

Abstract
Animal models treated with agricultural chemicals, such as rotenone, reproduce several degenerative features of human central nervous system (CNS) diseases. Glutamate is the most abundant excitatory amino acid transmitter in the mammalian central nervous system and its transmission is implicated in a variety of brain functions including mental behavior and memory. Dysfunction of glutamate neurotransmission in the CNS has been associated with a number of human neurodegenerative diseases, either as a primary or as a secondary factor in the excitotoxic events leading to neuronal death. Since many human CNS disorders do not arise spontaneously in animals, characteristic functional changes have to be mimicked by toxic agents. Candidate environmental toxins bearing any direct or indirect effects on the pathogenesis of human disease are particularly useful. The present longitudinal Magnetic Resonance Imaging (MRI) studies show, for the first time, significant variations in the properties of brain ventricles in a rotenone-treated (2mg/kg) mouse model over a period of 4 weeks following 3 days of rotenone treatment. Histopathological analysis reveals death of stria terminalis neurons following this short period of rotenone treatment. Furthermore, in vivo voxel localized 1H MR spectroscopy also shows for the first time significant bio-energetic and metabolic changes as well as temporal alterations in the levels of glutamate in the degenerating striatal region. These studies provide novel insights on the effects of environmental toxins on glutamate and other amino acid neurotransmitters in human neurodegenerative diseases.
Keywords: Rotenone, striatum, glutamate, neurodegeneration, MRI, 1H MRS, in vivo
Introduction
The contribution of environmental factors to the development of human disease has been gaining more recognition in addition to genetic and other lifestyle factors. Consistent findings of epidemiological studies associate between increased disease risk and such environmental factors as rural residence [Morano et al., 1994; Liou et al., 1997, Marder et al., 1998], farming [Semchuk et al., 1993; Liou et al., 1997; Gorell et al., 1998; Fall et al., 1999], the drinking of water from wells [Morano et al., 1994; Liou et al., 1997, Marder et al., 1998], and exposure to agricultural chemicals, pesticides, and/or herbicides [Semchuk et al., 1992; Semchuk et al., 1993; Liou et al., 1997; Gorell et al., 1998; Fall et al., 1999; Vanacore et al., 2002]. Recently, several models have been elaborated in which exposure of animals to various pesticide and herbicides has been used to induce Parkinsonism [Uversky, 2004, for review]. Rotenone is an agricultural chemical, which was originally employed by Indians as a fish poison, and today it is a commonly used pesticide and is also used in lakes and reservoirs to kill fish that are perceived as pests. Rotenone is extremely lipophilic, which makes it penetrate all cell types and move freely across cellular membranes independent of any reuptake transport mechanism [Talpade et al., 2000]. More importantly, rotenone easily crosses the blood-brain-barrier, enters the brain rapidly, and accumulates in subcellular organelles, such as mitochondria [Higgins & Greenamyre, 1996; Bertabet et al., 2000; Talpade et al., 2000], where it impairs oxidative phosphorylation by inhibiting complex I, also known as NADH-ubiquitinone reductase, the first enzyme complex of the electron transfer chain [Schuler & Casida, 2001; Sherer et al., 2003a].
Rotenone toxicity of the mitochondria may lead to major energy deficits in the cell. Complex I is the only point of entry for the major fraction of electrons that traverse the respiratory chain, it oxidizes NADH and transfers electrons to ubiquinone, thus providing a vital step in cellular energy supply. In addition, complex I translocates protons from the mitochondrial matrix to the intermembrane space, thus helping to establish the electrochemical gradient that is used to fuel ATP synthesis [Hatefi, 1985]. Therefore, inhibition of mitochondrial complex I results in a major cellular energy crisis, and may provide invaluable insights into the mechanisms of mitochondria-based human diseases. However, it is often argued that the bio-energetic defects alone cannot account for rotenone-triggered neurodegeneration, as rotenone toxicity also results in oxidative stress [Sherer et al., 2003b]. Chronic inhibition of complex I with rotenone produces progressive depletion of glutathione, oxidative damage to proteins and DNA, release of cytochrome c to cytosol, activation of caspase 3, mitochondrial depolarization, and eventually apoptosis [Greenamyre et al., 2001]. Additionally, the rotenone induced-degeneration of dopaminergic neurons in the nigrostriatal region may also be attributed to the activation of microglia, the resident macrophages in the brain, in response to the widespread oxidative damage provoked by rotenone [Gao et al., 2002; 2003; Sherer et al., 2003a]. In cell culture models, inhibition of complex I with rotenone causes cell death in a variety of cell types, including dopaminergic and non-dopaminergic neurons, via distinct mechanisms [Kweon et al., 2004]. The multiple effects caused by rotenone on cell survival and integrity triggers the assumption that the common use/exposure to rotenone may be a risk factor contributing to the development of neurodegenerative diseases.
Rotenone is a well studied compound in animal models of neurodegeneration, especially in Parkinson’s disease [Uversky, 2004]. Chronic administration of rotenone in rodents have been shown to cause complex I inhibition uniformly throughout the brain, but only dopaminergic neurons in the nigrostriatal region were particularly susceptible to rotenone toxicity [Bertabet et al., 2000; Alam & Schmidt, 2002]. Chronic inhibition of complex I with rotenone, caused striatal denegeration over a period of months [Greenamyre et al., 1999; Bertabet et al., 2000]. In addition to the extensive evidence on rotenone toxicity to dopaminergic neurons, which largely mimics Parkinsonism [Bertabet et al., 2000; Alam & Schmidt, 2002; Hoglinger et al., 2003; Sherer et al., 2003c; Perier et al., 2003; Lapointe et al., 2004], evidence also exists that rotenone causes striatal lesion of non-dopaminergic neurons [Hoglinger et al., 2003]. Chronic treatment of rats with rotenone leads to N-methyl-D-aspartate (NMDA)-receptor dysfunction, which leads to further mitochondrial impairment [Greenamyre et al., 1999]. In stark contrast with chronic treatment with rotenone, inhibition of mitochondrial complex I with a single systemic rotenone (acute) administration does not lead to neurotoxicity but rather to increased glutamate –induced dopamine release in rat striatum [Leng et al., 2003], alluding to a possible involvement of glutamate transmission in the early stages of the protracted death of striatal neurons. Taken together, these findings are indicative of a potential relationship between environmental toxins and glutamate transmission in the CNS, which is an area of research that needs further investigation.
Glutamate is the most abundant excitatory amino acid transmitter in the CNS [Curtis & Watkins, 1961; Curtis & Johnson, 1974; Roberts et al., 1981; Fonnum, 1984]. Glutamate-mediated excitotoxicity has been hypothesized to play a major role in various neurodegenerative diseases, including seizure and epilepsy [Avallone et al., 2006; Cepeda et al., 2006], post-traumatic brain injury and ischemia [Oechmichen & Meissner, 2006; Van Landeghem et al., 2006], amyotrophic lateral sclerosis (ALS) [Rothestein et al., 1995; Lin et al., 1998; Van Damme et al., 2005], Huntington’s disease (HD) [Azerberger et al., 1997; Cowan & Raymond, 2006], Parkinson’s disease (PD) [Gubellini et al., 2006], Alzheimer’s disease (AD) [Masliah et al., 1996; Scott et al., 2002; Hayashi et al, 2006; Kirvell et al., 2006], and in various animal models of neurological diseases [Brujin et al., 1997; Li et al., 1997; Trotti et al., 1999; Lievens et al., 2001]. Therefore, dysfunction of glutamate transmission, particularly glutamate transport, is a critical factor in many neurodegenerative diseases. Here we test the possibility that rotenone-induced neurotoxicity in the striatum is exacerbated by a feed-forward mechanism driven by glutamate, which leads to neurodegenerative death of striatal neurons over a protracted period of time.
Materials and Methods
Animals and treatment
A total of 8 male C57BL/6 mice received subcutaneous injections with 2mg/kg rotenone in DMSO once a day for 3 consecutive days at 2 months of age. An equal number of animals were injected with DMSO alone (sham-treated). Animals were studied following 3 days of rotenone treatment using MRI/MRS once a week for 4 weeks. Four weeks following in vivo analysis, all animals were sacrificed and perfused and the brain was dissected out for immunohistological analysis.
Brain Magnetic Resonance Imaging (MRI) and water suppression voxel localized 1H spectroscopy (MRS) in vivo
MRI brain imaging combined with in vivo 1H MRS was conducted on rotenone-treated mice to detect neurodegeneration by noting brain lesions or morphometric changes and bio-energetic or metabolic alterations, respectively. To image the brain, using a 7-Tesla Bruker BioSpec 20 cm horizontal bore (Bruker Biospin, Bellerica, MA), animals were anesthetized with Isoflurane using 4% for induction and 1.5% for maintenance of anesthesia, with 70% oxygen and 30% nitrous oxide mixture. After which the animals were placed in a stereotaxic holder [Fricke et al., 2004] where Isoflurane is maintained at 1.5%, the stereotaxic holder incorporates a warm water heating system that maintains the temperature at 37°C. Also integrated into the holder is an inductive respiratory monitoring system as well as a four channel thermal optical monitoring system used to monitor animal’s skin temperature, animal core temperature (rectal), ambient temperature and wall temperature of holder. MRS was conducted just prior to the imaging experiment. At the onset of the experiment the 1H frequency is used to find the center frequency and the magnetic field is shimmed over the brain region of the animal. 1H scout images are obtained in 3 orthogonal planes. The PRESS sequence used had echo time (TE) = 13.77 milliseconds, repetition time (TR) = 1.5 seconds, Spectral width = 3472.22 Hertz, Number of points acquired were 1024, Number of Averages (NA) = 512, gradient spoiling time = 0.5 milliseconds, excitation pulse length 10 μseconds, refocusing pulse length 1000 milliseconds. For whole brain imaging the voxel size used was 7mm × 5.5mm × 10mm. Shimming was preformed on this volume element until the full width half maximum H2O line width is 5 Hertz or smaller. Spectra were obtained after localizing the voxel (2mm × 2mm × 2mm) on the selected striatal brain region where the legion was observed, the field is shimmed and spectra were acquired. Spectra were processed with 6 Hertz line broadening. Automatic baseline adjustment was performed using XWINMR Version 6.2 software (Bruker BioSpin, Bellerica, MA), the NAA/NAAG peak was calibrated to 1 and set as an internal reference, and all other peaks were integrated relative to NAA/NAAG. Volumetric evaluation or segmentation of brain images was performed using Insight ITK-SNAP computer software [Yushkevich et al., 2006], which implements 3D active contour segmentation methods. Spherical bubbles were placed inside the ventricles as starting points for the snake evolution, and the software was allowed to run 50-60 iterations until the bubbles fully fill the ventricles, then the program was asked to do the statistics and numerical values were obtained.
Immunocytochemical and histological analysis of brain sections
Immunohistochemistry was performed on 20 micron-thick brain sections perfused with 4% paraformaldehyde (PFA) and compared between different treatment conditions. Mice were anesthetized with equithesin (3mg/kg) and perfused with 4% PFA in 1 × PBS. Brains were removed and submerged overnight in 4% PFA and then in 5, 10 and 20% sucrose and frozen at -80°C. 20 micron thick sections were cut on a cryostat, thaw-mounted onto positively charged slides (Fisher Scientific Pittsburgh, PA) and stored at -80°C until used. For tyrosine hydroxylase (TH)-staining, slides were washed 3×5 min in 1 × PBS, incubated for 1h in 1% BSA in 1 × PBS to block non-specific binding, and then incubated overnight at 4°C in anti-TH rabbit polyclonal antibody (1:1000). Slices were then washed 3×5 min in PBS and TH immunoreactivity was detected by Alexa-488 conjugated goat anti-rabbit secondary antibody (Invitrogen) at 1:1000 dilution. Slices were then washed 3×5 times in 1 × PBS, rinsed in distilled water and mounted with coverslips using Prolong Gold antifade reagent (Invitrogen). Further silver degenerating staining using FD NeuroSilver Kit (FD NeuroTechnologies, Inc) was performed to assess neurodegeneration in brain tissues. Standard Hematoxylin and Eosin (H&E) histological staining was also be performed.
Results
Brain MRI shows significant changes in the volume of ventricles of rotenone-treated mice
A total of 8 male C57BL/6 mice (2-months old) were treated with 2mg/kg rotenone in DMSO once a day for 3 consecutive days. Following treatment, 2 mice died on the second day of injection and another died on the third day. One of the surviving rotenone-treated mice developed a cataract in the right eye. All 8 sham-treated (with DMSO) animals survived with no obvious health problems. Brain MRI in the remaining 4 mice revealed significant enlargement of brain ventricles in the third and fourth week post-treatment in rotenone-treated animals in comparison to sham-treated (Fig. 1, arrows in top grid). To obtain a quantitative value of the overall changes in the volume of the ventricles, segmentation was performed and showed a significant increase in the volume of brain ventricles in week 3 (12.86±0.41mm3, Mean±SD) and week 4 (13.98±0.39mm3, Mean±SD) post-treatment with rotenone compared with week 3 (9.10±1mm3, Mean±SD) and week 4 (9.72±0.24mm3, Mean±SD, respectively, in sham-treated mice (Fig.1. histograms). Additionally, obvious degenerative changes were observed in periventricular/subventricular brain regions (Fig. 1, arrows in B2-E2 and D4-J4). The anatomical changes revealed by MRI are within the striatum, which is contiguous to the lateral ventricles (B2-E2) in the rotenone-treated mouse.
Figure 1.
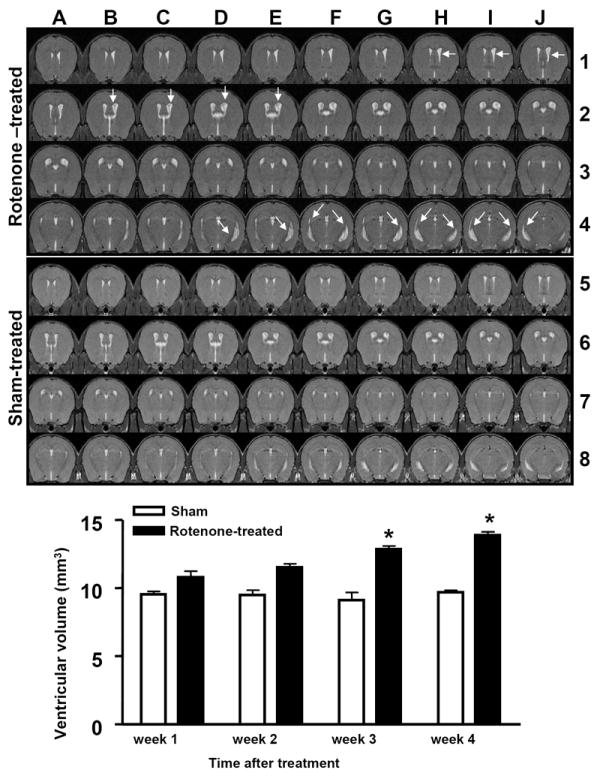
(A1-J4). In vivo brain MRI scans (100 micron thick) of mouse brain 4-weeks after rotenone treatment and (A5-J8) are scans of sham-treated control littermates. Histograms represent volumetric evaluation or segmentation of brain ventricular volume over a period of 4 weeks following treatment with rotenone. * indicates significantly different to control, n=4, P<0.05, Mann Whitney U, mean ±SD.
Rotenone treatment causes degenerative cell death in the mouse striatum
Immunohistopathology was conducted to confirm the anatomical changes observed in the MRI studies on the mouse striatum over a 4-week period post-treatment with rotenone. Hematoxylin and Eosin (H&E) staining were performed to detect histological changes in brain tissue. Tyrosine hydroxylase (TH) staining did not reveal any changes in dopaminergic nerve endings (Fig. 2A) in the striatum of rotenone-treated compared to sham-treated (DMSO) mice (Fig. 2B). No other brain regions including, the dopaminergic neurons of the substantia nigra (data not shown) showed any changes in TH-immunoreactivity, consistent with the MRI results that show no lesion in any brain region other than the striatum, as a result of rotenone treatment. H&E staining revealed major histological changes in the subventricular regions in the striatum (Fig, 2 C), which is contiguous to the ventricles, in the rotenone-treated mouse compared to sham-treated (Fig. 2D) animals. A striking reduction in overall tissue density is observed in the striatum of rotenone-treated mice and thinning of the tissue is particularly obvious in the subventricular zone (double arrow, Fig. 2C). The decrease in tissue density may be indicative of degenerative cell death in the striatum as neuronal cell bodies also seem to recede into what looks like cavities of degenerating tissue (single arrow, Fig. 2C). These changes were not observed in other brain regions, and were not even widespread but restricted to the striatum in rotenone-treated mice. Further histological analysis with silver staining (20×) to detect possible degenerative cell death showed a high number of positively silver-stained (black) neurons (arrows, Fig. 3E) in the same striatal region, where reduced tissue density was observed with H&E staining, compared to the same region in sham-treated animals (Fig. 6F). Higher magnification (40×) revealed silver staining of axonal fibers (double arrows, Fig. 2G) and cell bodies (single arrows), perhaps indicating that the degenerative cell death is restricted to resident cells of the striatum in rotenone-treated mice compared to control (Fig. 2H). No positively silver-stained neurons were observed in any other region of the brain in rotenone-treated animals, further suggesting that the rotenone-induced degenerative cell death in the striatum is perhaps due to the presence of a population of neurons that are specifically vulnerable to rotenone toxicity.
Figure 2.
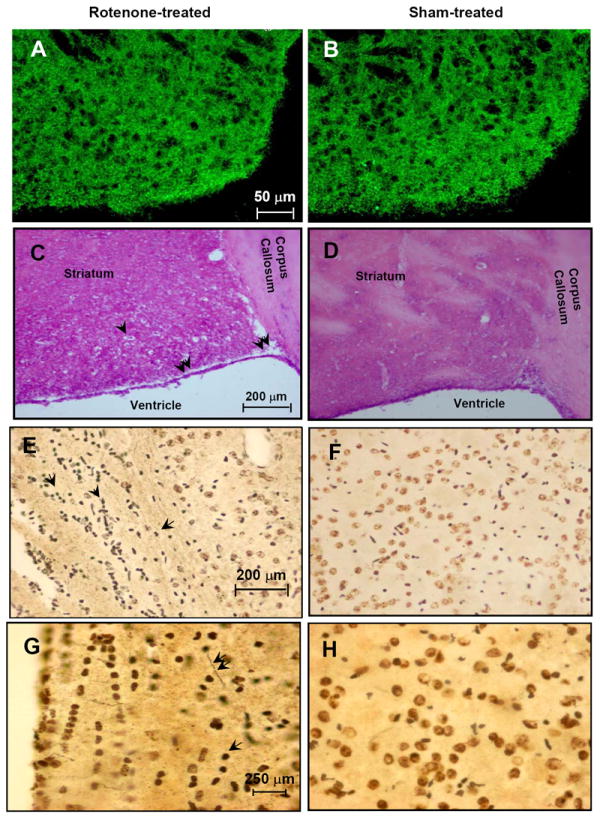
(A). TH-immunoreactivity of mouse striatum treated with rotenone and (B). sham-treated control littermates. (C). H&E staining of striatum in rotenone treated-mice and (D). sham-treated control littermates. Single arrows indicate neuronal cell body in a cavity of degenerating tissue. Double arrows indicate thinning of subventricular tissue. (E). 20× magnification of silver positive staining of neurons (arrows) in rotenone-treated mice and (F). sham-treated control littermates. (G). 40× magnification of silver stained axonal fibers (double arrows) and neuronal cell bodies (single arrow) in rotenone-treated mouse striatum compared to sham-treated control littermates (H).
Figure 3.
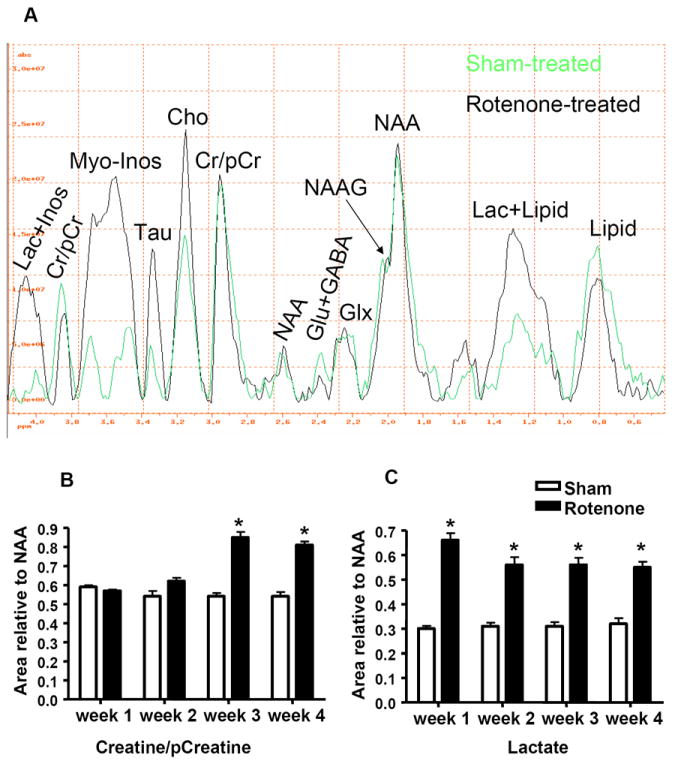
(A). In vivo 1H MRS spectra obtained from the striatum of rotenone- and sham-treated mice after 4-weeks of rotenone treatment. (B). Histograms represent the value of the area under the peak relative to NAA of Cr/pCr and (C). Lactate. NAA: N-Acetyl-Aspartate, Glu: Glutamate, GABA: Gamma-Aminobutyric Acid, Glx: glutamate and glutamine, Lac: Lactate, Cr: Creatine, pCr: phosphor-creatine, Inos: Inositol, Tau: Taurine, Cho: Choline. * indicates significantly different to control, n=4, Mann Whitney U, P<0.05, mean ±SD.
Voxel localized in vivo 1H spectroscopy reveals significant bioenergetic changes in the rotenone-treated mouse striatum
Voxel localized In vivo 1H spectroscopy in the subventricular zone including the striatum revealed significant bio-energetic changes, consistent with the inhibition of mitochondrial complex I with rotenone, and impairment of striatal glutamate transmission in the rotenone-treated mice compared to sham-treated control. In addition to major metabolic and bio-energetic fluctuations in rotenone-treated mice, a significant surge in choline (Cho) levels was apparent (Fig. 3A, 3.1-3.2 ppm), suggestive of cell membrane breakdown in the striatal region. A significant increase in taurine (Tau) levels was also observed in rotenone-treated mice, indicative of possible osmo-regulatory or electrolytic changes in the mouse brain as a result of rotenone treatment. Taurine is also an inhibitory neurotransmitter in the CNS. Quantitative analysis of the spectral peaks at 2.9 and 3.85 ppm, which designate the Cr/pCr levels relative to NAA/NAAG (NAA=1 was used as an internal reference), showed, interestingly, a significant increase in the levels of Cr and pCr (Fig. 3B) in week 3 (0.84±0.05, Mean±SD) compared to sham-treated animals (0.54±0.03, Mean±SD) and this increase remained at high levels through week 4 (0.82±0.03, Mean±SD) in rotenone-treated compared to sham-treated animals (0.533±0.045, Mean±SD).
A significant surge in lactate levels (1.3 and 4 ppm, Fig 3) was observed in rotenone-treated animals (Fig. 3C) as early as week 1 (0.65±0.05, Mean±SD) compared to sham-treated animals (0.3±0.023, Mean±SD), and the levels of lactate remained significantly high up until week 4 (Fig.4B). The separation of lactate from either inositol or total lipid is rather methodologically difficult with in vivo 1H MRS. However, since MRS was conducted on both rotenone- and sham-treated animals under identical experimental conditions (30-45 min with 1.5% isoflurane), lactate measurement is likely due to rotenone treatment. Levels of myo-inositol (Fig. 4 A) were also significantly increased (P<0.05, Mann Whitney U, n = 4), in week 2 (0.356±0.04,Mean±SD) in rotenone-treated (Fig. 4C) compared to sham-treated (0.19±0.02,Mean±SD) animals. A five-fold increase in the levels of myo-inositol was observed in week 3 (1.47±0.02, Mean±SD) and 4 (Fig. 4A) in rotenone-treated mice.
Figure 4.
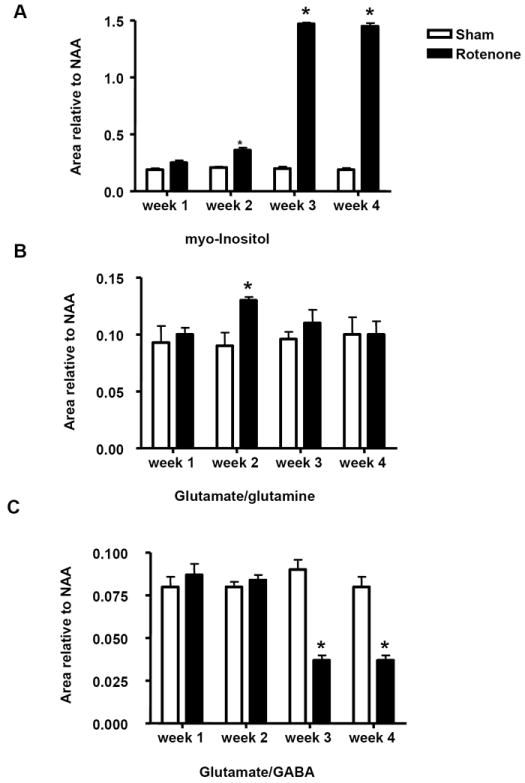
(A). Histograms represent the value of the area under the peak relative to NAA of myo-inositol, (B). glutamate and glutamine (Glx), and (C). glutamate and GABA, over a period of 4-week treatment with rotenone. * indicates significantly different to control, n=4, Mann Whitney U, P<0.05, mean ±SD.
Impairment of mitochondrial oxidative phosphorylation via complex I inhibition with rotenone induces profound changes in glutamate neurotransmission
The levels of glutamate and glutamine (Glu/Gln), which is the total pool of these amino acids (not the ratio), were significantly increased in week 2 (0.13±0.005, Mean±SD) in rotenone-treated (Fig. 4B) compared to sham-treated (0.093±0.015, Mean±SD) mice. The levels of glutamate/glutamine (Glx) reverted to control levels in week 3 and 4 (Fig. 3 and 4B). This change in glutamate/glutamine levels in week 2 was followed by a massive decrease in the total pool levels of glutamate and GABA (glutamate/GABA) in week 3 (0.036±0.005, Mean±SD) and week 4 (0.036±0.005, Mean±SD) in rotenone –treated (Fig. 3 and 4C) compared to sham-treated animals in week 3 (0.09±0.01, Mean±SD) and 4 (0.08±0.01, Mean±SD).
Results
The present work indicates anatomical, bio-energetic and biochemical disturbances in striatum of rotenone-treated mice. The significant changes in ventricular volumes and the degeneration of contiguous striatal nuclei as revealed by MRI are novel findings pertaining to striatal damage following a short term (3 injections over 3 days) treatment with rotenone. Voxel localized In vivo 1H spectroscopy in the subventricular zone and the striatum revealed significant bio-energetic changes, consistent with the inhibition of mitochondrial complex I with rotenone, and impairment of striatal glutamate transmission in the rotenone-treated mice compared to sham-treated control. More importantly, analysis of changes in indicators of energy substrates such as the creatine and phosphocreatine-containing substrate levels (Cr/pCr), myo-Inositol-containing substrates (myo-Inos) and lactate (Lac) is in agreement with the expected changes that result from energy deficits as a result of inhibition of complex I of the mitochondrial electron transfer chain with rotenone. The observed increase in these substrates may be a compensatory cellular mechanism to account for loss of ATP energy production following an insult to oxidative phosphorylation, one of the major sources of cellular energy supply. The creatine-phosphocreatine conversion pathway by the enzyme creatine kinase is one source from which the cell derives its ATP energy supply, so the level of these compounds (Cr/pCr) are indicative of cellular mechanisms that are activated to compensate for ATP energy loss as a result of rotenone-induced impairment of oxidative phosphorylation via inhibition of complex I of the mitochondria. Another compensatory mechanism for loss of mitochondrial ATP is the production of cytosolic energy from lactate in the form of NADH. Myo-inositol is another energy reservoir capable of producing cellular energy. Therefore, the surge in myo-inositol levels may also be indicative of cellular mobilization of energy reserves following rotenone treatment. These changes in cellular bio-energetics are indicative of the profound effects of rotenone treatment on the mouse striatum.
Inhibition of the first complex of the mitochondrial electron transfer chain (complex I) may be predicted to lead to major energy deficits, but the observed alterations to glutamate transmission, the most abundant excitatory amino acid in the CNS, are striking. The increase in glutamate-glutamine levels in week 2 that precedes the significant 2-fold decrease in glutamate-GABA levels observed in week 3 and 4, has significant ramifications for cell survival and homeostasis, and may serve to explain some very profound changes in rotenone-induced neurodegeneration. The inhibition of mitochondrial complex I and oxidative phosphorylation with rotenone may lead to an increase in the rate of the tricarboxylic acid (TCA) cycle (Fig 5), the second major mitochondrial pathway for maintenance of cellular energy supply [Moussa et al., 2002; Rae et al., 2005; Rae et al., 2006; Moussa et al., 2007]. Therefore, the increase in glutamate and glutamine pools may be indicative of changes in the glutamate-glutamine cycle, which is a major source of glutamate production and detoxification in the cell [Van der Berg & Garfinkel, 1971; Reijnierse et al., 1975; Bradford & Ward, 1976; Hamberger et al., 1983; Hamberger & Nystrom, 1984; Lehre et al., 1995; Schmidt et al., 1996; Danbolt 2000 and 2001], and this increase in glutamate level may result in excitotoxic death of neurons. In addition, an increase in the rate of TCA cycle to re-compensate for failure of oxidative phosphorylation also results in increased glutamate levels via the conversion of α-ketoglutarate (an intermediate of the TCA cycle) to glutamate [Reijnierse et al., 1975; Bradford & Ward, 1976; Lehre et al., 1995; Schmidt et al., 1996; Danbolt 2000 and 2001], which may cause further toxicity and cell death. Thus, the decreased levels of glutamate in week 3 and 4 may be the result of cytotoxic death due to the increase of glutamate levels, which is observed in week 2. Therefore, the effects of rotenone treatment on the CNS, primarily in relation to glutamate transmission, may be contributing factors to several neurodegenerative diseases. These studies provide novel insights on the effects of rotenone-induced changes on glutamate transmission, and other transmitter systems such as GABA, in the mouse striatum; and delineate possible mechanisms involved in the initiation of excitotoxic death of neurons as a result of exposure to environmental toxins such as rotenone.
Figure 5.
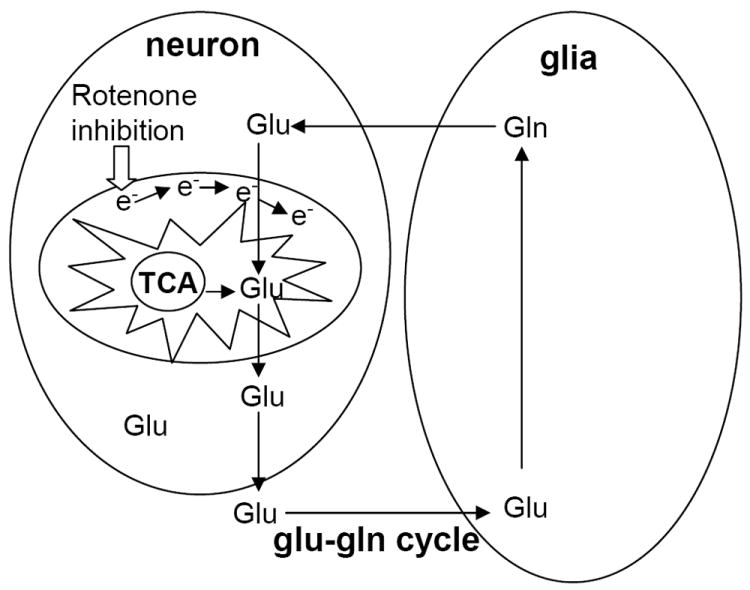
Schematic representation shows the intimate relationship between mitochondrial function (TCA cycle) and glutamate-glutamine (glu-gln) cycle.
Rotenone is extensively studied in animal models of Parkinson’s disease [Uversky, 2004]; where chronic rotenone treatment causes complex I inhibition uniformly throughout the brain [Bertabet et al., 2000; Alam & Schmidt, 2002], and cell death in the striatum [Greenamyre et al., 1999; Bertabet et al., 2000], therefore rotenone-induced changes seem to originate in the striatum and this is agreement with our results. In addition to the extensive evidence on rotenone toxicity to dopaminergic neurons [Bertabet et al., 2000; Alam & Schmidt, 2002; Hoglinger et al., 2003; Sherer et al., 2003c; Perier et al., 2003; Lapointe et al., 2004], evidence also exists that rotenone causes striatal lesion of non-dopaminergic neurons [Hoglinger et al., 2006], and glutamatergic dysfunction, which leads to further mitochondrial impairment [Greenamyre et al., 1999]. In comparison with chronic treatment with rotenone, a single systemic rotenone (acute) administration leads to increased glutamate-induced dopamine release in rat striatum [Leng et al., 2003], further supporting our hypothesis about the involvement of glutamate transmission in the early stages of the protracted death of striatal neurons after a short term treatment with rotenone. Taken together, the present studies and previous findings suggest that there is a relationship between rotenone toxicity and glutamate transmission in the CNS, which is an area of major interest in several human neurodegenerative diseases. These studies provide novel insights on the role of rotenone-induced changes to glutamate transmission in the CNS, and delineate possible mechanisms involved in the initiation of excitotoxic death of neurons as a result of exposure to environmental toxins such as rotenone. Furthermore, MRI/MRS studies are still very useful in the delineation of possible early chemical and anatomical bio-makers, such as neurotransmitters (MRS) and ventricular volumes (MRI), as a result of exposure to agricultural chemicals including rotenone and other environmental toxins.
Acknowledgments
These studies were supported in part by NIH grants NS04326 and AGO28108 [To Anita Sidhu], American Parkinson’s Disease Association [to Charbel E-H Moussa]; and NIH-National Center for Research Resources 5M01RR020359-03 (PI Batshaw M.L., Core Director Fricke S.T.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Azerberger T, Krampfl K, Leimgruber S, Weindl A. Changes of NMDA receptor subunit (NR1NR2B) and glutamate transporter (GLT-1) mRNA expression in Huntington’s disease-an in situ hybridization study. J Neuropathol Exp Neurol. 1997;56:440–454. doi: 10.1097/00005072-199704000-00013. [DOI] [PubMed] [Google Scholar]
- 2.Alam ZI, Schmidt WJ. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res. 2002;136:914–921. doi: 10.1016/s0166-4328(02)00180-8. [DOI] [PubMed] [Google Scholar]
- 3.Avallone J, Gashi E, Magrys B, Friedman LK. Distinct regulation of metabotropic glutamate receptor (mGluR1 alpha) in the developing limbic system following multiple early-life seizures. Exp Neurol. 2006;202:100–111. doi: 10.1016/j.expneurol.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 4.Bradford HF, Ward HK. On glutaminase in mammalian synaptosomes. Brain Res. 1976;110:115–125. doi: 10.1016/0006-8993(76)90212-2. [DOI] [PubMed] [Google Scholar]
- 5.Bismuth C, Garnier R, Baud FJ, Muszynski J, Keyes C. Paraquat poisoning. An overview of the current status. Drug Saf. 1990;5:243–251. doi: 10.2165/00002018-199005040-00002. [DOI] [PubMed] [Google Scholar]
- 6.Brujin LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD-1 containing inclusions. Neuron. 1997;18:327–328. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 7.Bertabet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 8.Curtis DR, Watkins JC. The chemical excitation of spinal neurons by certain amino acids. Journal of physiology (London) 1961;166:1–14. [Google Scholar]
- 9.Curtis DR, Johnson GAR. Amino acid transmitters in the mammalian central nervous system. Ergebn Physiol. 1974;69:94–188. doi: 10.1007/3-540-06498-2_3. [DOI] [PubMed] [Google Scholar]
- 10.Cleveland DW, Rothstein JD. From Charcot to LouGehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:1664. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 11.Cepeda C, Andre VM, Levine MS, Salamon N, Miyata H, Vinters HV, Mathern GW. Epileptogenesis in pediatric cortical displasia: the dysmature cerebral developmental hypothesis. Epilepsy Behav. 2006;9(2):219–235. doi: 10.1016/j.yebeh.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 12.Cowan CM, Raymond LA. Selective neuronal degeneration in Huntington’s disease. Curr Top Dev boil. 2006;75:25–41. doi: 10.1016/S0070-2153(06)75002-5. [DOI] [PubMed] [Google Scholar]
- 13.Danbolt NC. In: Sodium- and potassium-dependent amino acid transporters in brain plasma membrane. Bjorklund A, Hokfelt T, editors. 2000. pp. 231–254. [Google Scholar]; Ottersen OP, Strom-Mathisen J, editors. Handbook of chemical neuroanatomy. 18 [Google Scholar]; Glutamate. Elsevier; Amesterdam, Lausanne, New York, Oxford Shannon, Singapore, Tokyo: [Google Scholar]
- 14.Danbolt NC. Glutamate uptake (review) Progress Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 15.Fonnum F. Glutamate: a neurotransmitter in the mammalian brain (review) Journal of neuroschemistry. 1984;42(1):1–11. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- 16.Fall PA, Fredrikson M, Axelson O, Granerus AK. Nutritional and occupational factors influencing the risk of Parkinson’s disease: a case-control study in southeastern Sweden. Mov Disord. 1999;14:28–37. doi: 10.1002/1531-8257(199901)14:1<28::aid-mds1007>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 17.Fricke ST, et al. Consistent and reproducible slice selection in rodent brain using a novel stereotaxic device for MRI. J Neurosci Methods. 2004;136(1):99–102. doi: 10.1016/j.jneumeth.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ. The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50:1346–1350. doi: 10.1212/wnl.50.5.1346. [DOI] [PubMed] [Google Scholar]
- 19.Greenamyre JT, MacKenzie G, Peng TI, Stephans SE. Mitochondrial dysfunction in Parkinson’s disease. Biochem Soc Symp. 1999;66:85–97. doi: 10.1042/bss0660085. [DOI] [PubMed] [Google Scholar]
- 20.Greenamyre JT, Sherer TB, Betarbet R, Panov AV. Complex I and Parkinson’s disease. IUBMB Life. 2001;52:135–141. doi: 10.1080/15216540152845939. [DOI] [PubMed] [Google Scholar]
- 21.Gao HM, Hong JS, Zhang W, Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2002;22:782–790. doi: 10.1523/JNEUROSCI.22-03-00782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao HM, liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2003;23:6181–6187. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gubellini P, Eusebio A, Oueslati A, Melon C, Kerkerian-Le Goff L, Salin P. Chronic high frequency stimulation of the sub-thalamic nucleus and L-DOPA treatment in experimental parkinsonism; effects of motor behaviour and striatal glutamate transmission. Eur J Neuro Sci. 2006;24:1802–1814. doi: 10.1111/j.1460-9568.2006.05047.x. [DOI] [PubMed] [Google Scholar]
- 24.Hamberger A, Berthold CH, Karlsson B, Lehmann A, Nystrom B. Extracellular GABA, glutamate, and glutamine in vivo-perfusion dialysis of rabbit hippocampus. Neurol Neurobiol. 1983;7:473–492. [Google Scholar]
- 25.Hamberger A, Nystrom B. Extra- and intracellular amino acids in the hippocampus during development of hepatic encephalopathy. Neurochem Res. 1984;9:1181–1192. doi: 10.1007/BF00973033. [DOI] [PubMed] [Google Scholar]
- 26.Hatefi Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu Re Biochem. 1985;54:1015–1069. doi: 10.1146/annurev.bi.54.070185.005055. [DOI] [PubMed] [Google Scholar]
- 27.Higgins DS, Jr, Greenamyre JT. [3]Dihydrorotenone binding to NADH;ubiquitinone reductase (complex I) of the electron transfer chain: an autoradiographic study. J Neurosci. 1996;16:3807–3816. doi: 10.1523/JNEUROSCI.16-12-03807.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoglinger GU, Feger J, Prigent A, Michel PP, Parain K, Champy P, Ruberg M, Oertel WH, Hirsch EC. Chronic systemic complex I inhibition induces a hypojinetic mutlisystem degeneration in rats. J Neurochem. 2003;84:491–502. doi: 10.1046/j.1471-4159.2003.01533.x. [DOI] [PubMed] [Google Scholar]
- 29.Hayashi T, Shoji M, Abe K. Molecular mechanisms of ischemic neuronal cell death with relevance to Alzheimer’s disease. Curr Alzheimer Res. 2006;3:351–358. doi: 10.2174/156720506778249498. [DOI] [PubMed] [Google Scholar]
- 30.Kweon GR, Marks JD, Krencik R, Leung EH, Schumacker PT, Hyland K, Kang UJ. Distinct mechanisms of neurodegeneration induced by complex I inhibition in dopaminergic and non-dopaminergic cells. J boil Chem. 2004;279(50):51783–51792. doi: 10.1074/jbc.M407336200. [DOI] [PubMed] [Google Scholar]
- 31.Kirvell SL, Esiri M, Francis PT. Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer’s disease. J Neurochem. 2006;98:939–950. doi: 10.1111/j.1471-4159.2006.03935.x. [DOI] [PubMed] [Google Scholar]
- 32.Lehre KP, Levy LM, Otterson OP, Strom-Mathisen J, Danbolt NC. Differential expression of two glial glutamate tansporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, Mallory M, Alford M, Tanaka S, Masliah E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol. 1997;56:901–911. doi: 10.1097/00005072-199708000-00008. [DOI] [PubMed] [Google Scholar]
- 34.Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, Chen RC. Environmental risk-factors and Parkinson’s disease: a case-control study in Taiwan. Neurology. 1997;48:1583–1588. doi: 10.1212/wnl.48.6.1583. [DOI] [PubMed] [Google Scholar]
- 35.Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, Rothstein JD. Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron. 1998;20:589–602. doi: 10.1016/s0896-6273(00)80997-6. [DOI] [PubMed] [Google Scholar]
- 36.Lievens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol Dis. 2001;8:807–821. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- 37.Leng A, Feldon J, Ferger B. Rotenone increases glutamate-induced dopamine release but does not affect hydroxyl-free radical formation in rat striatum. Synapse. 2003;50:240–250. doi: 10.1002/syn.10260. [DOI] [PubMed] [Google Scholar]
- 38.Lapointe N, St Hilaire M, Martinoli MG, Blanchet J, Gould P, Rouillard C, Cicchetti F. Rotenone induces non-specific central nervous system and systemic toxicity. FASEB J. 2004;18:117–119. doi: 10.1096/fj.03-0677fje. [DOI] [PubMed] [Google Scholar]
- 39.Morano A, Jimenez-Jimenez FJ, Molina JA, Antolin MA. Risk-factors for Parkinson’s disease: case-control study in the province of Caceres, Spain. Acta Neurol Scand. 1994;89:164–170. doi: 10.1111/j.1600-0404.1994.tb01655.x. [DOI] [PubMed] [Google Scholar]
- 40.Masliah E, Alford M, De Teresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol. 1996;40:759–766. doi: 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- 41.Marder k, Logroscino G, Alfaro B, Mejia H, Halim A, Louis E, Cote L, Mayeux R. Environmental risk factors for Parkinson’s disease in an urban multiethnic community. Neurology. 1998;50:279–281. doi: 10.1212/wnl.50.1.279. [DOI] [PubMed] [Google Scholar]
- 42.Moussa E-HC, Mitrovic AD, Vandenberg RJ, Provis T, Rae C, Bubb WA, Balcar VJ. Effects of L-glutamate transport inhibition by a conformationally restricted glutamate analogue (2S, 1’S, 2’S)-2-(carboxycyclopropyl)glycine (L-CCGIII) on metabolism in brain tissue in vitro analyzed by NMR spectroscopy. Neurochem Res. 2002;27:27–35. doi: 10.1023/a:1014842303583. [DOI] [PubMed] [Google Scholar]
- 43.Moussa E-HC, Rae C, Bubb WA, Griffin JL, Deters NA, Balcar VJ. Inhibitors of glutamate transport modulate distinct patterns in brain metabolism. J Neurosci Res. 2007;85:342–350. doi: 10.1002/jnr.21108. [DOI] [PubMed] [Google Scholar]
- 44.Oechmichen M, Meissner C. Cerebral hypoxia and ischemia: the forensic point of view: a review. J Forensic Sci. 2006;51:880–887. doi: 10.1111/j.1556-4029.2006.00174.x. [DOI] [PubMed] [Google Scholar]
- 45.Perier C, Bove J, Vila M, Przedborski S. The rotenone model of Parkinson’s disease. Trends Neurosci. 2003;26:345–346. doi: 10.1016/S0166-2236(03)00144-9. [DOI] [PubMed] [Google Scholar]
- 46.Reijnierse GLA, Veldstra H, Van den Berg CJ. subcellular localization of gamma-aminobutyric transaminase and glutamate dehydrogenase in adult rat brain. J Neurochem. 1975;152:469–475. doi: 10.1042/bj1520469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roberts PJ, Storm-Mathesin J, Johnson GAR. Glutamate transmitter in the central nervous system. John Wiley and Sons; Chichester: 1981. [Google Scholar]
- 48.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 49.Rae C, Moussa E-HC, Griffin JL, Bubb WA, Wallis T, Balcar VJ. Group I and II metabotropic glutamate receptors alter brain cortical metabolic and glutamate/glutamine cycle activity: a 13C NMR spectroscopy and metabolomic study. J Neurochem. 2005;92:405–4016. doi: 10.1111/j.1471-4159.2004.02880.x. [DOI] [PubMed] [Google Scholar]
- 50.Rae C, Moussa E-HC, Griffin JL, Parekh SB, Bubb WA, Hunt NH, Balcar VJ. A metabolomic approach to ionotropic glutamate receptor subtype function: a nuclear magnetic resonance in vitro investigation. J Cereb Blood Flow Metab. 2006;26:1005–1017. doi: 10.1038/sj.jcbfm.9600257. [DOI] [PubMed] [Google Scholar]
- 51.Semchuk KM, Love EJ, Lee RG. Parkinson’s disease and exposure to agricultural work and pesticide chemicals. Neurology. 1992;42:1328–1335. doi: 10.1212/wnl.42.7.1328. [DOI] [PubMed] [Google Scholar]
- 52.Semchuk KM, Love EJ, Lee RG. Parkinson’s disease: a test of the mutlifactorial etiologic hypothesis. Neurology. 1993;43:1173–1180. doi: 10.1212/wnl.43.6.1173. [DOI] [PubMed] [Google Scholar]
- 53.Schmitt A, Asan E, Puschel B, Jons T, Kugler P. Expression of the glutamate transporter GLT1 in neuronal cells of the rat central nervous system: non-radioactive in situ hybridization and comparative immunohistochemistry. Neurosci. 1996;71:789–1004. doi: 10.1016/0306-4522(95)00477-7. [DOI] [PubMed] [Google Scholar]
- 54.Schuler F, Casida JE. Functional coupling of PSST and ND1 subunits in NADH;ubiquitinone oxireductase established by photoaffinity labeling. Biochem Biophys Acta. 2001;1506:79–87. doi: 10.1016/s0005-2728(01)00183-9. [DOI] [PubMed] [Google Scholar]
- 55.Scott HL, Pow DV, Tannenberg AE, Dodd PR. Aberrant expression of the glutamate transporter excitatory amino acid transporter 1 (EAAT1) in Alzheimer’s disease. J Neurosci. 2002;22:RC206. doi: 10.1523/JNEUROSCI.22-03-j0004.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sherer TB, Betarbet R, Kim JH, Greenamyre JT. Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci Lett. 2003a;341:87–90. doi: 10.1016/s0304-3940(03)00172-1. [DOI] [PubMed] [Google Scholar]
- 57.Sherer TB, Betarbet R, Tesla CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanisms of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 2003b;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003c;179:9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- 59.Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2:427–433. doi: 10.1038/8091. [DOI] [PubMed] [Google Scholar]
- 60.Talpade DJ, Greene JG, Higgins DS, Jr, Greenamyre JT. In vivo labeling of mitochondrial complex I (NADH:ubiquitinone oxireductase) in rat brain using [(3)]dihydrorotenone. J Neurochem. 2000;75:2611–2621. doi: 10.1046/j.1471-4159.2000.0752611.x. [DOI] [PubMed] [Google Scholar]
- 61.Uversky NV. Neurotoxicant-induced animal models of Parkinson’s disease: understanding the role of rotenone, maneb and paraquat in neurodegeneration. Cell and Tissue Res. 2004:441–476. doi: 10.1007/s00441-004-0937-z. [DOI] [PubMed] [Google Scholar]
- 62.Van den Berg CJ, Garfinkel D. A stimulation study of brain compartments-metabolism of glutamate and related substances in mouse brain. J Biochem. 1971;123:211–218. doi: 10.1042/bj1230211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vanacore N, Nappo A, Gentile M, Brustolin A, Palange S, Liberati A, Di Rezze S, Caldora G, Gasparini M, Benedetti F, Bonifati V, Forastiere F, Quercia A, Meco G. Evaluation risk of Parkinson’s disease in a cohort licensed pesticide users. Neurol Sci. 2002;(suppl 2):S119–120. doi: 10.1007/s100720200098. [DOI] [PubMed] [Google Scholar]
- 64.Van Landeghem FK, Weiss T, Oechmichen M, von Deimling A. Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J Neurotrauma. 2006;23:1518–1528. doi: 10.1089/neu.2006.23.1518. [DOI] [PubMed] [Google Scholar]
- 65.Van Damme P, Dewil M, Robberecht W, Van Den Bosch L. Excitotoxicity and amyotrophic lateral sclerosis. Neurodgener Dis. 2005;2:147–159. doi: 10.1159/000089620. [DOI] [PubMed] [Google Scholar]
- 66.Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, Gerig G. User-guided 3D active contour segmentation of anatomical structures: Significantly improved efficiency and reliability. Neuroimage. 2006 doi: 10.1016/j.neuroimage.2006.01.015. Article In Press. [DOI] [PubMed] [Google Scholar]