

Abstract
Nitric oxide (NO) regulates vascular smooth muscle cell (VSMC) structure and function, in part by activating soluble guanylate cyclase (sGC) to synthesize cGMP. The objective of this study was to further characterize the signaling mechanisms by which NO regulates VSMC gene expression using transcription profiling. DNA microarrays were hybridized with RNA extracted from rat pulmonary artery smooth muscle cells (RPaSMC) exposed to the NO donor compound, S-nitroso-glutathione (GSNO). Many of the genes, whose expression was induced by GSNO, contain a cAMP-response element (CRE), of which one encoded the inducible cAMP early repressor (ICER). sGC and cAMP-dependent protein kinase, but not cGMP-dependent protein kinase, were required for NO-mediated phosphorylation of CRE-binding protein (CREB) and induction of ICER gene expression. Expression of a dominant-negative CREB in RPaSMC prevented the NO-mediated induction of CRE-dependent gene transcription and ICER gene expression. Pre-treatment of RPaSMC with the intracellular calcium (Ca2+) chelator, BAPTA-AM, blocked the induction of ICER gene expression by GSNO. The store-operated Ca2+ channel inhibitors, 2-ABP and SKF-96365, reduced the GSNO-mediated increase in ICER mRNA levels, while 2-ABP did not inhibit GSNO-induced CREB phosphorylation. Our results suggest that induction of ICER gene expression by NO requires both CREB phosphorylation and Ca2+ signaling. Transcription profiling of RPaSMC exposed to GSNO revealed important roles for sGC, PKA, CREB, and Ca2+ in the regulation of gene expression by NO. The induction of ICER in GSNO-treated RPaSMC highlights a novel cross-talk mechanism between cGMP and cAMP signaling pathways.
Keywords: nitric oxide, vascular smooth muscle, cAMP-response element, cyclic GMP, protein kinase A
1. INTRODUCTION
Nitric oxide (NO) is a critical regulator of vascular remodeling via its effects on endothelial and vascular smooth muscle cell (VSMC) differentiation, proliferation, apoptosis and migration [1-3]. NO elicits its effects, in part, by stimulating soluble guanylate cyclase (sGC) to produce the intracellular second messenger cGMP. sGC is a heterodimer composed of β1 subunit and an α1 or α2 subunit, and the sGCα1β1 is the predominant isoform in the vasculature. cGMP, in turn, activates several intracellular target proteins including cGMP-dependent protein kinase (PKG), cGMP-gated channels, and cGMP-regulated phosphodiesterases (PDEs) [4], as well as under some circumstances, cAMP-dependent protein kinase (PKA) [5]. PKG phosphorylates serines and threonines on many cellular proteins, frequently resulting in changes in activity, function, or subcellular localization. In VSMC, the proteins that are phosphorylated by PKG regulate calcium homeostasis and calcium sensitivity of contractile proteins, as well as the expression of genes that modulate VSMC phenotype [4]. cGMP activates PDE2 and inhibits PDE3, thereby decreasing and increasing intracellular cAMP levels respectively [2], often in cell compartment-selective manner. The transcription factor, cAMP-response element (CRE)-binding protein (CREB) [6] is phosphorylated in response to a variety of stimuli including increases in the levels of intracellular calcium (Ca2+) and cyclic nucleotides. Phosphorylation of CREB at Ser133 by kinases, such as PKG and PKA [7], induces CREB to bind to CRE sequences in the promoters of multiple genes encoding c-fos, heme oxygenase 1 (HO1), hexokinase-2 (HK2), and inducible cAMP early repressor (ICER) [6, 8, 9]. The activity of CREB is limited, in part, by its induction of ICER, which binds to CRE sequences but lacks a transactivation domain [10].
In addition to activating gene transcription via cyclic nucleotide-stimulated phosphorylation of CREB, NO can modulate gene expression via Ca2+ –dependent mechanisms. Previous studies have identified several mechanisms by which NO can modulate intracellular Ca2+ levels including modulation of a variety of Ca2+ channels, for example voltage-dependent calcium channels (VDCC) and store-operated calcium channels (SOCC) [12]. NO regulates Ca2+ regulatory proteins and channels via cGMP/PKG-dependent mechanisms or, alternatively, via cGMP-independent, post-translational modifications [13, 14].
To further understand the mechanisms by which NO regulates gene expression in VSMC, we examined the transcriptional profile of rat pulmonary artery smooth muscle cells (RPaSMC) exposed to the NO-donor compound, S-nitroso-glutathione (GSNO). We observed that many of the genes induced by GSNO contain CRE sequences in their promoters. One of these CRE-containing genes, ICER, was markedly induced in RPaSMC exposed to GSNO and in lungs of mice breathing NO. We report here that NO increases ICER gene expression via mechanisms requiring sGC, PKA, and Ca2+, but not PKG. Moreover, NO-mediated phosphorylation of CREB is required, but not sufficient, for induction of ICER gene expression by NO.
2. MATERIALS AND METHODS
All experimental studies using animals were approved by the MGH Subcommittee on Research Animal Care.
2.1) Cell culture
Cultures of primary RPaSMC were prepared as described previously [15].
2.2) Microarray analysis
Two independent preparations of RPaSMCs were untreated or exposed to GSNO (100 μmol/L) for 1, 2, and 4 hours (h). RNA, cDNA, and labeled cRNA were generated as described in the online supplement. Fragmented cRNA was hybridized to Affymetrix rat U-34A GeneChips containing 8798 gene entries with the GeneChip Fluidics Station 450 and scanned with the GeneChip® Scanner 3000. Affymetrix GeneChip 5.0 software was used for data analysis according to Affymetrix protocols. The microarray data including information requested by the Microarray Gene Expression Data society can be accessed in the microarray database at NCBI Gene Expression Omnibus (GEO) using the following link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=thqflmmsosekire&acc=GSE19710.
GSNO was determined to alter gene expression if, in both RPaSMC preparations, there was a ≥ 2-fold change in gene expression and if the gene was described as present (P) by Affymetrix detection call after GSNO stimulation for genes whose expression was increased or before GSNO stimulation for genes whose expression was decreased. In a second approach, we organized gene expression data into microarray pathway profiles (MAPPs) that represent NO-regulated biological pathways [16]. To facilitate the identification of NO-responsive MAPPs, a less stringent criterion for change in gene expression (1.5-fold) was used.
CRE sequences were identified in genes upregulated after 4h of GSNO stimulation using the CRE sequence database (http://natural.salk.edu/CREB/) [17].
2.3) Measurement of gene expression
mRNA levels were measured using RNA blot hybridization and quantitative RT-PCR, as described in the online supplement.
2.4) Protein levels
Levels of ICER, total CREB, and phospho-CREB proteins were measured using immunoblot techniques as described in the online supplement.
2.5) NO exposure in vivo
Male WT mice (C57Bl/6) breathed air with or without 80 ppm NO for 2, 4, 8 and 24h in specially-designed chambers, as described previously [18]. Additionally, WT mice and mice deficient for the α1 subunit of sGC [19] breathed air with or without NO (80 ppm) for 2h. Lung tissue was harvested for measurement of ICER mRNA levels. Plasma nitrite and nitrate levels were measured as described in the online supplement.
2.6) CRE-dependent gene transcription
RPaSMC were plated in 6-well dishes (2×105 cells/ well) and transfected for 2h with a control plasmid (pcDNA3) or a plasmid encoding ICER plasmid directing expression of firefly luciferase under the control of a CRE-containing promoter and a plasmid directing expression of renilla luciferase (pRL-TK, Promega) to control for variations in transfection efficiency. After 24h, transfected cells were incubated in the presence and absence of GSNO with other agonists and antagonists as indicated in the Results section. After 4h, firefly and renilla luciferase activities were measured using the dual-luciferase reporter system (Promega). CRE-dependent gene transcription was estimated from the ratio of firefly luciferase activity to renilla luciferase activity.
2.7) Recombinant adenovirus generation
A replication-deficient adenovirus specifying rat ICER cDNA (Ad.ICER) was generated using the methods described by He and colleagues [20]. A full-length rat ICER cDNA was cloned into the HindIII and EcoRV restriction sites of the shuttle vector pAd.Track.CMV. A control adenovirus specifying β-galactosidase (Ad.βgal) generated using identical methods was provided by Dr. Anthony Rosenzweig (Beth Israel Deaconess Medical Center, Boston, Massachusetts). Both recombinant adenoviruses carry a second transcription cassette for green fluorescent protein (GFP). Wild-type adenovirus contamination was excluded by the absence of PCR-detectable E1 sequences.
The adenovirus carrying a dominant-negative CREB, in which Ser133 was mutated to alanine (Ad.CREBml), was provided by Dr. William Walker (University of Pittsburgh).
For adenovirus-mediated gene transfer, cells were infected with Ad.CREBml, Ad.ICER, or Ad.βgal for 2h in RPMI medium containing 10% NuSerum. Infected cells were then incubated in the same medium for 24h.
2.8) Statistics
All data are expressed as mean ± standard deviation (SD). Data were analyzed using the student t test, 1-way ANOVA with Bonferroni post-hoc test for multiple comparisons, or 2-way ANOVA for multiple comparisons including Bonferroni post-hoc test. Statistical significance was considered for p values <0.05 with statement of the exact p values. Guidelines for reporting statistics in journals published by the American Physiological Society were followed [21]. For additional details, see the supplemental information containing detailed Materials and Methods, Supplemental Figures and Tables.
3. THEORY/ CALCULATION
Nitric oxide (NO) regulates vascular smooth muscle cell (VSMC) structure and function. The objective of this study was to further characterize the signaling mechanisms by which NO regulates VSMC gene expression using transcription profiling. We characterized the transcriptional profile of rat pulmonary artery smooth muscle cells treated with and without nitric oxide in order to further elucidate the signaling mechanisms by which NO regulates VSMC structure and function. We identified several genes whose expression was up- or down-regulated by NO. We focused on one gene that was among the most markedly induced by GSNO, the inducible cAMP early repressor (ICER), to elucidate the mechanisms by which NO modulates gene transcription in RPaSMC.
4. RESULTS
4.1) Exposure to GSNO alters the transcriptional profile of RPaSMC
To examine the impact of NO on VSMC gene expression, we used DNA microarrays (>8000 gene entries) and RNA extracted from two independent isolates of RPaSMC (which contain abundant sGC [22]) exposed to GSNO (100 μmol/L) for 1, 2 and 4h. GSNO increased the expression of 65, 101, and 138 genes after 1, 2, and 4h, respectively. GSNO decreased expression of 18, 50, and 129 genes after 1, 2, and 4h, respectively (Supplemental Fig. 1, Supplemental Table 1). Genes, whose expression was increased by GSNO as detected by microarray analysis and confirmed by RNA blot hybridization, include inducible cAMP early suppressor (Fig. 1A, HO1, HK2, tissue plasminogen activator (tPA), metallothionein 1 (MT1), γ-glutamylcysteine synthetase heavy chain (GCS:hc) and light chain (GCS:lc), and p21(Waf1/Cip1) (Supplemental Fig. 2, Supplemental Table 2). Genes, whose expression was decreased by GSNO, include those encoding sGC α1 and β1 subunits, endothelin 1 (ET1), angiotensin II receptor (AT2R), and transforming growth factor-β3 (TGFβ3).
Fig. 1.
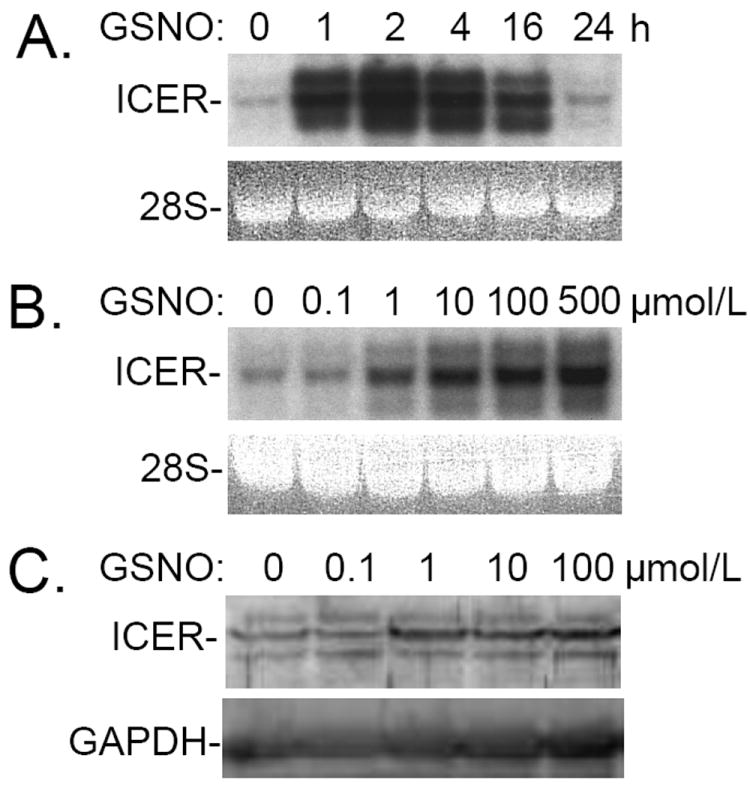
Panel A: Incubation with S-nitroso-L-glutathione (GSNO) induces ICER gene expression in rat pulmonary artery smooth muscle cells (RPaSMC) in a time- dependent manner. RNA was extracted from untreated RPaSMC and RPaSMC treated with GSNO for 1, 2, 4, 16, and 24h. RNA blot hybridization was performed using radiolabeled DNA probes specifying ICER. Ethidium bromide staining of 28S rRNA in the gel was scanned and confirmed the equal loading of RNA samples. A representative blot of at least 3 experiments is shown. Panel B: Incubation with S-nitroso-L-glutathione (GSNO) induces ICER gene expression in RPaSMC in a dose-dependent manner. RNA was extracted from untreated RPaSMC and RPaSMC exposed to varying concentrations of GSNO for 2h. RNA blot were hybridized with DNA probes specifying ICER. Ethidium bromide staining of 28S rRNA confirmed the equal loading of RNA samples. A representative blot of at least 3 experiments is shown. Panel C: Proteins were extracted from RPaSMC treated without or with 0.1, 1, 10, 100 μmol/L of GSNO for 4h. Immunoreactive ICER was detected using CREM-1 antibodies, and GAPDH was used as a loading control.
GenMAPP was used to organize gene expression data into MAPPs, that represent specific biological pathways and functionally grouped genes modulated by GSNO based on the gene ontology (GO) system [16]. NO-modulated MAPPs included pathways involved in oxidative stress, apoptosis, and glutathione biosynthesis (Supplemental Fig. 3A-C). Examination of the 5’ flanking sequences of genes, whose expression was induced by GSNO at 4h, revealed that 61% contain a cAMP-response element (10 occurrences of full site CREs, TGACGTCA, and 68 half site CREs, TGACG/CGTCA; data not shown) [17]. Subsequent studies focused on ICER gene expression as an example of a CRE-containing gene, whose expression is robustly induced by NO.
4.2) Nitric oxide increases ICER gene expression in RPaSMC
To validate the microarray data, ICER mRNA levels were measured in RPaSMC incubated with or without GSNO (100 μmol/L) for 1, 2, 4, 16, and 24h (Fig. 1A). ICER mRNA levels increased starting at 1h after GSNO stimulation, reaching maximum levels between 2 and 4h. ICER gene expression remained elevated for at least 16h and declined to basal levels at 24h. Incubation of RPaSMC with GSNO for as little as 2 hrs increased ICER protein levels (data not shown). The GSNO-mediated increase in ICER gene expression was dose-dependent: increased ICER mRNA levels were detected with exposure to as little as 1 μmol/L GSNO with maximal effects observed with 10 μmol/L (Fig. 1B). Similar dose-dependent effects of GSNO were observed when ICER protein levels were measured (Fig. 1C).
4.3) Cytokines increase ICER gene expression in RPaSMC
Exposure of RPaSMC to the cytokines interleukin 1β (IL-1β) and tumor necrosis factor α (TNFα) induces expression of NO synthase 2 (NOS2) resulting in increased NO levels [22]. To determine whether endogenous NO levels produced in response to cytokines are sufficient to induce ICER gene expression, NOS2 and ICER mRNA levels were measured in RPaSMC exposed to a combination of IL-1β and TNFα. Incubation of RPaSMC with IL-1β and TNFα for 4h induced both NOS2 and ICER gene expression. Pretreatment of RPaSMC with L-NIL (0.5 mol/L), a selective NOS2 inhibitor, blocked the ability of cytokines to induce ICER, but not NOS2 gene expression (Fig. 2A). These results suggest that endogenously-produced levels of NO are sufficient to induce ICER gene expression in RPaSMC.
Fig. 2.
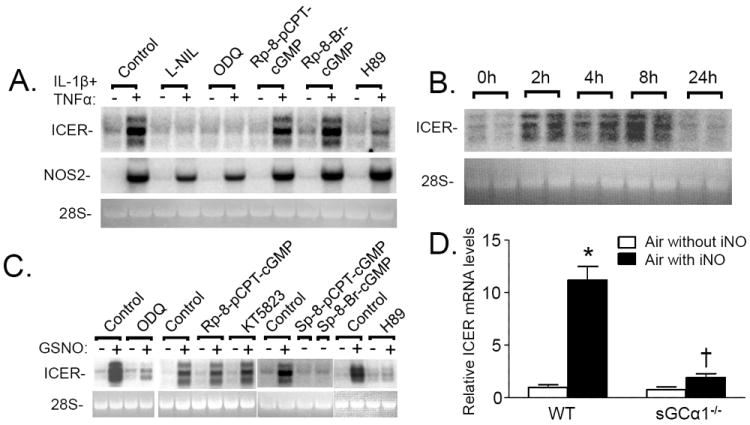
Panel A: Cytokines increase ICER gene expression in RPaSMC via NO- and cGMP-dependent mechanisms. RNA was extracted from untreated RPaSMC or RPaSMC incubated with IL-1β (20 ng/ml) and TNFα (100 ng/ml) for 4h. Similarly-treated RPaSMC were pretreated without or with the NOS2 inhibitor, L-NIL (0.5 mmol/L); the sGC inhibitor, ODQ (10 μmol/L); PKG antagonists, Rp-8-pCPT-cGMP (100 μmol/L) and Rp-8-Br-cGMP (100 μmol/L); or the PKA inhibitor, H89 (10 μmol/L). Membranes were hybridized with DNA probes specifying rat ICER and NOS2; the experiment shown is representative of 3 separate experiments. Panel B: Inhaled nitric oxide (NO) increases pulmonary ICER gene expression in vivo. RNA was extracted from lungs of wild-type mice that breathed air supplemented with NO gas (80 ppm) for 2, 4, 8, or 24h. Membranes were hybridized with DNA probes specifying ICER; the experiment shown is representative of 3 different experiments. Panel C: GSNO induces ICER in RPaSMC via PKA. RNA blots were prepared from RPaSMC pretreated with or without ODQ (10 μmol/L), PKG inhibitors, Rp-8-pCPT-cGMP (100 μmol/L) and KT5823 (1 μmol/L); H89 (10 μmol/L); or cGMP analogues, Sp-pCPT-cGMP (100 μmol/L) and Sp-8-Br-cGMP (100 μmol/L) for 30 min followed by incubation with and without GSNO (100 μmol/L) for 2h. Membranes were hybridized with DNA probes specifying ICER; the experiment shown is representative of 3 different experiments. Panel D: WT and sGCα1-/-mice breathed air supplemented with or without NO gas (80 ppm) for 2h. Pulmonary ICER mRNA levels were induced in WT mice breathing air supplemented with NO (*p<0.0001 lungs of WT mice breathing air vs breathing air supplemented with NO). sGCα1-/-mice showed a minor trend to induce ICER mRNA levels when breathing NO, that was significantly less than the induction of WT mice that breathed air supplemented with NO (†p<0.0001 sGCα1-/-mice breathing NO vs WT mice breathing air with iNO. WT mice increased hin=5 mice in each group; mean (SD).
4.4) Inhaled NO induces pulmonary ICER gene expression
To determine whether NO induces ICER gene expression in vivo, we measured pulmonary ICER mRNA levels in C57Bl/6 (WT) mice breathing air supplemented with NO (80 ppm) gas for 0, 2, 4, 8, and 24h. Increased pulmonary ICER mRNA levels were evident in WT mice within 2h of initiating NO inhalation, and elevated ICER mRNA levels persisted at least 8h (Fig. 2B). Interestingly despite continued NO inhalation, pulmonary ICER mRNA levels returned to baseline by 24h. Consistent with mRNA levels, pulmonary ICER protein levels in WT mice breathing NO for 2 hrs were greater than those in mice breathing air (Supplemental Fig. 4).
4.5) Mechanisms by which NO induces ICER gene expression
NO is known to regulate cellular functions via both cGMP-dependent and -independent mechanisms. To investigate the role of cGMP signaling in induction of ICER gene expression by GSNO, we studied RPaSMC incubated in the presence and absence of the sGC inhibitor ODQ. Pretreatment of RPaSMCs with ODQ markedly inhibited the ability of GSNO (Fig. 2C) or cytokines (Fig. 2A) to increase ICER mRNA levels. To confirm the importance of sGC in the upregulation of ICER gene expression by NO, we compared pulmonary ICER mRNA levels in WT and sGCα1-/- mice [19] breathing NO (Fig. 2D). Inhalation of NO (80 ppm) for 2 hours increased pulmonary ICER mRNA levels to a much greater extent in WT than in sGCα1-/- mice, whereas plasma nitrite and nitrate levels increased similarly in the two genotypes (Supplemental Fig. 5). These results suggest that sGCα1β1 is critical to the ability of NO to induce ICER gene expression.
To elucidate the mechanisms by which cGMP modulates ICER gene expression, we studied RPaSMC pretreated with the PKG inhibitors, Rp-8-Br-cGMPS, Rp-8-pCPT-cGMPS and KT5823. PKG inhibition did not alter the ability of GSNO or cytokines to increase ICER gene expression (Fig. 2A, C). In complementary studies, treatment of RPaSMC with membrane-permeable cGMP analogues that specifically activate PKG, Sp-8-pCPT-cGMP and Sp-8-Br-cGMP, did not augment ICER gene expression (Fig. 2C). The inability of PKG activators and inhibitors to modulate ICER gene expression was not attributable to the absence of PKG in RPaSMC, as the Iα isoform of PKG was readily detected in RPaSMC using RNA blot and immunoblot techniques (data not shown).
It is known that ICER gene expression is induced by cAMP signaling and PKA [7]. To test whether PKA activity is required for the induction of ICER gene expression by GSNO, we studied RPaSMC incubated in the presence and absence of H89, an inhibitor of PKA [23, 24]. Pretreatment with H89 prevented the increase in ICER mRNA levels induced by cytokines (Fig. 2A) and GSNO (Fig. 2C, Supplemental Fig. 6). Taken together, these results suggest that activation of sGC and PKA, but not PKG, is required for the induction of ICER gene expression by NO.
4.6) GSNO stimulates CRE-dependent gene transcription
Since ICER gene expression is known to be regulated by CRE-dependent transcription factors [25], and because the 5’ flanking sequences of many of the NO-inducible genes contain a cAMP-response element, we examined the ability of GSNO to activate CRE-dependent gene transcription in RPaSMC. RPaSMC were transiently transfected with a luciferase reporter plasmid containing an upstream CRE site and subsequently exposed to GSNO. Incubation with GSNO for 4h markedly increased CRE-dependent luciferase activity (Fig. 3A). Pretreatment with ODQ prevented the induction of CRE-dependent gene transcription by GSNO, whereas PKG inhibitors had no effect. Moreover, PKG agonists did not stimulate CRE-dependent gene transcription (Supplemental Fig. 7), whereas the cAMP analogue, 8-Br-cAMP, increased luciferase activity. On the other hand, inhibition of PKA with H89 blocked the ability of GSNO to induce CRE-dependent gene transcription (Fig. 3B). Taken together, these findings suggest that NO activates CRE-dependent gene transcription via a cGMP synthesis- and PKA-dependent mechanism.
Fig. 3.
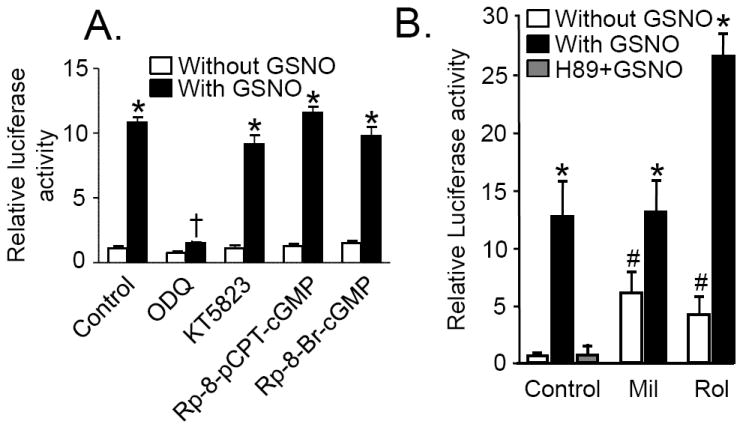
Panel A: GSNO mediated cAMP response element (CRE)-dependent gene transcription is sGC-dependent. RPaSMC transfected with CRE-luciferase reporter plasmid were preincubated with ODQ (10 μmol/L), KT5823 (1 μmol/L), Rp-8-pCPT-cGMP (100 μmol/L), or Rp-8-Br-cGMP (100 μmol/L) for 30min and then stimulated with or without GSNO (100 μmol/L) for 4h. The results are expressed as firefly luciferase activity normalized to control renilla luciferase activity. *p<0.0005 GSNO treated cells vs control cells, either unstimulated, or pretreated with KT5823, Rp-8-pCPT-cGMP or RP-8-Br-cGMP; and †p<0.0001 ODQ- vs vehicle-pretreated treated RPaSMC stimulated with GSNO. Panel B: RPaSMCs incubated with Rolipram (Rol) plus GSNO induce CRE-dependent gene transcription more effectively than RPaSMC treated with GSNO alone RPaSMCtreated with GSNO plus milrinone (mil). RPaSMC transfected with CRE-luciferase reporter plasmid were pretreated with H89 (10 μmol/L), Mil (30 μmol/L) or Rol (10 μmol/L) for 30 min and stimulated with or without GSNO (100 μmol/L) for 4h. Firefly and renilla luciferase activities were measured. *p<0.001 versus control, # p<0.001 vs GSNO only. n≥3 for each condition; mean (SD).
To investigate the mechanisms by which NO can activate CRE-dependent gene transcription via PKA, we studied the effects of milrinone, a PDE3 inhibitor, and rolipram, an inhibitor of the cAMP-specific PDE4. Incubation of CRE-luciferase-transfected RPaSMC with milrinone or rolipram augmented CRE-dependent gene transcription (Fig. 3B). However, milrinone did not augment the ability of GSNO to induce luciferase activity, whereas rolipram markedly augmented the induction of CRE-dependent gene transcription by GSNO. These findings suggest that NO/cGMP signaling and milrinone activate CRE-dependent gene transcription via the same mechanism, inhibition of PDE3. Moreover, the importance of cAMP signaling in the stimulation of CRE-dependent gene transcription by NO is highlighted by the ability of rolipram to augment the effects of GSNO.
4.7) Activation of CREB is required for GSNO-mediated ICER induction
When CREB undergoes phosphorylation at Ser133, it binds to CRE sequences and induces gene transcription [25]. Since we observed that GSNO induces CRE-dependent gene transcription, we examined whether GSNO can induce CREB phosphorylation. Incubation of RPaSMC with either GSNO or forskolin (an activator of adenylate cyclase) rapidly induced CREB phosphorylation (Fig. 4A). ODQ inhibited the ability of GSNO, but not forskolin, to induce CREB phosphorylation. In contrast, H89 inhibited phosphorylation of CREB by both GSNO and forskolin. These results suggest that PKA is required for GSNO-mediated CREB phosphorylation.
Fig. 4.
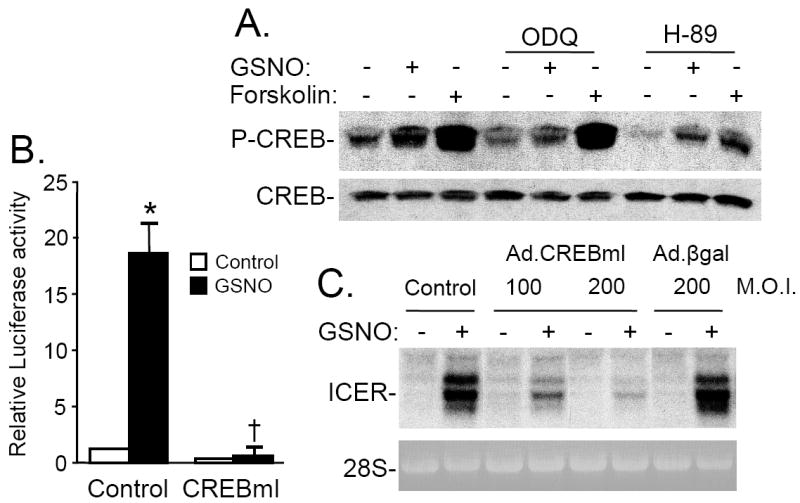
Panel A: GSNO mediated CREB phosphorylation is sGC- and PKA-dependent. RPaSMC preincubated without or with ODQ (10 μmol/L) or H-89 (10 μmol/L) were stimulated with GSNO or forskolin. Phosphorylated CREB (p-CREB) and total CREB were detected with immunoblotting. Panel B: Activation of CREB is required for induction of CRE luciferase activity in GSNO-stimulated RPaSMC. RPaSMC were transfected with a CRE-responsive luciferase plasmid without or with a plasmid specifying a dominant-negative CREB (CREBml) and stimulated with and without GSNO. Four hours later,firefly and renilla luciferase activities were measured. n=3; means (SD); *p<0.001 vs control, †p<0.001 vs GSNO control). Panel C: Activation of CREB is required for GSNO-mediated ICER induction. Uninfected cells and cells infected with Ad.CREB-ml (100 and 200 M.O.I.) or Ad.βgal (200 M.O.I.) were incubated with GSNO. Membranes were hybridized with DNA probes specifying ICER. Ethidium bromide staining of 28S rRNA confirmed the equal loading of RNA samples.
To further examine the role of CREB in the induction of CRE-dependent gene expression by GSNO, we transfected RPaSMC with a CRE-luciferase reporter plasmid together with a plasmid encoding CREBml, a dominant-negative CREB mutant in which Ser133 is replaced with an alanine [26]. Co-transfection with CREBml prevented the GSNO-mediated increase in CRE-luciferase expression (Fig. 4B). To test whether CREB activation was required for the induction of ICER gene expression by NO, RPaSMC were infected with an adenovirus specifying CREBml (Ad.CREBml) or a control adenovirus specifying β-galactosidase (Ad.βgal). Infection with Ad.CREBml, but not Ad.βgal, markedly attenuated the ability of GSNO to increase ICER mRNA levels (Fig. 4C). Taken together, these results suggest that NO-mediated phosphorylation of CREB is sGC- and PKA-dependent and that NO-mediated induction of CRE-dependent gene transcription, as well as ICER gene expression, requires phosphorylation and activation of CREB.
4.8) Induction of ICER gene expression by NO requires intracellular calcium
Previous studies found that the ability of NO to induce gene expression required the synergistic effects of PKA activation and an increase in Ca2+ signaling [2, 4, 11, 12]. Moreover, CREB is phosphorylated in response to increases in intracellular Ca2+ levels [27, 28]. To test whether intracellular Ca2+ was required for the induction of ICER mRNA levels by NO, we studied RPaSMC pretreated with the Ca2+ chelator, BAPTA-AM. Chelation of intracellular Ca2+ with BAPTA-AM markedly attenuated the ability of GSNO to induce ICER gene expression in RPaSMC (Fig. 5A). Similarly, 2-ABP and SKF-96365, inhibitors of SOCCs, blocked the induction of ICER mRNA levels by GSNO (Fig. 5B) and forskolin (Supplemental Fig. 8). In contrast, inhibition of the voltage-dependent Ca2+-channels (VDCCs) with nifedipine did not prevent the ability of GSNO to induce ICER gene expression (Fig. 5C). Our results suggest that Ca2+-mediated ICER induction requires SOCCs.
Fig. 5.
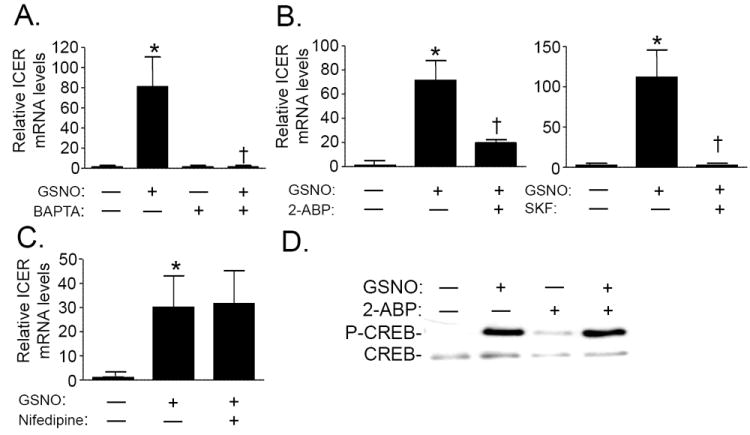
Intracellular calcium is required for GSNO-mediated ICER induction. RPaSMC were pretreated with the intracellular Ca2+-chelator, BAPTA-AM (50 μmol/L; Panel A) store-operated Ca2+inhibitors, 2-ABP and SKF-96365 (Panel B); or the voltage dependent Ca2+-channel inhibitor, nifedipine (100 μmol/L) (Panel C), followed by incubation with or without GSNO (100 μmol/L). ICER mRNA levels were detected using qRT-PCR. n=3; mean (SD). (Panel A)*p=0.041 control vs GSNO-treated cells, †p=0.042 vs GSNO-treated cells without BAPTA-AM. (Panel B)Left panel: *p=0.037 control vs GSNO-treated cells, †p=0.017 RPaSMC pretreated with 2-ABP followed by GSNO vs. GSNO-treated cells. Right panel: *p=0.007 control vs GSNO-treated cells, †p=0.008 RPaSMC pretreated with SKF followed by GSNO vs. GSNO-treated cells. (Panel C)*p=0.02 control vs GSNO treated cells. Incubation with nifedipine did not alter ICER mRNA levels. (Panel D)GSNO stimulates CREB phosphorylation in presence of 2-ABP. Cells were preincubated with or without 2-ABP for 30 min and exposed to GSNO (100 μmol/L) for 4hrs. p-CREB and CREB phosphorylation were detected using immunoblot. Incubation of RPaSMC with GSNO induced CREB phosphorylation. CREB phosphorylation was not prevented by preincubation with the store-operated Ca2*inhibitor 2-ABP. A representative result of 3 different experiments is shown.
To test whether Ca2+ is required for the activation of CREB in RPaSMC, we measured CREB phosphorylation in RPaSMC exposed to 2-ABP. In the presence of 2-ABP, GSNO was able to induce CREB phosphorylation (Fig. 5D). Together, these findings suggest that intracellular Ca2+ is required for the induction of ICER gene expression by GSNO. However, phosphorylation of CREB in response to GSNO does not require SOCC activity. Furthermore, CREB phosphorylation, alone, is not sufficient to induce ICER gene expression in RPaSMC.
4.9) ICER inhibits cAMP signaling in RPaSMC
To explore the impact of ICER gene expression on CRE-dependent gene transcription, we studied RPaSMC transiently transfected with the CRE-luciferase reporter plasmid and a plasmid encoding ICER (pcDNA3-ICER). Transfection of pcDNA3-ICER attenuated the ability of GSNO or forskolin to activate CRE-dependent luciferase expression (Fig. 6A). Moreover infection of RPaSMC with an adenovirus specifying ICER (Ad.ICER), but not with a control adenovirus Ad.βgal, inhibited the ability of forskolin to increase expression of genes encoding HK2 and GCS:hc (Fig. 6B). These results suggest that induction of ICER in RPaSMC inhibits cAMP-stimulated CRE-dependent gene transcription.
Fig. 6.
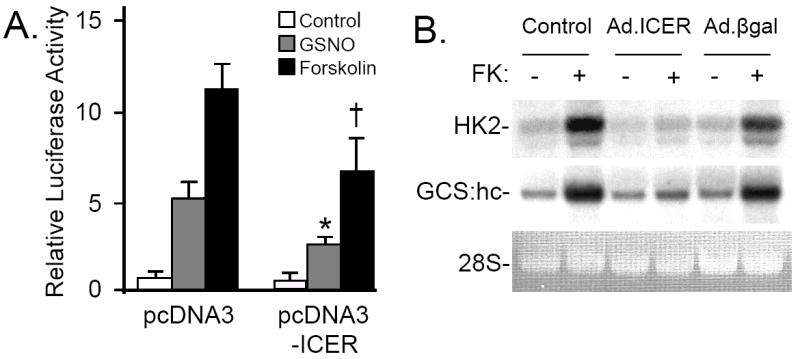
Panel A: ICER inhibits cAMP-dependent gene transcription. RPaSMC were transiently transfected with the CRE-luciferase reporter plasmid and a control plasmid (pcDNA3) or a plasmid encoding ICER (pcDNA3-ICER) for 24h. Transfected cells were treated with GSNO (100 μmol/L) or forskolin (FK; 10 μmol/L) for 4h. Firefly and renilla luciferase activities was measured. n=3, mean (SD); *p<0.001 RPaSMC transfected with pcDNA3 and stimulated with GSNO vs cells transfected with pcDNA3-ICER stimulated with GSNO, †p<0.001 RPaSMC transfected with pcDNA3 and stimulated with forskolin vs cells transfected with pcDNA3-ICER and stimulated with forskolin. Panel B: ICER inhibits cAMP-stimulated CRE-dependent gene expression. Uninfected RPaSMC and RPaSMC infected with Ad.ICER or Ad.βgal (100 M.O.I. each) were incubated with FK (10 μmol/L) for 2h. RNA blot hybridization was performed using radiolabeled DNA probes specifying hexokinase2 (HK2) and γ-glutamylcysteine synthetase heavy chain (GCS:hc) probes.
5. Discussion
Using transcription profiling, we found that exposure to NO for up to 4h dramatically changed expression of a large number of genes in RPaSMC (Supplemental Fig. 1, 2). The majority of the genes up-regulated by GSNO contained cAMP-response elements. One of the CRE-containing genes encodes ICER, which was induced more than 100-fold by GSNO. To further elucidate the pathways by which NO regulated the expression of CRE-containing genes in RPaSMC, we focused our studies on the ICER gene (Fig. 1). We found that induction of ICER gene expression by NO was mediated by a sGC- and cGMP-dependent mechanism (Fig. 2, 3), PKA signaling, and activation of the CRE-binding transcription factor, CREB (Fig. 4). The proposed mechanisms regulating ICER gene expression are summarized in Figure 7.
Fig. 7.
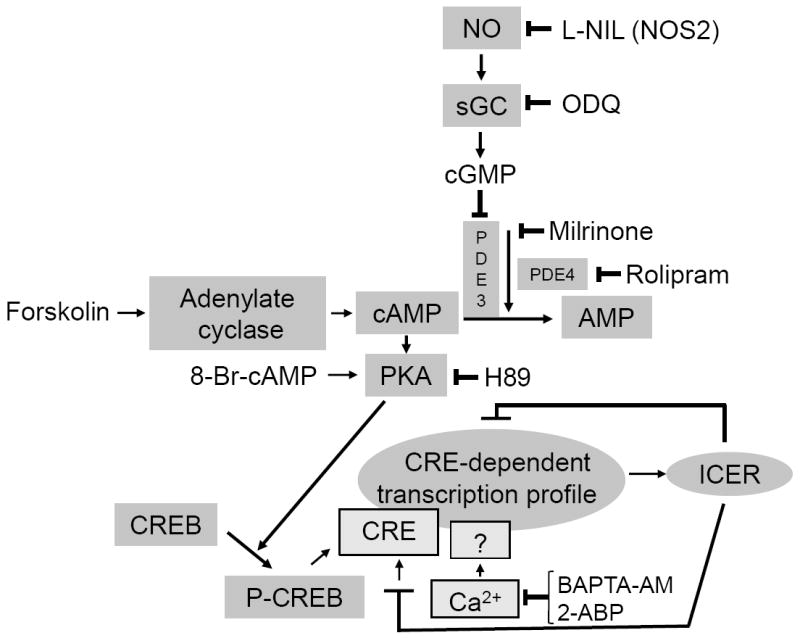
Proposed pathway of NO-mediated ICER induction. NO induces ICER expression in RPaSMC via cGMP–mediated inhibition of phosphodiesterase3 (PDE3), thereby elevating cAMP concentrations (likely in a specific intracellular compartment), leading to activation of PKA and CREB phosphorylation. Phosphorylated CREB binds to the cAMP-response element (CRE) leading to activation of gene transcription. Ca2+is also required for ICER gene induction by GSNO. Once the ICER gene is induced, it inhibits CRE-dependent transcription and its own expression via a negative feedback loop.
To further characterize the mechanisms by which NO regulates VSMC structure and function, we compared the transcription profile (~8000 gene elements) of RPaSMC incubated in the absence or presence of GSNO. Two approaches were used to analyze the microarray data. First, we looked for common transcription factor-binding sites in genes whose expression was changed by 2-fold or more. We found that ~100 genes were up-regulated by GSNO, and a similar number of genes were down-regulated (Supplemental Fig. 1). Of the genes whose expression was induced by NO, 61% contained CRE sequences in their promoter regions (Supplemental Table 2). In a second approach, we organized gene expression data into microarray pathway profiles (MAPPs) that represent NO-regulated biological pathways (Supplemental Fig. 3A-C).
The findings that multiple genes, whose expression is up-regulated by GSNO, share CRE regulatory sequences suggests that CREB plays an important role in the NO-mediated regulation of VSMC phenotype. To further characterize the role of CRE/CREB signaling in the regulation of RPaSMC gene expression by NO, we focused on the gene encoding ICER, which is highly responsive to NO and is known to be regulated by CREB [7, 10]. We used a high concentration of GSNO (100 μM) as a tool to identify genes that are modulated by NO. It is conceivable that the use of 100 μM GSNO might lead to the generation of non-physiological concentrations of NO during the initial few minutes of GSNO exposure. Once ICER was identified as a gene whose expression was markedly increased by high concentrations of GSNO, we studied the effects of lower concentrations of GSNO, as well as inhaled NO, at doses that induce vasorelaxation. We found that ICER mRNA levels were markedly increased in RPaSMC exposed to GSNO at 1 and 10 μM, in RPaSMC expressing NOS2 (Fig. 2A), and in lungs of WT mice breathing 80 ppm NO gas (Fig. 2B, D).
NO signals, in part, by activating sGC to synthesize cGMP. We observed that inhibition of sGC prevented the induction of ICER in RPaSMC exposed to GSNO, as well as in RPaSMC induced to express NOS2 (Fig. 2A). Moreover, the ability of inhaled NO to induce pulmonary ICER gene expression was markedly attenuated in mice lacking one sGC isoform, α1β1 (Fig. 2D) [19]. The modest residual induction of ICER gene expression in sGCα1-/- mice may attributable to the remaining sGC isoform, α2β1. These results in vitro and in vivo implicate sGC and cGMP signaling in the induction of ICER gene expression by NO.
cGMP is well known to modulate gene expression in VSMC via PKG [29, 30]. Pilz and colleagues reported that PKG activation induces gene expression in VSMC in a CRE- and CREB-dependent manner [6]. In contrast, Aizawa et al. [31] found that NO and cGMP can induce NFκB-dependent gene expression via a mechanism that does not require PKG. Similarly, we previously reported that NO/cGMP signaling induced PDE4B gene expression in RPaSMC via mechanisms that do not require PKG [32]. In the current study, we observed that PKG inhibitors did not block the ability of GSNO or endogenous NO to induce ICER gene expression in RPaSMC (Fig. 2A, C). In addition, PKG inhibition did not prevent the induction of CRE-dependent gene transcription by GSNO. Moreover, PKG-specific agonists were unable to induce ICER gene expression in these cells (Fig. 2C). Taken together these experiments suggest that sGC/cGMP signaling can regulate RPaSMC gene expression via a PKG-independent mechanism. We considered the possibility that PKG expression levels in RPaSMC may have been lost as the cells were passaged [33]: losing PKG activity would explain why compounds that modulate PKG activity had no effect in our experiments. However, PKG-Iα mRNA was readily detectable in the RPaSMC that we studied.
We present several lines of evidence suggesting that PDE3 and PKA are responsible for the induction of ICER gene expression by NO and cGMP. First, the PKA inhibitor, H89, inhibited NO-induced CRE-dependent gene transcription and ICER gene expression (Fig. 2A, C). Second, treatment of RPaSMC with rolipram, an inhibitor of the cAMP-specific PDE4, markedly potentiated the effect of GSNO on CRE-dependent gene transcription, whereas the PDE3 inhibitor, milrinone, did not (Fig. 3B). Our findings suggest that milrinone and GSNO acted on CRE-dependent gene transcription using the same mechanism, inhibition of PDE3. Moreover, the observation that rolipram augmented the effects of NO supports the concept that NO activates CRE-dependent gene transcription via cAMP. Recently, Kumar et al. reported that, in COS-7 cells, NO increased cAMP levels and induced GTP cyclohydrolase I gene expression in a PKA dependent manner [34]. However, we were unable to detect an increase in cAMP levels in RPaSMC exposed to GSNO. Yan and colleagues proposed that PDE3 inhibition by NO leads to localized increases of cAMP levels that were not detectable in whole cell extracts [35]. As further confirmation that NO increased cAMP levels in RPaSMC, we observed that GSNO induced phosphorylation of VASP at Ser157, the residue targeted by PKA, but not Ser239, the residue targeted by PKG [36] (data not shown). Taken together, these findings suggest that NO and cGMP signaling can regulate VSMC gene expression via cAMP-mediated PKA activation. cAMP activates gene transcription by phosphorylating the CRE-binding protein CREB at Ser133 [7, 37]. Others have observed that NO/sGC/cGMP signaling can induce CREB phosphorylation in a variety of cell types,, including glial cells of avian retina [38], neurons in the nucleus tractus solitarii [39], in sGC- and PKG-expressing BHK cells [6], and in COS-7 cells [34]. We observed that incubation of RPaSMC with GSNO led to CREB phosphorylation via sGC- and PKA-dependent mechanisms. Moreover, as we showed previously for PDE4B [32], expression of a dominant-negative CREB prevented GSNO induced CRE-dependent gene transcription and ICER gene expression.
In rat pheochromocytoma cells (PC12 cells), previous reports suggested that the ability of NO to induce gene expression required the synergistic effects of PKA activation and an increase in Ca2+ signaling [11]. We similarly found that chelation of intracellular Ca2+ with BAPTA-AM abrogated the ability of NO to induce ICER gene expression (Fig. 5A). Our results suggest that Ca2+ is required for NO to modulate ICER signaling in VSMC.
To identify the source of the Ca2+ participating in the NO response, we used selective Ca2+-channel inhibitors, 2-ABP and SKF, to block SOCCs and nifedipine to inhibit VDCCs. Importantly, our study suggests that the ability of NO to modulate ICER mRNA levels is dependent upon Ca2+ derived from SOCCs and not VDCCs (Fig. 5B-C). Prior studies have shown that Ca2+ signaling can induce CREB phosphorylation [40]. However, the ability of NO to phosphorylate CREB in RPaSMC was insensitive to SOCC inhibition (Fig. 5D). These results suggest that induction of ICER gene expression by NO requires both CREB phosphorylation and Ca2+ signaling. Prior studies have shown that Ca2+ has a critical role in regulating ICER gene transcription and potential downstream transcription factors have been identified, including thyrotropic embryonic factor (TEF) [41]. The identities of the transcription factors, in addition to CREB, that are responsible for activation of ICER gene transcription by NO in RPaSMC remain to be determined.
ICER is known to be a potent inhibitor of cAMP/PKA/CRE-dependent gene transcription [7]. Similarly, we found that transient expression of ICER in RPaSMC attenuated GSNO-dependent gene transcription. Moreover, that overexpression of ICER markedly inhibited forskolin-stimulated gene expression (Fig. 6). These results suggest that induction of ICER gene expression by NO signaling can modulate gene expression stimulated by subsequent exposure to cAMP.
NO and ICER share the ability to inhibit proliferation and increase apoptosis of VSMC, as well as to inhibit neointima formation in rat carotid arteries subjected to balloon injury [30, 42, 43]. It is conceivable that the induction of ICER expression by NO may be responsible, at least in part, for the ability of the latter to modulate VSMC functions. Additional in vitro and in vivo studies will be required to elucidate the role of ICER in the ability of NO to modulate VSMC phenotype.
In summary, we used transcription profiling to characterize the mechanisms by which NO regulates gene expression in RPaSMC (summarized in Fig. 7). NO induces expression of a wide variety of genes, many of which contain CRE regulatory sequences. By focusing on one of these NO-induced genes, ICER, critical roles were found for sGC, cGMP, and PDE3 in the regulation of CRE-dependent gene transcription. Unexpectedly, PKA, but not PKG, is required for the induction of CRE-dependent gene transcription, CREB phosphorylation, and ICER gene expression by NO. Moreover, a critical CREB-independent role for Ca2+ signaling in NO-induced ICER gene expression was identified. ICER induction represents a negative feedback loop for NO-stimulated gene expression and a novel “cross talk” mechanism between cGMP and cAMP signaling pathways.
Supplementary Material
Acknowledgments
The authors thank Jennifer Couget for assistance with transcriptional profiling studies, and Drs. Michael Costigan and Stuart Cooke for helpful advice on data analysis. We thank Drs. William H. Walker and John J. Lepore for kindly providing ICER and CREB plasmids and AdCREBml. Ad.βgal was kindly provided by Dr. Anthony Rosenzweig.
GRANTS
This work was supported by the German DFG grant SW 119-3/1 (AUS), a Scientist Development Grant 9930085N from the American Heart Association (HL), a Research Training Fellowship for Medical Students from the Howard Hughes Medical Institute (KJ), NHLBI T32HL007208(RM), a Scientist Development Grant 10SDG2610313 from the American Heart Association (ESB), grants from the FWO-Vlaanderen and the UGent-GOA programs (PB), and grants from the NHLBI, HL-42397 (WMZ) and HL-57172 (KDB).
Abbreviations
- NO
nitric oxide
- ICER
inducible cAMP early repressor
- RPaSMC
rat pulmonary artery smooth muscle cells
- VSMC
vascular smooth muscle cells
- PKG
cGMP-dependent protein kinase
- sGC
soluble guanylate cyclase
- sGCα1-/-
mice deficient for the soluble guanylate cyclase α subunit
- IL-1β
interleukin-1β
- CRE
cAMP-response element
- CREB
CRE-binding protein
- P-CREB
phosphorylated CREB
- CREM
cAMP-response element modulator
- PDE
phosphodiesterase
- SOCC
store-operated, non-voltage gated calcium channels
- VDCC
voltage-dependent calcium channels
- NOS2
nitric oxide synthase 2
- Ad
adenovirus
- βgal
beta-galactosidase
- M.O.I
multiplicity of infection
- KID
kinase-inducible domain
- VASP
vasodilator-stimulated phosphoprotein
- HO1
heme-oxygenase 1
- tPA
tissue plasminogen activator
- HK2
rat hexokinase 2
- MT1
metallothionein 1
- GCS:hc
γ-glutamylcysteine synthetase heavy chain
- GCS:lc
γ-glutamylcysteine synthetase light chain
- ET1
endothelin 1
- AT2R
angiotensin II receptor
- TGF-β3
transforming growth factor-β3
Footnotes
DISCLOSURES
KDB receives research grant support via a sponsored research agreement with INOTherapeutics and the Massachusetts General Hospital. WMZ receives royalties on patents licensed by Massachusetts General Hospital to Linde Corp (Munich, Germany) and Ikaria (Clinton, New Jersey) on inhaled NO.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Eigenthaler M, Lohmann SM, Walter U, Pilz RB. Signal transduction by cGMP-dependent protein kinases and their emerging roles in the regulation of cell adhesion and gene expression. Rev Physiol Biochem Pharmacol. 1999;135:173–209. doi: 10.1007/BFb0033673. [DOI] [PubMed] [Google Scholar]
- 2.Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res. 2003;93:280–91. doi: 10.1161/01.RES.0000087541.15600.2B. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt HH, Lohmann SM, Walter U. The nitric oxide and cGMP signal transduction system: regulation and mechanism of action. Biochim Biophys Acta. 1993;1178:153–75. doi: 10.1016/0167-4889(93)90006-b. [DOI] [PubMed] [Google Scholar]
- 4.Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525–63. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sausbier M, Schubert R, Voigt V, Hirneiss C, Pfeifer A, Korth M, Kleppisch T, Ruth P, Hofmann F. Mechanisms of NO/cGMP-dependent vasorelaxation. Circ Res. 2000;87:825–30. doi: 10.1161/01.res.87.9.825. [DOI] [PubMed] [Google Scholar]
- 6.Gudi T, Casteel DE, Vinson C, Boss GR, Pilz RB. NO activation of fos promoter elements requires nuclear translocation of G-kinase I and CREB phosphorylation but is independent of MAP kinase activation. Oncogene. 2000;19:6324–33. doi: 10.1038/sj.onc.1204007. [DOI] [PubMed] [Google Scholar]
- 7.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 8.Kronke G, Bochkov VN, Huber J, Gruber F, Bluml S, Furnkranz A, Kadl A, Binder BR, Leitinger N. Oxidized phospholipids induce expression of human heme oxygenase-1 involving activation of cAMP-responsive element-binding protein. J Biol Chem. 2003;278:51006–14. doi: 10.1074/jbc.M304103200. [DOI] [PubMed] [Google Scholar]
- 9.Osawa H, Robey RB, Printz RL, Granner DK. Identification and characterization of basal and cyclic AMP response elements in the promoter of the rat hexokinase II gene. J Biol Chem. 1996;271:17296–303. doi: 10.1074/jbc.271.29.17296. [DOI] [PubMed] [Google Scholar]
- 10.Molina CA, Foulkes NS, Lalli E, Sassone-Corsi P. Inducibility and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell. 1993;75:875–86. doi: 10.1016/0092-8674(93)90532-u. [DOI] [PubMed] [Google Scholar]
- 11.Peunova N, Enikolopov G. Amplification of calcium-induced gene transcription by nitric oxide in neuronal cells. Nature. 1993;364:450–3. doi: 10.1038/364450a0. [DOI] [PubMed] [Google Scholar]
- 12.Lincoln TM, Dey N, Sellak H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J Appl Physiol. 2001;91:1421–30. doi: 10.1152/jappl.2001.91.3.1421. [DOI] [PubMed] [Google Scholar]
- 13.Trebak M, Ginnan R, Singer HA, Jourd’heuil D. Interplay between calcium and reactive oxygen/nitrogen species: an essential paradigm for vascular smooth muscle signaling. Antioxid Redox Signal. 2010;12:657–74. doi: 10.1089/ars.2009.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 15.Filippov G, Bloch DB, Bloch KD. Nitric oxide decreases stability of mRNAs encoding soluble guanylate cyclase subunits in rat pulmonary artery smooth muscle cells. J Clin Invest. 1997;100:942–8. doi: 10.1172/JCI119610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Chen X, Gong X, Liu Y, Feng H, Qiu L, Hu Z, Zhang J. A transcript profiling approach reveals the zinc finger transcription factor ZNF191 is a pleiotropic factor. BMC Genomics. 2009;10:241. doi: 10.1186/1471-2164-10-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–54. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 18.Nagasaka Y, Fernandez BO, Garcia-Saura MF, Petersen B, Ichinose F, Bloch KD, Feelisch M, Zapol WM. Brief periods of nitric oxide inhalation protect against myocardial ischemia-reperfusion injury. Anesthesiology. 2008;109:675–82. doi: 10.1097/ALN.0b013e318186316e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buys ES, Sips P, Vermeersch P, Raher MJ, Rogge E, Ichinose F, Dewerchin M, Bloch KD, Janssens S, Brouckaert P. Gender-specific hypertension and responsiveness to nitric oxide in sGCalpha1 knockout mice. Cardiovasc Res. 2008;79:179–86. doi: 10.1093/cvr/cvn068. [DOI] [PubMed] [Google Scholar]
- 20.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curran-Everett D, Benos D. Guidelines for reporting statistics in journals published by the American Physiological Society. Physiol genomics. 2004;18:249–251. doi: 10.1152/physiolgenomics.00155.2004. [DOI] [PubMed] [Google Scholar]
- 22.Takata M, Filippov G, Liu H, Ichinose F, Janssens S, Bloch DB, Bloch KD. Cytokines decrease sGC in pulmonary artery smooth muscle cells via NO-dependent and NO-independent mechanisms. Am J Physiol Lung Cell Mol Physiol. 2001;280:L272–8. doi: 10.1152/ajplung.2001.280.2.L272. [DOI] [PubMed] [Google Scholar]
- 23.Bundgaard H, Liu CC, Garcia A, Hamilton EJ, Huang Y, Chia KK, Hunyor SN, Figtree GA, Rasmussen HH. {beta}3 Adrenergic Stimulation of the Cardiac Na+-K+ Pump by Reversal of an Inhibitory Oxidative Modification. Circulation. 2010;122:2699–708. doi: 10.1161/CIRCULATIONAHA.110.964619. [DOI] [PubMed] [Google Scholar]
- 24.Sobolewski A, Jourdan KB, Upton PD, Long L, Morrell NW. Mechanism of cicaprost-induced desensitization in rat pulmonary artery smooth muscle cells involves a PKA-mediated inhibition of adenylyl cyclase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L352–9. doi: 10.1152/ajplung.00270.2003. [DOI] [PubMed] [Google Scholar]
- 25.Andrisani OM. CREB-mediated transcriptional control. Crit Rev Eukaryot Gene Expr. 1999;9:19–32. [PubMed] [Google Scholar]
- 26.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–80. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 27.Enslen H, Soderling TR. Roles of calmodulin-dependent protein kinases and phosphatase in calcium-dependent transcription of immediate early genes. J Biol Chem. 1994;269:20872–7. [PubMed] [Google Scholar]
- 28.Sheng M, Thompson MA, Greenberg ME. CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–30. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 29.Cornwell TL, Arnold E, Boerth NJ, Lincoln TM. Inhibition of smooth muscle cell growth by nitric oxide and activation of cAMP-dependent protein kinase by cGMP. Am J Physiol. 1994;267:C1405–13. doi: 10.1152/ajpcell.1994.267.5.C1405. [DOI] [PubMed] [Google Scholar]
- 30.Pilz RB, Casteel DE. Regulation of gene expression by cyclic GMP. Circ Res. 2003;93:1034–46. doi: 10.1161/01.RES.0000103311.52853.48. [DOI] [PubMed] [Google Scholar]
- 31.Aizawa T, Wei H, Miano JM, Abe J, Berk BC, Yan C. Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ Res. 2003;93:406–13. doi: 10.1161/01.RES.0000091074.33584.F0. [DOI] [PubMed] [Google Scholar]
- 32.Busch CJ, Liu H, Graveline AR, Bloch KD. Nitric oxide induces phosphodiesterase 4B expression in rat pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L747–L753. doi: 10.1152/ajplung.00298.2005. [DOI] [PubMed] [Google Scholar]
- 33.Lincoln TM, Dey NB, Boerth NJ, Cornwell TL, Soff GA. Nitric oxide--cyclic GMP pathway regulates vascular smooth muscle cell phenotypic modulation: implications in vascular diseases. Acta Physiol Scand. 1998;164:507–15. doi: 10.1111/j.1365-201x.1998.tb10700.x. [DOI] [PubMed] [Google Scholar]
- 34.Kumar S, Sun X, Sharma S, Aggarwal S, Ravi K, Fineman JR, Black SM. GTP cyclohydrolase I expression is regulated by nitric oxide: role of cyclic AMP. Am J Physiol Lung Cell Mol Physiol. 2009;297:L309–17. doi: 10.1152/ajplung.90538.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan C, Kim D, Aizawa T, Berk BC. Functional interplay between angiotensin II and nitric oxide: cyclic GMP as a key mediator. Arterioscler Thromb Vasc Biol. 2003;23:26–36. doi: 10.1161/01.atv.0000046231.17365.9d. [DOI] [PubMed] [Google Scholar]
- 36.Zhuang S, Nguyen GT, Chen Y, Gudi T, Eigenthaler M, Jarchau T, Walter U, Boss GR, Pilz RB. Vasodilator-stimulated phosphoprotein activation of serum-response element-dependent transcription occurs downstream of RhoA and is inhibited by cGMP-dependent protein kinase phosphorylation. J Biol Chem. 2004;279:10397–407. doi: 10.1074/jbc.M313048200. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto KK, Gonzalez GA, Biggs WH, 3rd, Montminy MR. Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature. 1988;334:494–8. doi: 10.1038/334494a0. [DOI] [PubMed] [Google Scholar]
- 38.Socodato RE, Magalhaes CR, Paes-de-Carvalho R. Glutamate and nitric oxide modulate ERK and CREB phosphorylation in the avian retina: evidence for direct signaling from neurons to Muller glial cells. J Neurochem. 2009;108:417–29. doi: 10.1111/j.1471-4159.2008.05778.x. [DOI] [PubMed] [Google Scholar]
- 39.Chan SH, Chang KF, Ou CC, Chan JY. Nitric oxide regulates c-fos expression in nucleus tractus solitarii induced by baroreceptor activation via cGMP-dependent protein kinase and cAMP response element-binding protein phosphorylation. Mol Pharmacol. 2004;65:319–25. doi: 10.1124/mol.65.2.319. [DOI] [PubMed] [Google Scholar]
- 40.Zanassi P, Paolillo M, Feliciello A, Avvedimento EV, Gallo V, Schinelli S. cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J Biol Chem. 2001;276:11487–95. doi: 10.1074/jbc.M007631200. [DOI] [PubMed] [Google Scholar]
- 41.Krueger DA, Warner EA, Dowd DR. Involvement of thyrotroph embryonic factor in calcium-mediated regulation of gene expression. J Biol Chem. 2000;275:14524–31. doi: 10.1074/jbc.275.19.14524. [DOI] [PubMed] [Google Scholar]
- 42.Jeremy JY, Dashwood MR, Mehta D, Izzat MB, Shukla N, Angelini GD. Nitric oxide, prostacyclin and cyclic nucleotide formation in externally stented porcine vein grafts. Atherosclerosis. 1998;141:297–305. doi: 10.1016/s0021-9150(98)00183-x. [DOI] [PubMed] [Google Scholar]
- 43.Ohtsubo H, Ichiki T, Miyazaki R, Inanaga K, Imayama I, Hashiguchi Y, Sadoshima J, Sunagawa K. Inducible cAMP early repressor inhibits growth of vascular smooth muscle cell. Arterioscler Thromb Vasc Biol. 2007;27:1549–55. doi: 10.1161/ATVBAHA.107.145011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.