.5 mg/kg, i.m.) and xylazine (5 mg/kg, i.m.). Ketorolac (3 mg/kg, i.p.) was administered for analgesia. For catheter implantation, SILASTIC tubing (Dow Corning) was implanted into the jugular vein. The tubing exited the vein and traveled subcutaneously to exit on the back. The tubing was attached to a stainless-steel cannulae that was held within a harness (Instech). For microdialysis, cannulas (20 gauge; Plastics One) were implanted bilaterally, aimed at the NA core [AP, +1.4 mm; ML, ±2.5 mm at 6° angle; DV, −5.0 mm; all coordinates from Paxinos and Watson (2005)]. Catheters were flushed with heparin (100 U/ml) and timentin (25 mg/d) for 10 d after surgery and then heparin alone throughout the remainder of self-administration.
Self-administration and extinction procedures.
Animals self-administered cocaine in a two-lever operant chamber (Med Associates). Yoked-saline controls received saline infusions when their cocaine counterpart received cocaine. Cocaine was available on an FR-1 schedule of reinforcement in which each press on the active lever (always the right lever) resulted in the delivery of 0.25 ml of cocaine, the illumination of the stimulus light above the active lever, and the delivery of a tone (2900 Hz). The light signified the length of the time-out period (20 s) during which presses on the active lever were recorded but did not result in delivery of drug. To be included in the study, rats were required to earn at least 10 infusions/2 h session for 12 d. Animals that failed to meet this criterion were eliminated from the study. Subsequent to the completion of the cocaine self-administration phase of the experiment, animals underwent daily extinction training during which time responses on the previously active lever no longer produced cocaine or drug-paired cues. Animals underwent extinction training for 2–3 weeks before being used in microdialysis, uptake, or electrophysiological studies, as described below.
Microdialysis and HPLC for quantification of glutamate.
Hollow fiber microdialysis tubing was affixed to a 24 gauge cannula (Plastics One). The end of the tubing was sealed with epoxy, leaving 2 mm of active membrane between the glued membrane and the end of the cannula. The other end of the cannulae was threaded with the inlet and outlet tubing made of fused silica.
For microdialysis procedures during reinstatement testing, subjects were implanted with a microdialysis probe and placed into the self-administration chamber the night before testing. Subjects were perfused overnight with artificial CSF (aCSF) composed of the following (in mm): 5 d-glucose, 2.7 KCl, 140 NaCl, 1.2 MgCl2, and 1.4 CaCl2, pH 7.2, at a rate of 0.2 μl/min. Food and water were available overnight. The next morning, the flow rate was increased to 2 μl/min for 2 h followed by collection of 12 10 min baseline samples. Animals received an injection of cocaine (10 mg/kg, i.p.) and the levers were extended for the reinstatement trial. Samples were collected at 10 min intervals during the 2 h reinstatement test, yielding 12 samples.
For no-net flux microdialysis, animals were probed and placed into a Plexiglas chamber overnight, and dialysis was performed as described above. After collection of baseline samples for 2 h, the no-net flux procedure began by changing the perfusate to dialysis buffer containing 2.5, 5, or 10 μm glutamate. Liquid switches were used to minimize the pressure fluctuations while dialysis buffers containing varying concentrations of glutamate were perfused. Four samples were collected at 20 min intervals for each concentration.
Glutamate levels were analyzed using an HPLC system with electrochemical detection. The mobile phase consisted of MeOH (15% v/v), acetylnitrile (2.5% v/v), and 100 mm sodium dihydrogen phosphate monobasic, pH 6.0. Precolumn derivatization with o-phthalaldehyde was performed using an ESA Model 540 autosampler. Separation was done with a Shiseido column and glutamate was detected by a spectrophotometer (ESA). The glutamate content in each sample was analyzed by area under the curve and compared with an external standard curve for quantification.
3H glutamate uptake.
Glutamate uptake was measured using an in vitro slice preparation as described previously (Knackstedt et al., 2010). Briefly, rats were decapitated and bilateral punches of the NA core were dissected and sliced into 250 × 250 μm sections using a McIllwan tissue chopper. Slices were incubated at 37°C in either oxygenated Krebs–Ringer's solution phosphate buffer (in mm: 140 NaCl, 1.3 CaCl2, 1.2 KH2PO4, 5 HEPES, 10 glucose, and 1 MgCl2, pH 7.4) to quantify sodium-dependent uptake, or an identical buffer in which NaCl was replaced with 140 mm choline chloride to measure sodium-independent uptake. Uptake was stimulated by the addition of l-[3H] glutamate (40 nm, 51 Ci/mm; PerkinElmer) to slices in the presence of 3 μm l-glutamate. The reaction was conducted at 37°C for 15 min and terminated by ice-cold, sodium-free buffer. Slices were solubilized with 1% SDS and radioactivity determined using a liquid scintillation counter. Protein content was measured using a bicinchoninic acid assay protein assay (Pierce) and counts per million per milligram of protein calculated.
Electrophysiology.
Rats were anesthetized with isoflurane and decapitated. Brains were removed and immersed in cold (4°C) oxygenated aCSF composed of the following (in mm): 200 sucrose, 1.9 KCl, 33 Na2HCO3, 6 MgCl2, 0.5 CaCl2, 10 glucose, and 0.4 ascorbic acid. Coronal slices (300 μm) containing the accumbens were obtained with a vibratome (VT1000S; Leica) and incubated for 1 h at room temperature in aCSF containing the following (in mm): 126 NaCl, 2.5 KCl, 25 NaHCO3, 10 glucose, 4 MgCl2, 1 CaCl2, and 3 kynurenic acid. Slices were immersed in a recording solution composed of the following (in mm): 126 NaCl, 3 KCl, 26 NaHCO3, 1 MgCl2, 1.3 CaCl2, and 10 glucose, maintained at 28–32°C at a perfusion rate of 1.5–2 ml/min and viewed by differential interference contrast optics. The core compartment of the nucleus accumbens was localized using the anterior commissure as a landmark.
Borosilicate pipettes were filled with the following (in mm): 135 CsCl, 2 MgCl2, 10 HEPES, 1 EGTA, 4 NaCl, 2 Na-ATP, and 0.3 Tris-GTP. QX-314 Cl− (2%) was added to pipettes during the evoked experiment to block voltage-sensitive sodium channels from generating action potentials. Pipettes were connected to the headstage of a Multiclamp 700B amplifier (Molecular Devices) with Ag/AgCl wire. An Ag/AgCl reference wire or pellet was placed in the bath and, by using offset, Vm shifts were corrected. Amplifier was controlled by Multiclamp Commander (Molecular Devices). Signals were digitized and recorded on a Windows-based PC running Axograph X software. Voltage-clamp recordings were obtained in continuous voltage-clamp mode and filtered at 2.9 kHz. Series resistance and capacitance were compensated automatically by Multiclamp Commander and optimized manually. Access resistance was monitored continuously, and a 15% change was deemed acceptable. Spontaneous EPSCs (sEPSCs) and evoked EPSCs (eEPSCs) were collected in the presence of picrotoxin (100 μm). Data were analyzed using Minianalysis software (Synaptosoft). A bipolar stimulating electrode (World Precision Instruments) was placed medial to the lateral ventricle in the prelimbic cortex and responses were recorded in the accumbens. Approximately 20 min after break-in, eEPSC data were collected with 10 sweeps at 60 s intervals. The pulse was 0.1 ms and its intensity was set to yield a stable response equal to ∼75% of maximum amplitude (derived from the input–output function). For paired-pulse analysis, a set of two pulses was delivered with an interpulse interval of 50 ms, and the pairs of pulses were applied every 60 s.
Results
Cocaine self-administration
Animals self-administered cocaine for 12 d and subsequently underwent extinction training. During extinction training, animals were treated with either vehicle (0.3 ml, i.p.) or ceftriaxone (200 mg/kg, i.p.) for 5 d before microdialysis procedures or death for uptake assays or electrophysiology. Vehicle- and ceftriaxone-treated animals displayed similar levels of cocaine intake (Fig. 1A) and extinction responding (Fig. 1B).
Figure 1.
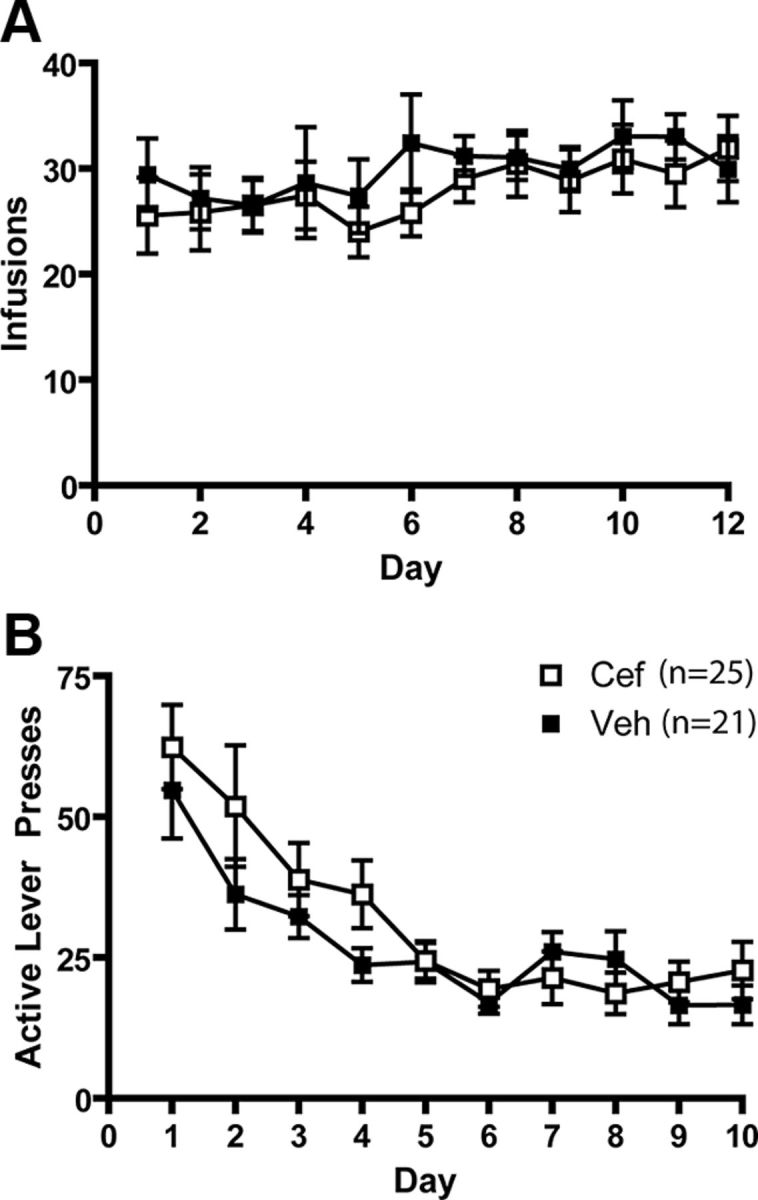
Cocaine self-administration and extinction training. A, The mean number of infusions attained by animals later treated with ceftriaxone (Cef) and vehicle (Veh) did not differ significantly between groups. B, There were no group differences in extinction responding. n = 21–25/group.
Ceftriaxone reversed cocaine-induced deficits in both glutamate uptake and export
We have previously shown that sodium-dependent glutamate uptake is reduced following cocaine self-administration and extinction training, accompanied by a decrease in Vmax but not Km (Knackstedt et al., 2010). Here, we incubated NA core slices in a concentration of glutamate in which uptake was previously found to be reduced (3 μm). In agreement with our previous results, we found a significant decrease in glutamate uptake in tissue taken from animals with a history of cocaine self-administration; ceftriaxone prevented this decrease (F(1,27) = 10.066, p < 0.01; Fig. 2A). When conducted in sodium-free conditions, a [3H]-glutamate uptake assay can accurately assess the activity of system xC− (Melendez et al., 2005). This assay revealed that ceftriaxone also reversed a deficit in NA core system xC− activity produced by cocaine (F(1,3) = 7.534, p < 0.05; Fig. 2B).
Figure 2.

Ceftriaxone normalized deficits in glutamate uptake. A, Cocaine (Coc) animals displayed a significant decrease in sodium-dependent glutamate uptake that was normalized by ceftriaxone (Cef). B, Sodium-independent uptake was reduced by cocaine. *p < 0.05 compared with vehicle-saline (Veh-Sal) using a Bonferroni post hoc test. n = 4–10/group.
Ceftriaxone treatment normalizes both synaptically released glutamate and nonsynaptic glutamate
A two-way repeated-measures ANOVA conducted on the no-net-flux dialysis data revealed a significant Group × Concentration interaction (F(3,30) = 4.614, p < 0.001), a significant effect of Group (F(3,30) = 8.737, p < 0.001), and a significant effect of Concentration (F(3,90) = 11.871, p < 0.001). Using the point of y = 0 to determine basal glutamate, ceftriaxone increased basal glutamate relative to vehicle-treated animals (F(1,3) = 5.207, p < 0.05; Fig. 3B). A two-way ANOVA determined that the slope of the regression line was significantly different between the ceftriaxone- and vehicle-treated groups (F(1,3) = 15.865, p < 0.001; Fig. 3C). A two-way ANOVA revealed that vehicle-treated animals displayed a significant increase in glutamate during a cocaine-primed reinstatement test while ceftriaxone-treated animals did not (Fig. 3D). This test revealed a significant Time × Group interaction (F(8,88) = 3.118, p < 0.01), effect of Group (F(1,11) = 4.72, p = 0.05), and Time (F(8,88) = 2.530, p < 0.05). There was a significant reduction in active lever presses during the reinstatement test in ceftriaxone-treated animals (ceftriaxone = 35.43, vehicle = 81.6; t(11) = 4.158, p = 0.002). Figure 3E indicates the placement of microdialysis probes for animals used in the microdialysis studies. Accurate placement was achieved when two-thirds of the active dialysis membrane was within the NA core. Data from animals with misplaced cannulae are not shown and were excluded from analysis.
Figure 3.

Ceftriaxone attenuates synaptically released glutamate during reinstatement and increases basal glutamate in the NA core. A, Ceftriaxone (Cef) restores basal glutamate levels in cocaine self-administering rats. B, Ceftriaxone-treated animals displayed increased basal glutamate relative to vehicle (Veh)-treated controls. C, Ceftriaxone-treated animals display reduced slope of the line of regression relative to vehicle-treated controls. D, Vehicle-treated animals displayed a significant increase in glutamate during a cocaine (Coc)-primed reinstatement test while ceftriaxone-treated animals did not. E, Diagrams showing dialysis probe tracks through the NA core. #p < 0.05 in comparison with vehicle-saline (Sal); *p < 0.05, comparing ceftriaxone with vehicle; both using a Bonferroni post hoc. n = 5–10/group.
Ceftriaxone normalized NA core excitatory transmission following cocaine self-administration
Two-way ANOVAs revealed that both sEPSC frequency and amplitude were increased following cocaine self-administration, and ceftriaxone reversed this effect (Fig. 4). These tests revealed a significant Ceftriaxone × Cocaine interaction (F(1,4) = 15.423, p < 0.001) for sEPSC frequency (Fig. 4A) as well as sEPSC amplitude (F(1,4) = 4.452, p < 0.05; Fig. 4B). Cocaine self-administration also increased the amplitude of synaptic evoked EPSCs, an effect that was normalized by ceftriaxone (F(1,4) = 8.1, p < 0.01; Fig. 4C). Thus, we observed identical effects under stimulated (evoked) and basal (spontaneous) conditions. We found no significant effect on paired-pulse facilitation (F(1,4) = 0.348, n.s.; data not shown).
Figure 4.

Excitatory synaptic transmission is increased following cocaine self-administration and normalized by ceftriaxone. A, sEPSC frequency is increased following cocaine (Coc) self-administration; this effect is reversed by ceftriaxone (Cef) treatment. B, sEPSC amplitude is increased following cocaine self-administration, ceftriaxone reverses this effect. C, Sample sEPSCs. Black traces show representative samples from control animals; gray traces are from cocaine-treated animals. D, Cocaine self-administration increases synaptic evoked EPSCs, an effect that is prevented by ceftriaxone. Representative traces shown in black are from control animals, gray traces are from cocaine-treated animals. *p < 0.05, compared with vehicle-cocaine using a Bonferroni post hoc test. Veh, Vehicle; Sal, saline. n = 8 cells/group.
Discussion
We have previously shown that ceftriaxone restores GLT-1 expression following cocaine self-administration while attenuating cue- and cocaine-primed reinstatement (Knackstedt et al., 2010). Ceftriaxone may exert protection against reinstatement by increasing GLT-1 expression and function, thus increasing glutamate uptake and preventing the overflow of synaptically released glutamate during reinstatement. In support of this hypothesis, ceftriaxone restored sodium-dependent glutamate uptake in the NA core following cocaine self-administration (Fig. 2A). Furthermore, ceftriaxone attenuated cocaine-induced glutamate overflow during a reinstatement test (Fig. 3C), an effect that may be the result of increased uptake.
The therapeutic effects of ceftriaxone may also derive from its ability to increase system xC− activity (Lewerenz et al., 2009; Knackstedt et al., 2010). Accordingly, here we found that ceftriaxone increased basal glutamate in the NA core of cocaine self-administering animals (Fig. 3A,B). However, reports exist that indicate that ceftriaxone decreases glutamate in the striatum (Miller et al., 2008; Rasmussen et al., 2011), a discrepancy that may arise from differences in species and technique. Miller and colleagues (2008) sampled from a larger striatal region in the mouse, whereas in the rat, our probes were localized to the NA core where system xC− strongly contributes to basal glutamate levels (Baker et al., 2002). Rasmussen and colleagues (2011) used qualitative microdialysis but did not take into account probe recovery; estimates of basal neurotransmitter levels obtained via this method can be misleading (for review, see Chefer et al., 2009).
In light of the reports of reduced basal glutamate following ceftriaxone, we conducted a [3H]-glutamate uptake assay to measure the activity of system xC− following cocaine and ceftriaxone. When conducted in sodium-free conditions, this assay characterizes the activity of system xC− (Melendez et al., 2005). We found that ceftriaxone reverses cocaine-induced deficits in system xC− activity (Fig. 2B). Furthermore, the pattern of ceftriaxone and cocaine-induced changes in basal glutamate (Fig. 3B) and system xC− activity (Fig. 2B) are similar, providing support for the idea that an increase in system xC− activity is the mechanism by which ceftriaxone restores basal glutamate. Furthermore, as a ceftriaxone-induced increase in NA core GLT-1 activity (Fig. 2A) is not accompanied by reduced basal glutamate levels (Fig. 3), it can be surmised that upregulated GLT-1 is not sufficient to decrease basal glutamate levels.
The slope of the line of regression for no-net-flux dialysis plots reflects the extraction fraction (Ed), which is potentially an estimate of neurotransmitter clearance (Bungay et al., 2003). The slope has been empirically shown to be a measure of clearance for the neurotransmitters dopamine (Smith and Justice, 1994) and acetylcholine (Vinson and Justice, 1997) following infusion of inhibitors of uptake, such that a lower Ed indicates less clearance. A study of this sort has not yet been conducted for the neurotransmitter glutamate. Here we found a reduction in slope following ceftriaxone treatment (Fig. 3A,C), indicating that glutamate uptake may be reduced by ceftriaxone. Obviously, this interpretation does not fit with our results from the sodium-dependent glutamate uptake assay, which indicated that ceftriaxone restored uptake. Chen (2006) reported that while recovery may be altered due to uptake inhibition, recovery is also dependent on the ratio of neurotransmitter release to uptake. In the case of an increase in basal glutamate arising from a decrease in glutamate uptake, slope may be an accurate indicator of clearance. However, ceftriaxone increases both glutamate uptake (Fig. 2A) and export (Fig. 2B), resulting in a more complicated relationship between glutamate uptake and the point of no-net-flux of glutamate, and thereby preventing slope from serving as an accurate estimate of uptake (for further discussion, see Chen, 2006; Pendyam et al., 2009).
N-acetylcysteine serves as an artificial source of cysteine, which drives the exchanger to export glutamate, thus increasing basal glutamate levels (Baker et al., 2003; Moussawi et al., 2011). Because mGluR2/3 antagonism prevents N-acetylcysteine from attenuating reinstatement (Moran et al., 2005), it has been proposed that an increase in basal glutamate restores tone to release-regulating mGluR2/3 autoreceptors, thereby dampening the synaptically released glutamate that drives reinstatement. The data presented here (Fig. 3) support a similar mechanism of action for ceftriaxone to attenuate reinstatement by increasing basal glutamate levels and restoring tone on mGluR2/3 autoreceptors, reducing glutamate release during reinstatement (Fig. 3D). N-acetylcysteine also upregulates GLT-1 expression (Knackstedt et al., 2010), an adaptation that may also serve to dampen glutamate release during reinstatement. At this time, the relative importance of increased basal glutamate and increased GLT-1 function is not known.
Cocaine self-administration produces enduring synaptic potentiation in the nucleus accumbens, evidenced by increased AMPA/NMDA ratio (Conrad et al., 2008; Moussawi et al., 2011). Additionally, cocaine self-administration and extinction training result in an inability to induce LTP by stimulating PFC afferents to the NA core, indicating that the accumbens neurons are already in an LTP-like state (Moussawi et al., 2009). Together, these results suggest that cocaine self-administration potentiates glutamatergic transmission from the PFC to the NA core, a neural pathway that has been shown to underlie the behavior of cocaine reinstatement (McFarland et al., 2003). Here we confirmed this potentiation in cocaine self-administering animals and found that it was reversed by ceftriaxone (Fig. 4). The observed cocaine-induced increase in both sEPSC amplitude and frequency supports the hypothesis that there are both presynaptic and postsynaptic changes in glutamate transmission caused by cocaine self-administration. As discussed above, ceftriaxone may normalize presynaptic modulation of glutamate release by increasing basal glutamate, thereby restoring tone to mGluR2/3 receptors. The restoration of basal glutamate by ceftriaxone may also normalize postsynaptic determinants of excitability, such as AMPA receptor subunit composition. The failure of either cocaine or ceftriaxone to alter paired-pulse facilitation supports our conclusion that there are both presynaptic and postsynaptic alterations induced by these treatments. In agreement with the ability of ceftriaxone to normalize synaptic physiology after cocaine, N-acetylcysteine has been demonstrated to normalize the AMPA/NMDA ratio and restore the ability to induce LTP following cocaine self-administration and extinction (Moussawi et al., 2009, 2011).
In conclusion, we have confirmed previous studies that showed that cocaine self-administration and extinction training result in a reduction of basal glutamate levels, diminished glutamate transport and export, and enduring synaptic potentiation in the nucleus accumbens core. We show that ceftriaxone normalizes these pathologies. At this time, it is not known whether all of these neuroadaptations must be reversed to attenuate the reinstatement of cocaine seeking. Further studies will be done to assess the relative importance of glutamate export by system xC− and glutamate uptake by GLT-1 in mediating the therapeutic effects of ceftriaxone.
Footnotes
This work was supported by NIH Grant DA026010 (to L.A.K.) and by NIH Grants DA003906, DA015369, and DA012513 (to P.W.K.).
References
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- Bungay PM, Newton-Vinson P, Isele W, Garris PA, Justice JB. Microdialysis of dopamine interpreted with quantitative model incorporating probe implantation trauma. J Neurochem. 2003;86:932–946. doi: 10.1046/j.1471-4159.2003.01904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chefer VI, Thompson AC, Zapata A, Shippenberg TS. Overview of brain microdialysis. Curr Protoc Neurosci Chapter. 2009;7 doi: 10.1002/0471142301.ns0701s47. Unit7 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KC. Effects of tissue trauma on the characteristics of microdialysis zero-net-flux method sampling neurotransmitters. J Theor Biol. 2006;238:863–881. doi: 10.1016/j.jtbi.2005.06.035. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Melendez RI, Kalivas PW. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol Psychiatry. 2010;67:81–84. doi: 10.1016/j.biopsych.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SG, Su ZZ, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem. 2008;283:13116–13123. doi: 10.1074/jbc.M707697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Albrecht P, Tien ML, Henke N, Karumbayaram S, Kornblum HI, Wiedau-Pazos M, Schubert D, Maher P, Methner A. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem. 2009;111:332–343. doi: 10.1111/j.1471-4159.2009.06347.x. [DOI] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez RI, Hicks MP, Cagle SS, Kalivas PW. Ethanol exposure decreases glutamate uptake in the nucleus accumbens. Alcohol Clin Exp Res. 2005;29:326–333. doi: 10.1097/01.alc.0000156086.65665.4d. [DOI] [PubMed] [Google Scholar]
- Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR, Kennedy RT, Rebec GV. Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington's disease phenotype in the R6/2 mouse. Neuroscience. 2008;153:329–337. doi: 10.1016/j.neuroscience.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. 2005;25:6389–6393. doi: 10.1523/JNEUROSCI.1007-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, Kalivas PW. N-acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–189. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Zhou W, Shen H, Reichel CM, See RE, Carr DB, Kalivas PW. Reversing cocaine-induced synaptic potentiation provides enduring protection from relapse. Proc Natl Acad Sci U S A. 2011;108:385–390. doi: 10.1073/pnas.1011265108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 5. Amsterdam: Elsevier Academic; 2005. [Google Scholar]
- Pendyam S, Mohan A, Kalivas PW, Nair SS. Computational model of extracellular glutamate in the nucleus accumbens incorporates neuroadaptations by chronic cocaine. Neuroscience. 2009;158:1266–1276. doi: 10.1016/j.neuroscience.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen BA, Baron DA, Kim JK, Unterwald EM, Rawls SM. Beta-lactam antibiotic produces a sustained reduction in extracellular glutamate in the nucleus accumbens of rats. Amino Acids. 2011;40:761–764. doi: 10.1007/s00726-010-0589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Sari Y, Smith KD, Ali PK, Rebec GV. Upregulation of GLT1 attenuates cue-induced reinstatement of cocaine-seeking behavior in rats. J Neurosci. 2009;29:9239–9243. doi: 10.1523/JNEUROSCI.1746-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD, Justice JB. The effect of inhibition of synthesis, release, metabolism and uptake on the microdialysis extraction fraction of dopamine. J Neurosci Methods. 1994;54:75–82. doi: 10.1016/0165-0270(94)90161-9. [DOI] [PubMed] [Google Scholar]
- Sondheimer I, Knackstedt LA. Ceftriaxone prevents the induction of cocaine sensitization and produces enduring attenuation of cue- and cocaine-primed reinstatement of cocaine-seeking. Behav Brain Res. 2011;225:252–258. doi: 10.1016/j.bbr.2011.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Vinson PN, Justice JB., Jr Effect of neostigmine on concentration and extraction fraction of acetylcholine using quantitative microdialysis. J Neurosci Methods. 1997;73:61–67. doi: 10.1016/s0165-0270(96)02213-3. [DOI] [PubMed] [Google Scholar]