

Abstract
Background
Helicobacter pylori infection is associated with a lower risk of chronic autoimmune diseases including IBD. H. pylori modulates the gastric immune response, decreasing the local inflammatory response to itself. In mice, chronic Salmonella typhimurium infection induces colitis similar to Crohn’s disease characterized by inflammation which progresses towards fibrosis. The aim of this study was to determine whether prior H. pylori infection acts at a distance to modulate the immune response of S. typhimurium-induced colitis.
Methods
Mice were infected with the mouse-adapted strain of H. pylori (SS1), followed by infection with S. typhimurium. The effect of H. pylori on colitis was determined by gross pathology, histopathology, cytokine response, and development of fibrosis in the cecum. Gastritis and systemic immune response was measured in response to infection.
Results
H. pylori suppresses the Th17 response to S. typhimurium infection in the mouse cecum, but does not alter the Th2 or Treg response or the development of fibrosis. H. pylori infection induces IL-10 in the mesenteric lymph nodes, suggesting an extra-gastric mechanism for immunomodulation. H. pylori/S. typhimurium co-infection decreases inflammation in both the cecum and the stomach.
Conclusions
This study demonstrates a potential mechanism for the negative association between H. pylori and IBD in humans. H. pylori represses the lower gastrointestinal tract Th17 response to bacterially induced colitis via extra-gastric immunomodulatory effects, illustrating immunological crosstalk between the upper and lower gastrointestinal tract.
Keywords: Inflammatory bowel disease, Helicobacter pylori, Salmonella typhimurium, colitis, gastritis
INTRODUCTION
Helicobacter pylori (H. pylori) is a gram-negative bacterium which frequently and chronically colonizes the human gastric mucosa. H. pylori’s capacity to cause chronic infection stems from its ability to modulate the gastric immune response. Recent data demonstrate that H. pylori up-regulates the production of anti-inflammatory T-regulatory cells in the stomach, thereby decreasing the inflammatory response against the bacteria1. In fact, H. pylori-colonized patients express higher levels of gastric FoxP3, a T-regulatory cell marker2. Whether this up-regulation affects systemic immune responses in addition to the local response against H. pylori is unknown. However, colonization with H. pylori has been inversely associated with certain chronic inflammatory diseases, such as asthma and Lupus3. This suggests that immune regulation by H. pylori may have systemic effects.
Inflammatory bowel disease (IBD) is a chronic inflammatory condition of the intestinal tract, resulting in part from a dysregulated immune response. Previous work in animal models has illustrated the importance of T-regulatory cells in the pathogenesis of colitis. For example, mice deficient in IL-10, a key regulatory cytokine of T-regulatory cells, develop spontaneous colitis 4. Additionally, adoptive transfer of T-regulatory cells can inhibit the development of colitis in certain animal models 5-7. Furthermore, a meta-analysis of studies investigating the relation between H. pylori colonization and IBD found a significant inverse association between the two, suggesting a possible protective role for H. pylori 8colonization against the development of IBD. However, these clinical data have various confounders, limiting the certainty of the findings.
In this study, we aimed to examine the affect of H. pylori on an experimental model of colitis and further define the relationship between H. pylori and IBD. We used the Salmonella typhimurium model of colitis in order to illustrate the effect of gastric H. pylori colonization on a distant bacterial-host immune system interaction in the colon 9. We found that H. pylori infection decreased cecal inflammatory cytokine expression in response to S. typhimurium infection. We also found that prior H. pylori infection influences distal mucosal immune responses as evidenced by an increased IL-10 expression measured in the mesenteric lymph nodes of mice infected with H. pylori. Our data provide the first evidence that H. pylori infection in the stomach alters the immunological environment of the lower GI tract providing mechanistic support for the epidemiological observation of a negative association between H. pylori status and risk of IBD.
MATERIALS AND METHODS
Mice
Female 8-12 week old C57/BL6 mice (Jackson Laboratories, Bar Harbor, ME) were infected with either H. pylori SS1, S. typhimurium ΔaroA or H. pylori SS1 + S. typhimurium ΔaroA. Twenty-four hours prior to oral infection with 3×106 colony-forming units (CFU) of S. typhimurium strain ΔaroA in 100 μl 0.1M HEPES buffer (pH = 8.0), mice received 20 mg of streptomycin in 0.1 M HBSS by oral gavage. All animal experiments were conducted with the approval and oversight of the University of Michigan UCUCA (University Committee on Use and Care of Animals).
Bacterial Strains
Salmonella typhimurium strain ΔaroA (a gift from Guntram Grassl, University of British Columbia, Vancouver, Canada) which is naturally resistant to streptomycin was grown in LB broth containing 100 μg/ml streptomycin at 37°C. The mouse adapted Helicobacter pylori SS1 strain (a gift from Kathryn Eaton, University of Michigan, Ann Arbor, MI) was grown on Campylobacter-selective agar (BD Diagnostics, Bedford, MA) supplemented with 5% sterile horse blood, trimethoprim (5 μg/mL), vancomycin (10 μg/mL), and nystatin (10 μg/mL) for 2 days at 37 °C in a humidified microaerophilic chamber (BBL Gas System, with CampyPak Plus packs, BD Microbiology, Sparks, MD).
Animal Studies
Mice were infected with H. pylori SS1 with an oral gavage of 108 CFU of live organisms thrice over 1 week. Control animals received HBSS by oral gavage. Thirty-four days after the H. pylori infection, half of the H. pylori animals (H. pylori + S. typhimurium) received 20 mg streptomycin by oral gavage followed by infection with 3×106 cfu S. typhimurium ΔaroA in 100λ 0.1M HEPES buffer (pH = 8.0) (Supplemental figure 1). The other half of the H. pylori-infected cohort (H. pylori group) was not infected with S. typhimurium but received streptomycin and HBSS on the same schedule. Of the animals uninfected with H. pylori, half received streptomycin and subsequent S. typhimurium infection (S. typhimurium group). Negative control (HBSS group) animals were not infected with S. typhimurium but received streptomycin and HBSS. Mice were euthanized 21 days post S. typhimurium infection.
Gross Pathology and Tissue Collection
Mice were euthanized at 21 days post S. typhimurium infection. Cecum and distal colon were collected, photographed, measured, and weighed. Cecal area was determined from digital photographs using ImageJ and a defined region of interest (ROI) to delineate the cecum. To control for differences in images, the cecal area was normalized against a 1 cm marker in the photographic image. Tissues were snap-frozen in liquid nitrogen and stored at −80°C prior to molecular analysis. Cecal contents were collected and serially diluted before plating onto LB-streptomycin plates to determine bacterial titers.
Histology
Formalin-fixed and paraffin-embedded tissues were stained with hematoxylin and eosin (H&E, inflammatory histology) and Masson’s trichrome (fibrosis) by the University of Michigan CCGC Research Histology and Immunoperoxidase Laboratory (Ann Arbor, MI). Digital photomicrographs of tissue sections were taken with an Olympus BX microscope (University of Michigan Microscopy and Image Analysis Laboratory). Histological scoring was performed by two blinded observers [JK, JL]. Tissue measurements were quantitated with ImageJ analysis software (NIH, Bethesda, MD).
Histological scoring
Gastric tissue was scored for inflammation on a scale of 0=none to 3=marked by the Modified Sydney Approach.10 Cecal inflammation was determined using a separate scoring system 11(0) no inflammation, (1) low level of inflammation with scattered infiltrating mononuclear cells, (2) moderate inflammation with multiple foci (3) high level of inflammation with increased vascular density and marked wall thickening, (4) maximal severity of inflammation with transmural leukocyte infiltration and loss of goblet cells.
Immunoblotting
Immunoblotting was utilized for the detection of α-smooth muscle actin. Whole tissue was pulverized under liquid nitrogen and lysed in ice-cold RIPA buffer with a cocktail of proteinase inhibitors (Roche, Indianapolis, IN). Protein content was determined using a modified Bradford assay kit (BioRad, Hercules, CA). Total protein was separated by SDS polyacrylamide gel electrophoresis and transferred to PVDF membranes (Amersham Biosciences, Piscataway, NJ). Membranes were blocked in 5% milk solution for one hour at room temperature or overnight at 4°C. α-smooth muscle actin was detected by incubating the membrane overnight at 4°C with mouse anti-human monoclonal antibody (Sigma, St. Louis, MO) at 1: 5000 dilution in 5% milk/TBST. As a loading control, a mouse antibody for GAPDH (Chemicon, Temecula, CA) was used after a stripping procedure. Secondary antibody anti-mouse IgG+ HRP (Amersham, Piscataway, NJ) was incubated for one hour at room temperature and the signal was detected by the Pierce detection system (Pierce, Rockford, IL). Autoradiographs were scanned and quantitated using Image J analysis software (NIH).
Meta-analysis
The relative risk (RR) of H. pylori infection in IBD versus controls was determined from 26 observational studies as previously described 8. Briefly, RR was used to describe the ratio of the probability of the H. pylori infection occurring in IBD patients versus the controls. The RR was calculated with a 95% confidence interval (CI) based on a random-effects model as described by Mantel-Haenszel12. Meta-analysis was performed with the metan command in Stata 10.1 (StataCorp, College Station, TX).
Real-time Quantitative PCR
RNA was extracted from the cecum using the RNeasy kit (Qiagen, Valencia, CA). Reverse transcription of 2 μg of total RNA was performed with the Superscript First Strand RT kit (Invitrogen, Carlsbad, CA). Quantitative real-time PCR (QPCR) of IL-1β, TNF-α, IFN-γ, IL-17, IL-6, IL-13, IL-12p40, FoxP3, IL-10, IL-4, TGF-β and GAPDH was performed with the TaqMan gene expression assays (ABI, Foster City, CA). QPCR was performed using a Stratagene Mx3000P real-time PCR system (Stratagene, La Jolla, CA). Cycling conditions were 95° C 10 minutes, followed by 40 cycles of 95° C 15 seconds and 62° C 60 seconds. ΔΔCt were calculated from GAPDH expression.
Statistical Analysis
Comparisons between several groups of mice and associated tissues or RNA expression were analyzed with ANOVA, while pairwise comparisons of two groups were performed with Student’s t test.
RESULTS
H. pylori infection is negatively associated with the relative risk for developing IBD in human populations
An update of our previously published meta-analysis of 24 observational studies was performed to include an additional 2 new studies.1 The overall relative risk (RR) of developing IBD in patients infected with H. pylori was 0.63 (95% confidence interval [CI]; 0.54-0.73, p <0.001, Figure 1). This observation suggests that H. pylori infection is protective against the development of IBD.
Figure 1. H. pylori infection is negatively associated with the relative risk for developing IBD in human populations.

The summary relative risk (RR) was calculated from 26 studies based on a random effects model. Horizontal lines delineate the upper and lower confidence limits with diamonds indicating the point estimates. The area of the gray rectangular boxes corresponds to the weight of the study in the meta-analysis. Relative risk is shown in the right column with upper and lower 95% confidence intervals in parentheses. The pooled RR estimate (Overall) is shown at bottom (P <0.0001). The solid vertical line represents no effect.
H. pylori infection suppresses the Th17 cytokine response in the cecum
To examine the effect of H. pylori gastric infection on distal gut inflammation, H. pylori-infected mice were co-infected with S. typhimurium. Chronic infection with S. typhimurium is characterized by a dramatic reduction in cecal size and a concurrent increase in tissue inflammation.9 Mice infected with either S. typhimurium or co-infected with H. pylori/S. typhimurium developed marked changes in cecal appearance compared to either the uninfected or H. pylori-infected mice (Figure 2A). H&E histological sections of cecal tissue from S. typhimurium or H. pylori/S. typhimurium-infected animals exhibited extensive crypt loss, inflammatory infiltration, transmural inflammation, and hypertrophy in the muscularis propria, findings that were not present in uninfected or H. pylori-infected controls (Figure 2B). S. typhimurium infection elicits both innate (IL-1β and TNF-α) and adaptive (IL-12p40, IL-6, IFN-γ and IL-17) responses in the cecum (Figure 2C-H). H. pylori/S. typhimurium co-infection significantly repressed IL-6 and IL-17 mRNA expression (3-fold and 5.3-fold respectively) in the cecum, with a trend toward lower IL-1β and TNF-α in the cecum compared to infection by S. typhimurium alone. As expected, H. pylori infection alone did not significantly induce these pro-inflammatory cytokines in the cecum except for a small but significant rise in IFN-γ expression. Overall, gastric H. pylori infection significantly down-regulates the cecal Th17 response to S. typhimurium infection.
Figure 2. H. pylori infection suppresses the Th17 cytokine response in the cecum.

A. Representative gross appearance of the cecum and distal colon of H. pylori-infected mice compared to uninfected (no Tx), S. typhimurium-infected (St) and H. pylori/S. typhimurium (St+Hp) infected with a 1 cm reference ruler. B. H&E stained histological sections (100x magnification). Infection with S. typhimurium induces substantial submucosal expansion, collagen deposition, epithelial destruction, and inflammatory infiltration. Scale bar represents 200 μm. (C-H) qRT-PCR gene expression of Th1 and inflammatory cytokines in the cecum of uninfected (no Tx) compared to H. pylori (Hp), S. typhimurium (St) and H. pylori/S. typhimurium (St+Hp) infected mice. The markers of innate immune response IL-1β (C) and TNFα (D) are compared to markers of the adaptive immune response IL-12p40 (E), IL-6 (F), IFN-γ (G) and IL-17 (H). Gene expression was normalized to GAPDH expression. The results are from 2 independent animal experiments. Numbers (n) below the graph labels represent number of animals per experimental group. (ns = not significant, * P<0.05, ** P<0.01, ***P <0.001)
H. pylori gastric colonization and S. typhimurium cecal colonization are not altered by co-infection
To examine whether H. pylori suppression of S. typhimurium colitis is caused by a reduction in the level of S. typhimurium cecal colonization, the effect of H. pylori infection upon S. typhimurium cecal colonization was determined. S. typhimurium heavily colonizes the mouse cecum, persisting at day 21 post-infection. Luminal cecal contents were isolated from uninfected, S. typhimurium, H. pylori, and H. pylori/S. typhimurium co-infected mice. Despite prior H. pylori infection, S. typhimurium cecal colonization did not differ between S. typhimurium-infected and H. pylori/S. typhimurium co-infected mice (Figure 3). Therefore H. pylori suppression of the Th17 response to S. typhimurium was not due to a decrease in S. typhimurium colonization of the cecum.
Figure 3. H. pylori gastric colonization and S. typhimurium cecal colonization are not altered by co-infection.
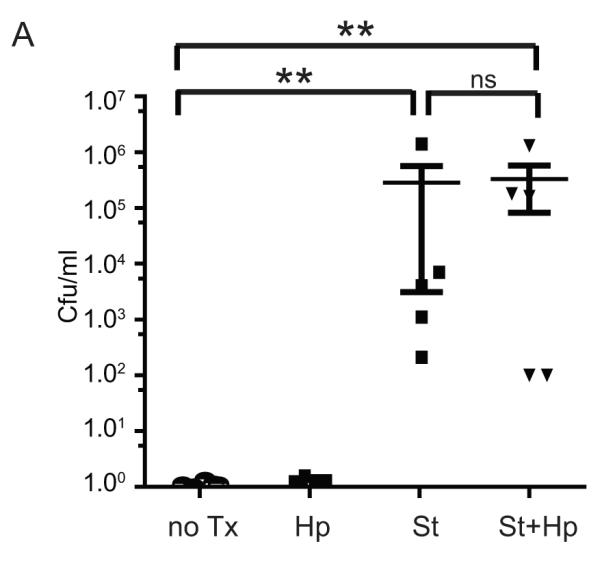
A. S. typhimurium colonizes the mouse cecum, irrespective of H. pylori co-infection. Colonization was determined from serial dilutions of 100 μl of cecal contents of uninfected (no Tx), H. pylori (Hp), S. typhimurium (St), and H. pylori/S. typhimurium-infected mice cultured on LB/streptomycin plates. The results are from 2 independent animal experiments. Statistical comparisons are made to the uninfected group. (ns = not significant,** P<0.01)
To investigate further the mechanism of H. pylori’s effect on distal GI tract, we studied whether H. pylori could colonize the mouse cecum, allowing it to directly suppress local mucosal immune responses. First, levels of gastric H. pylori colonization were compared and no difference was found between H. pylori infection alone vs co-infection with S. typhimurium by qRT-PCR (Supplemental Figure 2A). Our data also showed that H. pylori did not colonize the cecum as cecal tissue samples had undetectable levels of H. pylori urease A measured by quantitative RT-PCR from either H. pylori or H. pylori/S. typhimurium co-infected mice (Supplemental Figure 2B).
H. pylori infection does not alter S. typhimurium-induced Th2 or Treg cytokine responses in the mouse cecum
Another potential mechanism of immune suppression by H. pylori is by induction of Th2 or Treg responses in the cecum. H. pylori infection alone did not induce significant cecal expression of Th2 cytokines IL-4 and IL-13, but did induce higher levels of IL-10 and Foxp3 in the cecum (Figure 4). During co-infection with S. typhimurium, however, the H. pylori co-infection was not associated with an increased cecal IL-10 expression and but a trend towards increased Foxp3 was observed (Figure 4A & B). H. pylori infection alone induced significant increases in the expression of IL-10 and Foxp3 in the cecum, and a mild increase in IL-13, but had no effect on IL-4. During co-infection with H. pylori and S. typhimurium, there was not a further increase in cecal IL-10 and FoxP3. Addition of S. typhimurium did reverse the induction of IL-13 by H. pylori, to either mono-infection with H. pylori or S. typhimurium. These findings suggest that gastric H. pylori’s immunosuppressive effect at a distance in the cecum may be related to the immunoregulatory modulation of the Th17 cecal mucosal immune response.
Figure 4. H. pylori infection does not alter S. typhimurium-induced Th2 or Treg cytokine responses in the mouse cecum.

A. qRT-PCR gene expression of Th2 cytokines, IL-13 (A) and IL-4 (B) in the cecum of uninfected (no Tx) mice compared to H. pylori (Hp), S. typhimurium (St), and H. pylori/S. typhimurium (St+Hp) infected mice. The qRT-PCR expression of Treg cytokines IL-10 (C) and FoxP3 (D) in the cecum. Gene expression was normalized to GAPDH expression. Results are from 2 independent animal experiments. (ns = not significant,*P<0.05, ** P<0.01, ***P <0.001)
H. pylori infection induces IL-10 expression in the mesenteric lymph nodes
To examine whether the immunoregulatory effect of H. pylori infection in the cecum also occurs in the secondary draining lymph nodes, mesenteric lymph nodes (MLNs) were collected and cytokine profiles compared. As shown in Figure 5A, H. pylori infection increased IL-10 mRNA expression in the mesenteric lymph nodes compared to uninfected mice. During S. typhimurium infection, the levels of MLN IL-10 decreased and the levels of IL-17 and IFN-γ were increased compared to uninfected mice (Figure 5B and C). No significant differences in these cytokine levels were measured with H. pylori/S.typhimurium co-infection suggesting a greater influence of H. pylori infection in the cecum than in the mesenteric lymph nodes. This could be due to the presence of H. pylori antigen traveling through the intestinal lumen, and augmenting the immunoregulatory effect of H. pylori at the cecal mucosa.
Figure 5. H. pylori infection induces IL-10 expression in the mesenteric lymph nodes.

qRT-PCR expression of IL-10 (A), IL-17 (B), and IFN-γ (C) in H. pylori-infected mice (Hp) compared to uninfected (no Tx), S. typhimurium-infected (St) and H. pylori/S. typhimurium (St+Hp) infected. Results are from 5 animals per experimental group. Cytokine expression was normalized to GAPDH expression. (ns = not significant, *P<0.05, ** P<0.01).
H. pylori infection does not affect S. typhimurium-induced cecal fibrosis
Chronic S. typhimurium infection produces an intestinal fibrosis phenotype similar to human Crohn’s disease9. Fibrosis is largely confined to the cecum and is characterized by excessive extracellular matrix deposition, submucosal hypertrophy, and myofibroblast activation. S. typhimurium infection induces a marked increase in extra-cellular collagen deposition as determined by trichrome staining of cecal tissue compared to uninfected or H. pylori-infected mice (Figure 6A). H. pylori/S. typhimurium co-infection induces a similar response. Cecal submucosal expansion, a measure of both edema and fibrosis, was induced 4.7-fold by S. typhimurium infection compared to uninfected mice (Figure 6B). H. pylori/S. typhimurium co-infection induced a similar 4.5-fold increase in submucosal thickness, indicating H. pylori did not alter the fibrogenic response induced by S. typhimurium (p=0.01 vs uninfected), while H. pylori infection alone had no effect. Collagen deposition is effected by activated resident myofibroblasts which express α-smooth muscle actin (αSMA) that is mediated by TGF-β during the wound healing response. S. typhimurium induces higher αSMA protein and TGF-β mRNA expression compared to uninfected mice as determined by Western blotting (Figure 6C and 6D). H. pylori/S. typhimurium co-infection induces similar levels of αSMA protein TGF-β mRNA expression, comparable to S. typhimurium alone. Therefore, gastric H. pylori co-infection does not reduce S. typhimurium-induced intestinal fibrosis.
Figure 6. H. pylori infection does not affect S. typhimurium-induced cecal fibrosis.

A. Trichrome stained histological sections (5x magnification). Infection with S. typhimurium (St) or H. pylori/S. typhimurium (St+Hp) induces substantial increase in the extracellular matrix characterized by collagen deposition (blue staining) compared to uninfected (no Tx) or H. pylori-infected (HP). The scale bar represents 2 mm. B. Cecal submucosal thickness measurements. The submucosal thickness was determined at three reference points from photomicrographs above. Each point represents an individual animal. Horizontal bars indicate the average submucosal thickness for each experimental group. C. Representative Western blot illustrating αSMA protein expression in cecal extracts. GAPDH protein expression was used as a loading control. D. qRT-PCR gene expression of TGF-β in the cecum of uninfected (no Tx) compared to H. pylori (Hp), S. typhimurium (St) and H. pylori/S. typhimurium (St+Hp) infected. Gene expression was normalized to GAPDH expression. Results are from 2 independent animal experiments. (ns = not significant, *P<0.05, ** P<0.01)
H. pylori and S. typhimurium co-infection decreases inflammation in both the stomach and cecum
H. pylori infection is characterized by gastric hyperplasia and inflammatory infiltration in the gastric mucosa. H&E stained sections of stomach from H. pylori-infected mice, exhibit hyperplasia and leukocyte infiltration compared to uninfected mice (Figure 7A). S. typhimurium infection does not induce gastritis. To assess the extent of H. pylori induced gastritis, H&E stained sections of mouse stomach were scored by two blinded observers using the Modified Sydney Approach for inflammation10. Mice infected with H. pylori developed significant gastritis compared to uninfected mice (2.0 vs 0 points, p <0.01). H. pylori/S. typhimurium co-infected mice have a lower gastritis score than H. pylori-infected mice (Figure 7B).
Figure 7. H. pylori and S. typhimurium co-infection decreases inflammation in both the stomach and cecum.

A. H&E histological sections (100x magnification). Infection with H. pylori (Hp) induces gastric hyperplasia and inflammatory infiltration compared to uninfected (no Tx), S. typhimurium (St) and H. pylori/S. typhimurium (St+Hp) infected mice. Scale bar represents 200 μm. B. H. pylori induced inflammation in the stomach is repressed by S. typhimurium infection. Gastritis was scored from H&E stained sections of stomach using the Modified Sydney Approach on a scale of 0 (normal) to 3 (marked inflammation). C. H. pylori infection represses S. typhimurium induced inflammation in the cecum. Cecal inflammation was determined from H&E stained sections of cecum were scored (0) no inflammation, (1) low level of inflammation with scattered infiltrating mononuclear cells, (2) moderate inflammation with multiple foci (3) high level of inflammation with increased vascular density and marked wall thickening, (4) maximal severity of inflammation with transmural leukocyte infiltration and loss of goblet cells. Results are from two independent animal experiments. (ns = not significant,*P<0.05, ** P<0.01, ***P <0.001)
While H. pylori infection induces gastritis, H. pylori effects on the lower gastrointestinal tract are unknown. H&E stained cecal tissue sections were scored by a blinded observer on a 0 to 4 scale11 for mononuclear infiltration, vascular density, wall thickening, transmural leukocyte infiltration, and loss of goblet cells (Figure 7C). Surprisingly, H. pylori infection alone induces mild cecal inflammation compared to uninfected mice (2.4 vs 3.6, p = 0.0009). While mice infected with S. typhimurium (3.6 vs0.4, p <0.0001) or H. pylori /S. typhimurium (2.4 vs 0.4, p = 0.004) developed marked cecal inflammation compared to uninfected controls, H. pylori/S. typhimurium co-infected mice exhibited less cecal inflammation than S. typhimurium-infected alone (2.4 vs 3.6, p = 0.009). This indicates immunological crosstallk between the upper and lower gastrointestinal tract.
DISCUSSION
The incidence of H. pylori infection is highest among individuals from developing countries and lower in developed countries. The incidence of IBD has an opposite trend, raising the possibility of immunological crosstalk between the stomach and the distal gastrointestinal tract. In fact, an update of our previously published meta-analysis8 indicates the overall relative risk (0.63) of developing IBD in patients infected with H. pylori remains significantly less than uninfected individuals. To investigate the mechanism for the inverse association observed between H. pylori status and the risk of developing inflammatory bowel disease, we examined the effect of H. pylori infection on the immunological responses in the lower GI tract and in a mouse model of S. typhimurium typhlitis. We showed that mice chronically infected with H. pylori had a significantly elevated IL-10 mRNA expression in the cecum and in the mesenteric lymph nodes indicating H. pylori induces immunoregulatory responses in the lower GI tract. Additional supporting evidence is the finding that a lower cecal Th17 response and a repressive trend in the innate immune response was measured in mice co-infected with H. pylori and S. typhimurium compared to infection with S. typhimurium alone. This was not due to a difference in Salmonella colonization of the cecum nor to differences in IL-13, IL-4, or IL-10 cytokine levels. No significant difference in fibrosis was detected between the two groups suggesting selective immunomodulation. Of note, we did observe decreased gastric inflammation in mice co-infected with H. pylori and Salmonella indicating a potential crosstalk between distant gastrointestinal sites colonized by different bacteria.
The ability of H. pylori to colonize the stomach depends on its expression of BabA which enables it to anchor on gastric epithelium 13. There is no evidence that H. pylori colonizes the intestine, except in regions with gastric heterotypia 14, which further supports the gastric specificity of H. pylori colonization. We further verified by PCR the absence of H. pylori in the cecal specimen of H. pylori colonized mice. Thus, we speculate that H. pylori potentially influences the distal gastrointestinal tract through systemic immunomodulation. Epidemiological data also supports this concept as a negative association between H. pylori status and asthma has been reported 15.
A potential mechanism for the observed H. pylori immune modulation in the distal gut is the regulation of mucosal immune response owing to the tolerogenic properties of H. pylori1. H. pylori-infected patients were found to have higher gastric expression of Foxp3, a regulatory T cell marker, than uninfected individuals 2. In fact, H. pylori-infected children with a lower degree of gastritis expressed higher levels of Foxp3 than infected adults with more severe gastritis 16 indicating that the H. pylori induced regulatory T cell response might play a critical in down-modulating mucosal inflammation. This response is not limited to the gastric mucosa as it has been shown to play a critical role in peripheral T cell anergy in H. pylori-infected patients 17. Our study further supports the systemic nature of H. pylori induced Treg responses by showing an increased IL-10 expression in the mesenteric lymph nodes of H. pylori-infected mice. This provides the critical missing mechanism to explain the observed negative association between H. pylori status and the development of IBD. We showed further that H. pylori infection reduces S. typhimurium induced cecal inflammatory cytokines and histologic inflammation, but a histological difference in fiborsis was difficult to demonstrate using this chronic model, especially in a model that induced fibrosis.
As H. pylori was shown to influence distant intestinal immune responses, S. typhimurium numerically reduced the severity of gastritis in H. pylori-infected hosts. This is consistent with the report by Lemke, et. al.18 that a lower bowel bacterium can influence gastritis. Whether this represents a systemic endotoxic tolerance induced by S. typhimurium remains to be seen. Another potential mechanism is the alteration of the gut microbiota by S. typhimurium which could alter the immune responses at a different site. This concept has been previously demonstrated, in that antibiotic-induced dysbiosis can alter pulmonary responses to mucosal pathogens 19. This crosstalk may be historically and evolutionarily important, as mono-infection is uncommon in areas with poor public hygiene, and colonization with multiple organisms is the rule, rather than the exception. In fact, colonization with H. pylori might have conferred a selective advantage in the millennium when a pandemic of typhoid fever shifted the Greek balance of power from Athens to Sparta20, and the conquests of Alexander the Great may have come to an end due to typhoid21. The potential power of the evolutionary selective pressure of Salmonella typhi is demonstrated by the prevalence of heterozygosity for CFTR mutations, as the heterozygous state for cystic fibrosis confers a selective advantage through protection from S. typhi22. It is speculative, but attractive, to consider the possibility that lethal gut infections like Salmonella may have selected for the survival of humans who were colonized with and immunologically tolerant to H. pylori.
In summary, our study demonstrated a potential mechanism to explain the observed negative association between H. pylori and IBD. H. pylori-infected hosts demonstrated an increased level of IL-10 in mesenteric lymph nodes and a repressed lower gastrointestinal Th17 response to S. typhimurium. These findings provide mechanistic support for the protective role of H. pylori, but prospective studies of the effect of H. pylori eradication on the risk of developing IBD will be required to prove the beneficial role of H. pylori for IBD prevention.
Supplementary Material
Supplemental figure 1. Schemata of H. pylori and S. typhimurium infection. In two independent experiments, female C57/BL6 mice were infected with H. pylori with an oral gavage of 108 cfu/ml of live organisms three times over 1 week. Control animals received HBSS by oral gavage. 34 days after the H. pylori infection, half of the H. pylori animals (St+Hp) received 20 mg streptomycin (strep) by oral gavage to disrupt the cecal flora followed by infection with 3×106 cfu S. typhimurium ΔaroA in 100 μl 0.1M HEPES buffer (pH = 8.0). The other half of the H. pylori-infected cohort (Hp) was not infected with S. typhimurium but received streptomycin and HBSS on the same schedule. As H. pylori is streptomycin resistant, streptomycin treatment does not affect H. pylori colonization. Of the animals uninfected with H. pylori, half received streptomycin and subsequent S. typhimurium infection (St). Negative control (no Tx) animals were not infected with S. typhimurium but received streptomycin and HBSS. Mice were euthanized 21 days post S. typhimurium infection. Boxes represent the four experimental groups.
Supplemental figure 2. H. pylori infects the stomach but not the cecum. A. H. pylori colonizes the stomach as determined by QPCR of H. pylori urease A in stomach of uninfected (no Tx) compared to H. pylori (Hp), S. typhimurium (St) and H. pylori/S. typhimurium (St+Hp) infected mice. Diamonds represent individual mice (n = 5 per experimental group), horizontal bar represents the average for each group. Dashed line represents assay limit of detection. B. H. pylori does not colonize the cecum as determined by QPCR of H. pylori urease A of uninfected (no Tx) compared to H. pylori (Hp), S. typhimurium (St) and H. pylori/S. typhimurium (St+Hp) infected mice. Diamonds represent individual mice (n = 5 per experimental group), horizontal bar represents the average for each group. Dashed line represents assay limit of detection. (** P<0.01, ***P <0.001)
REFERENCES
- 1.Kao JY, Zhang M, Miller MJ, et al. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138:1046–54. doi: 10.1053/j.gastro.2009.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rad R, Brenner L, Bauer S, et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology. 2006;131:525–37. doi: 10.1053/j.gastro.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Sawalha AH, Schmid WR, Binder SR, Bacino DK, Harley JB. Association between systemic lupus erythematosus and Helicobacter pylori seronegativity. J Rheumatol. 2004;31:1546–50. [PubMed] [Google Scholar]
- 4.Leach MW, Davidson NJ, Fort MM, Powrie F, Rennick DM. The role of IL-10 in inflammatory bowel disease: “of mice and men”. Toxicol Pathol. 1999;27:123–33. doi: 10.1177/019262339902700124. [DOI] [PubMed] [Google Scholar]
- 5.De Winter H, Cheroutre H, Kronenberg M. Mucosal immunity and inflammation. II. The yin and yang of T cells in intestinal inflammation: pathogenic and protective roles in a mouse colitis model. Am J Physiol. 1999;276:G1317–21. doi: 10.1152/ajpgi.1999.276.6.G1317. [DOI] [PubMed] [Google Scholar]
- 6.Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol Rev. 2006;212:256–71. doi: 10.1111/j.0105-2896.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 7.Gad M. Regulatory T cells in experimental colitis. Curr Top Microbiol Immunol. 2005;293:179–208. doi: 10.1007/3-540-27702-1_9. [DOI] [PubMed] [Google Scholar]
- 8.Luther J, Dave M, Higgins PD, Kao JY. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm Bowel Dis. 2010;16:1077–84. doi: 10.1002/ibd.21116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grassl GA, Valdez Y, Bergstrom KS, Vallance BA, Finlay BB. Chronic enteric salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology. 2008;134:768–80. doi: 10.1053/j.gastro.2007.12.043. [DOI] [PubMed] [Google Scholar]
- 10.Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996;20:1161–81. doi: 10.1097/00000478-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–6. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 12.Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 1959;22:719–48. [PubMed] [Google Scholar]
- 13.Aspholm-Hurtig M, Dailide G, Lahmann M, et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305:519–22. doi: 10.1126/science.1098801. [DOI] [PubMed] [Google Scholar]
- 14.Harris AW, Baron JH. Helicobacter pylori and gastric metaplasia of the duodenum. Gut. 1995;37:446. doi: 10.1136/gut.37.3.446-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reibman J, Marmor M, Filner J, et al. Asthma is inversely associated with Helicobacter pylori status in an urban population. PLoS One. 2008;3:e4060. doi: 10.1371/journal.pone.0004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris PR, Wright SW, Serrano C, et al. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology. 2008;134:491–9. doi: 10.1053/j.gastro.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS. Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infection and immunity. 2003;71:1755–62. doi: 10.1128/IAI.71.4.1755-1762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lemke LB, Ge Z, Whary MT, et al. Concurrent Helicobacter bilis infection in C57BL/6 mice attenuates proinflammatory H. pylori-induced gastric pathology. Infection and immunity. 2009;77:2147–58. doi: 10.1128/IAI.01395-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noverr MC, Falkowski NR, McDonald RA, McKenzie AN, Huffnagle GB. Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin-13. Infection and immunity. 2005;73:30–8. doi: 10.1128/IAI.73.1.30-38.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papagrigorakis MJ, Yapijakis C, Synodinos PN, Baziotopoulou-Valavani E. DNA examination of ancient dental pulp incriminates typhoid fever as a probable cause of the Plague of Athens. Int J Infect Dis. 2006;10:206–14. doi: 10.1016/j.ijid.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Oldach DW, Richard RE, Borza EN, Benitez RM. A mysterious death. N Engl J Med. 1998;338:1764–9. doi: 10.1056/NEJM199806113382411. [DOI] [PubMed] [Google Scholar]
- 22.Pier GB, Grout M, Zaidi T, et al. Salmonella typhi uses CFTR to enter intestinal epithelial cells. Nature. 1998;393:79–82. doi: 10.1038/30006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1. Schemata of H. pylori and S. typhimurium infection. In two independent experiments, female C57/BL6 mice were infected with H. pylori with an oral gavage of 108 cfu/ml of live organisms three times over 1 week. Control animals received HBSS by oral gavage. 34 days after the H. pylori infection, half of the H. pylori animals (St+Hp) received 20 mg streptomycin (strep) by oral gavage to disrupt the cecal flora followed by infection with 3×106 cfu S. typhimurium ΔaroA in 100 μl 0.1M HEPES buffer (pH = 8.0). The other half of the H. pylori-infected cohort (Hp) was not infected with S. typhimurium but received streptomycin and HBSS on the same schedule. As H. pylori is streptomycin resistant, streptomycin treatment does not affect H. pylori colonization. Of the animals uninfected with H. pylori, half received streptomycin and subsequent S. typhimurium infection (St). Negative control (no Tx) animals were not infected with S. typhimurium but received streptomycin and HBSS. Mice were euthanized 21 days post S. typhimurium infection. Boxes represent the four experimental groups.
Supplemental figure 2. H. pylori infects the stomach but not the cecum. A. H. pylori colonizes the stomach as determined by QPCR of H. pylori urease A in stomach of uninfected (no Tx) compared to H. pylori (Hp), S. typhimurium (St) and H. pylori/S. typhimurium (St+Hp) infected mice. Diamonds represent individual mice (n = 5 per experimental group), horizontal bar represents the average for each group. Dashed line represents assay limit of detection. B. H. pylori does not colonize the cecum as determined by QPCR of H. pylori urease A of uninfected (no Tx) compared to H. pylori (Hp), S. typhimurium (St) and H. pylori/S. typhimurium (St+Hp) infected mice. Diamonds represent individual mice (n = 5 per experimental group), horizontal bar represents the average for each group. Dashed line represents assay limit of detection. (** P<0.01, ***P <0.001)