

Abstract
Acidianus two-tailed virus (ATV) infects crenarchaea of the genus Acidianus living in terrestrial thermal springs at extremely high temperatures and low pH. ATV is a member of the Bicaudaviridae virus family and undergoes extra-cellular development of two tails, a process that is unique in the viral world. To understand this intriguing phenomenon, we have undertaken structural studies of ATV virion proteins and here we present the crystal structure of one of these proteins, ATV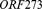 . ATV
. ATV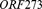 forms tetramers in solution and a molecular envelope is provided for the tetramer, computed from small-angle X-ray scattering (SAXS) data. The crystal structure has properties typical of hyperthermostable proteins, including a relatively high number of salt bridges. However, the protein also exhibits flexible loops and surface pockets. Remarkably, ATV
forms tetramers in solution and a molecular envelope is provided for the tetramer, computed from small-angle X-ray scattering (SAXS) data. The crystal structure has properties typical of hyperthermostable proteins, including a relatively high number of salt bridges. However, the protein also exhibits flexible loops and surface pockets. Remarkably, ATV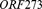 displays a new
displays a new 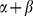 protein fold, consistent with the absence of homologues of this protein in public sequence databases.
protein fold, consistent with the absence of homologues of this protein in public sequence databases.
Introduction
Viruses are key components of biogeochemical cycles: they are the most abundant biological entities in the oceans [1] and probably on the planet. Thus, it has been estimated that every day, viruses kill about 20% of the oceanic biomass [2]. Viruses also represent a major genetic asset for the biosphere. Indeed, all organisms from each Domain of Life are likely to be infected by viruses. Although viruses infecting archaea are known since the early 1970s [3], they have only been studied in detail very recently. The notion that these viruses constitute a variety of bacteriophages with head and tail (Caudovirales), reinforced by the initial findings, was challenged by the analyses of samples isolated by Zillig and co-workers from extreme environments, rich in hyperthermophilic archaea, including the Icelandic solfatara [4]. These analyses revealed the presence of a large diversity of viral morphotypes, including viruses of linear, spindle-shaped, spherical and more exotic forms, such as drops and bottle-shapes. These viruses infect archaea living in such extreme environments, which mostly belong to the crenarchaeal orders Sulfolobales and Thermoproteales. They have been classified into eight viral families, primarily on the basis of their unusual morphotypes, subsequently backed by genomic analyses with a few viruses remaining unclassified.
Generally, crenarchaeal viruses display non-lytic life cycles, a strategy that would allow them to minimise contact to the extreme conditions of their environment. With the exception of the recently discovered Aeropyrum coil-shaped virus (ACV) [5], which has a single-stranded DNA genome, crenarchaeal viruses present double-stranded DNA genomes. Examination of the genomic sequences obtained so far shows that most of these viruses are unrelated to any other known viruses and that they probably have different evolutionary origins [6]. In spite of their interest from an evolutionary viewpoint, the biology of crenarchaeal viruses remains largely unexplored. This situation mirrors the fact that between 50% and 90% of the open reading frames (ORFs) predicted in the genomes of these viruses have no unambiguous functional annotations [6]. Structural analysis has been a useful tool for establishing evolutionary relationships amongst viruses [7], especially those infecting Archaea [8]. In the absence of sequence similarity to annotated proteins, structure similarity might provide insights into protein function [9]. Thus, we have worked on the structure determination of selected function–orphan crenarchaeal viral proteins with a view to obtain clues to their biological function [9]–[13].
The Acidianus two-tailed virus (ATV) was originally discovered in 2003 in Pozzuoli, Italy. It was isolated from a spring with temperatures higher than 85°C and at a pH of 1.5, where its host, the crenarchaeon Acidianus convivator, thrives [14], [15]. ATV is a member of the Bicaudaviridae family of crenarchaeal viruses. It has a circular double-stranded DNA genome of 62,730 base pairs, including 72 predicted open reading frames (ORFs), most without bona fide homologues in public sequence databases. Exceptionally for a crenarchaeal virus, ATV is known to undergo both lysogenic and lytic life cycles. Lysogeny can be interrupted and transformed into a lytic pathway by environmental stress factors. For example, ATV lytic propagation can be induced by lowering the temperature of the cultures from 85°C to 75°C [14].
Released ATV virions are initially spindle-shaped particles. At striking variance with all other known viruses, ATV undergoes an extracellular morphological transformation: between one hour and a few days, two tails develop irreversibly at each end of the particle. Tail development seems to depend solely on temperature, which must be close to that of the host's habitat (75°C to 90°C) [14]. Although infection of host cells by ATV has not yet been directly observed, it is possible that the tails facilitate or mediate the attachment of virions to host membranes.
The mechanism behind ATV's ability to grow bipolar tails has yet to be understood. At least 11 proteins have been identified in ATV virion preparations [15]. ATV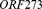 is the fourth most abundant of these proteins [15]. Notably, ATV
is the fourth most abundant of these proteins [15]. Notably, ATV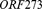 has no homologues in the distantly related virus Sulfolobus tengchongensis Spindle-shaped Virus 1 (STSV1), a virus that does not undergo cell–independent tail development [16]. To gain insight into this unique biological phenomenon, we have characterised the structure and the behaviour in solution of the ATV
has no homologues in the distantly related virus Sulfolobus tengchongensis Spindle-shaped Virus 1 (STSV1), a virus that does not undergo cell–independent tail development [16]. To gain insight into this unique biological phenomenon, we have characterised the structure and the behaviour in solution of the ATV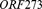 protein.
protein.
Results and Discussion
ATV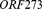 structure belongs to a new fold
structure belongs to a new fold
ATV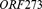 is an acidic protein (theoretical pI = 4.8) with a molar mass of 32154 Da. This protein (UniProt:Q3V4T6) was isolated from ATV virions and identified by N′-terminal Edman degradation from an SDS-PAGE band migrating with an apparent molecular weight of 38 kDa [15]. All the work reported here was carried out using a recombinant ATV
is an acidic protein (theoretical pI = 4.8) with a molar mass of 32154 Da. This protein (UniProt:Q3V4T6) was isolated from ATV virions and identified by N′-terminal Edman degradation from an SDS-PAGE band migrating with an apparent molecular weight of 38 kDa [15]. All the work reported here was carried out using a recombinant ATV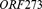 protein (molar mass 32977 Da, theoretical pI = 5.0) expressed in E. coli T7 Iq pLysS cells (New England Biolabs). Recombinant ATV
protein (molar mass 32977 Da, theoretical pI = 5.0) expressed in E. coli T7 Iq pLysS cells (New England Biolabs). Recombinant ATV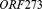 migrates on SDS polyacrylamide gels with an apparent 39 kDa molecular weight.
migrates on SDS polyacrylamide gels with an apparent 39 kDa molecular weight.
Two crystal forms of recombinant ATV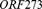 were obtained belonging to the tetragonal space group
were obtained belonging to the tetragonal space group 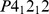 but with different cell parameters (Table 1). Crystals of the first form diffracted to 3.85 Å resolution and included one monomer in the asymmetric unit. The crystals of the second form reached a 2.15 Å diffraction limit and comprised two monomers in its asymmetric unit. Since ATV
but with different cell parameters (Table 1). Crystals of the first form diffracted to 3.85 Å resolution and included one monomer in the asymmetric unit. The crystals of the second form reached a 2.15 Å diffraction limit and comprised two monomers in its asymmetric unit. Since ATV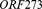 contains a single, N′-terminal methionine a triple mutant introducing three methionine residues was produced to facilitate SeSAD phasing. Thus, residues Leu31, Leu117 and Leu240 were selected for mutation into methionines based on their similar properties (bulky, hydrophobic) and on their predicted positioning in ordered
contains a single, N′-terminal methionine a triple mutant introducing three methionine residues was produced to facilitate SeSAD phasing. Thus, residues Leu31, Leu117 and Leu240 were selected for mutation into methionines based on their similar properties (bulky, hydrophobic) and on their predicted positioning in ordered  -helices by PSIPRED [17]. The SeMet substituted triple mutant protein ATV
-helices by PSIPRED [17]. The SeMet substituted triple mutant protein ATV was also produced in E. coli, purified, and the incorporation of SeMet was checked by mass spectrometry. The SeMet protein produced crystals of the second form with cell parameters isomorphous to those of the corresponding native crystals (Table 1).
was also produced in E. coli, purified, and the incorporation of SeMet was checked by mass spectrometry. The SeMet protein produced crystals of the second form with cell parameters isomorphous to those of the corresponding native crystals (Table 1).
Table 1. Summary of data collection, phasing and refinement statistics.
| Form 1 | native Form 2 | SeSAD Form 2 | |
| PDB code | 4ats | 4art | |
| Data collection a | |||
| Beamline (synchrotron) | ID14eh4 (ESRF) | ID29 (ESRF) | Proxima1 (Soleil) |
| Wavelength (Å) | 1.1810 | 0.9184 | 0.9791 |
| Space group |
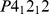
|
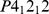
|
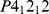
|
| Unit cell axes (Å) | a = b = 101.0, c = 53.0 | a = b = 78.6, c = 189.4 | a = b = 78.9, c = 188.9 |
| Resolution (Å) | 71.43 – 3.85 (3.86 – 3.85) | 50.00 – 2.15 (2.20 – 2.15) | 45.00 – 2.77 (2.85 – 2.77) |
| Unique reflections | 2843 | 32549 | 28786 |
| Multiplicity | 11.0 (11.9) | 12.8 (13.4) | 7.6 (7.2) |
| Completeness (%) | 98.6 (100.0) | 97.9 (99.9) | 99.8 (98.1) |
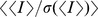
|
20.5 (6.8) | 24.0 (3.7) | 16.9 (3.1) |
R
 b
b
|
15.1 (65.9) | 7.7 (77.4) | 10.0 (74.4) |
| Phasing | |||
| Anom. corr. (%) c | 44 (3.74) | ||
| Phasing power d | 1.01 (3.74) | ||
| Refinement a | |||
| Reflections used e | 2819 (459) | 32529 (1660) | |
| Protein atoms | 1960 | 3917 | |
| Solvent atoms | 0 | 281 | |
R
 /R
/R
 (%)f (%)f
|
27.1/29.5 (29.1/32.1) | 20.2/23.4 (22.9/25.5) | |
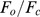 correlation coefficient
g
correlation coefficient
g
|
0.861 (0.838) | 0.947 (0.933) | |
| r.m.s.d. (bonds) h | 0.009 | 0.010 | |
| r.m.s.d. (angles) h | 1.08 | 1.05 | |
| Average isotropic B factors (Å 2 ) | |||
| Main chain | 132.7 | 44.5 | |
| Side chains | 148.8 | 54.8 | |
| Solvent | 51.9 | ||
| Ramachandran plot statistics (%) | |||
| Residues in favoured regions | 223 (97.4) | 453 (99.3) | |
| Residues in allowed regions | 6 (2.6) | 3 (0.7) | |
| Outliers | 0 (0) | 0 (0) | |
| Molprobity score (percentile) i | 1.91 (100) | 1.48 (98) |
Values in parentheses are for the highest resolution shell.
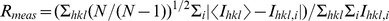 is the multiplicity (N) independent R
is the multiplicity (N) independent R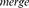 .
.
Mean correlation factor between two random subsets of anomalous intensity differences. In parenthesis, resolution at which anomalous correlation drops below 35%.
Value in parenthesis: resolution for which phasing power drops below 1.0.
In parenthesis, number of reflections randomly assigned to the test set.
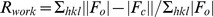 . R
. R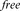 is defined as R
is defined as R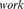 for the test set.
for the test set.
The value in parentheses corresponds to the test set reflections.
Root mean square deviation from the standard values.
As reported by the Molprobity server [57]. In parenthesis, the percentile of this value among structures of comparable resolution.
The SeMet data extended to 2.77 Å resolution. Its structure was solved by the SAD method with data collected at the selenium anomalous peak. The data were of sufficient quality to locate the 6 Se sites present in the two molecules in the asymmetric unit. The current model is refined against native data to 2.15 Å, resulting in an R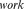 /R
/R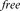 factors of 20.2%/23.4% (Table 1) and includes residues 22 to 270 in monomer A and 25 to 269 in monomer B. Two loops could not be built in the model owing to the lack of supporting electron density. The first of these loops comprises residues Ser46 to Thr53 in monomer A and Arg44 to Ile54 in monomer B, whereas the second missing loop encompases residues Gly148 to Arg151 in both monomers. The monomer A from this model was then used to solve the structure of the first crystal form by molecular replacement. Refinement against these data produced a new model, at 3.85 Å, with R
factors of 20.2%/23.4% (Table 1) and includes residues 22 to 270 in monomer A and 25 to 269 in monomer B. Two loops could not be built in the model owing to the lack of supporting electron density. The first of these loops comprises residues Ser46 to Thr53 in monomer A and Arg44 to Ile54 in monomer B, whereas the second missing loop encompases residues Gly148 to Arg151 in both monomers. The monomer A from this model was then used to solve the structure of the first crystal form by molecular replacement. Refinement against these data produced a new model, at 3.85 Å, with R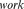 and R
and R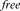 factors of 27.1% and 29.5%, respectively (Table 1). The second crystal form being solved at higher resolution, the rest of the structural description will be based on this current model, unless otherwise stated.
factors of 27.1% and 29.5%, respectively (Table 1). The second crystal form being solved at higher resolution, the rest of the structural description will be based on this current model, unless otherwise stated.
The structure of an ATV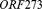 monomer is made up of 10 helices and two
monomer is made up of 10 helices and two 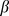 -sheets, one composed of seven strands and the other consisting of two short parallel strands (Figure 1). The major
-sheets, one composed of seven strands and the other consisting of two short parallel strands (Figure 1). The major 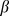 -sheet is mainly antiparallel, but strands
-sheet is mainly antiparallel, but strands 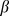 3 and
3 and 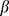 5 are parallel (Figure 1a). This
5 are parallel (Figure 1a). This 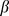 -sheet forms a half-barrel with the first
-sheet forms a half-barrel with the first  -helix (
-helix ( 1) packed onto its concave side. The rest of the helices and the small
1) packed onto its concave side. The rest of the helices and the small 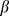 -sheet are arranged at the other side of the major
-sheet are arranged at the other side of the major 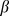 -sheet (Figure 1b). The monomer can be described as a disk with overall dimensions 59 Å
-sheet (Figure 1b). The monomer can be described as a disk with overall dimensions 59 Å 58 Å
58 Å 38 Å. The protein N′-terminus protrudes on one side of the disk, whereas the surface of the other side, lined by a high number of acidic residues (Figure 1c), is concave. The two unmodelled loops face each other at the periphery of the disk, with the visible extremes pointing towards the concave side (Figure 1a). This concave face (Figure 1c) includes two deep pockets with volumes of
38 Å. The protein N′-terminus protrudes on one side of the disk, whereas the surface of the other side, lined by a high number of acidic residues (Figure 1c), is concave. The two unmodelled loops face each other at the periphery of the disk, with the visible extremes pointing towards the concave side (Figure 1a). This concave face (Figure 1c) includes two deep pockets with volumes of  575 Å3 and
575 Å3 and  400 Å3, respectively, as calculated by the Relibase+ [18] implementation of LIGSITE [19]. Other smaller pockets are distributed across the protein surface. Analysis of these cavities with the SUMO server [20] did not suggest any clear-cut ligands matching them.
400 Å3, respectively, as calculated by the Relibase+ [18] implementation of LIGSITE [19]. Other smaller pockets are distributed across the protein surface. Analysis of these cavities with the SUMO server [20] did not suggest any clear-cut ligands matching them.
Figure 1. Crystal structure of ATV.

 .
a) Ribbon representation of the structure of a monomer of ATV
.
a) Ribbon representation of the structure of a monomer of ATV . Its
. Its 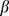 -strands are labelled. The secondary structure elements are colored from the N′- (blue) to the C′-terminus (red). The loops that were not modelled are represented by dashed, black lines. b) A view orthogonal to the previous one, showing the arrangement of the
-strands are labelled. The secondary structure elements are colored from the N′- (blue) to the C′-terminus (red). The loops that were not modelled are represented by dashed, black lines. b) A view orthogonal to the previous one, showing the arrangement of the  -helices. This view shows part of the concave face of the monomer (top, right) c) The solvent-accessible surface of the ATV
-helices. This view shows part of the concave face of the monomer (top, right) c) The solvent-accessible surface of the ATV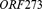 monomer concave face, colored by its electrostatic potential (red: −52 mV, blue: 52 mV), calculated at pH 7 and 150 mM NaCl with APBS [56]. Two cavities are marked with yellow open stars. d) A detail view of monomer B showing the salt bridge network formed by residues Glu29/Asp33/Lys260 as well as the disulphide bond established by cysteine residues 250 and 263. Amino acid residues are labelled in one-letter code; the secondary structure elements to which they belong are also labelled.
monomer concave face, colored by its electrostatic potential (red: −52 mV, blue: 52 mV), calculated at pH 7 and 150 mM NaCl with APBS [56]. Two cavities are marked with yellow open stars. d) A detail view of monomer B showing the salt bridge network formed by residues Glu29/Asp33/Lys260 as well as the disulphide bond established by cysteine residues 250 and 263. Amino acid residues are labelled in one-letter code; the secondary structure elements to which they belong are also labelled.
As expected for a hyperthermostable protein [21], the ATV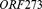 monomer is stabilised by a relatively high number of salt bridges (5 in monomer A and 8 in monomer B; to be considered as engaged in a salt bridge, the centroids of the side-chain charged groups and at least a pair of side-chain nitrogen and oxygen atoms of the ion-pairing residues must be within a 4 Å distance [22]). Some of these salt bridges form networks, namely the triplets Glu29/Asp33/Lys260 (Figure 1d) and Glu252/Lys254/Lys265 in monomer B, another feature related to hyperthermostability [21]. Furthermore, a disulphide bond is observed between cysteines 250 and 263 in both monomers (Figure 1d).
monomer is stabilised by a relatively high number of salt bridges (5 in monomer A and 8 in monomer B; to be considered as engaged in a salt bridge, the centroids of the side-chain charged groups and at least a pair of side-chain nitrogen and oxygen atoms of the ion-pairing residues must be within a 4 Å distance [22]). Some of these salt bridges form networks, namely the triplets Glu29/Asp33/Lys260 (Figure 1d) and Glu252/Lys254/Lys265 in monomer B, another feature related to hyperthermostability [21]. Furthermore, a disulphide bond is observed between cysteines 250 and 263 in both monomers (Figure 1d).
Structural similarity searches performed with Dali [23] and PDBeFold [24] produced no significant hits. The highest Z-scores were below 5 for Dali and below 4 for PDBeFold, corresponding to high r.m.s.d. values (worse than 2.7 Å) over a small number of residues ranging from 60 to 120 amino-acids. All these hits were essentially overlapping with the major 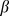 -sheet and some included the
-sheet and some included the 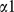 helix that is packed against this sheet. Thus, we conclude that the ATV
helix that is packed against this sheet. Thus, we conclude that the ATV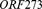 structure defines a new
structure defines a new 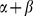 fold.
fold.
ATV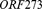 can form different types of oligomers
can form different types of oligomers
The elution of the recombinant ATV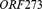 protein from the size-exclusion chromatography column used for its purification was compatible with the protein forming trimers or tetramers at pH 8.5. To define more accurately the stoichiometry of this oligomer, we analysed the purified protein by the MALS/SEC method (Figure 2). With the protein injected at 4 mg/mL (121
protein from the size-exclusion chromatography column used for its purification was compatible with the protein forming trimers or tetramers at pH 8.5. To define more accurately the stoichiometry of this oligomer, we analysed the purified protein by the MALS/SEC method (Figure 2). With the protein injected at 4 mg/mL (121 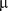 M), these experiments showed that ATV
M), these experiments showed that ATV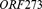 forms a tetramer (132.9
forms a tetramer (132.9 1.3 kDa) at pH 7.4 but a dimer (67.7
1.3 kDa) at pH 7.4 but a dimer (67.7 3.4 kDa) at pH 3.6. Hydrodynamic radius calculation performed with the ASTRA software yielded values of 4.84
3.4 kDa) at pH 3.6. Hydrodynamic radius calculation performed with the ASTRA software yielded values of 4.84 0.27 nm and 3.54
0.27 nm and 3.54 0.30 nm, respectively.
0.30 nm, respectively.
Figure 2. Oligomeric state of ATV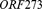 in solution.
in solution.

About 120  g of purified ATV
g of purified ATV were subjected to size-exclusion chromatography coupled to MALS/RI/UV detectors as described in the Experimental section. Two chromatograms/mass analyses are combined in this figure, showing the elution of the tetrameric (left) and dimeric (right) forms of ATV
were subjected to size-exclusion chromatography coupled to MALS/RI/UV detectors as described in the Experimental section. Two chromatograms/mass analyses are combined in this figure, showing the elution of the tetrameric (left) and dimeric (right) forms of ATV , obtained at pH 7.4 and 3.6, respectively. The molar mass (dotted lines), derived from refractive index measurements, and the absorption at 280 nm (full line) were plotted as functions of the elution volume around the peaks. The weight-averaged molar mass (Mw) values determined by the ASTRA software are indicated.
, obtained at pH 7.4 and 3.6, respectively. The molar mass (dotted lines), derived from refractive index measurements, and the absorption at 280 nm (full line) were plotted as functions of the elution volume around the peaks. The weight-averaged molar mass (Mw) values determined by the ASTRA software are indicated.
The analysis of the crystal interfaces present in the second crystal form with the PISA software [25] suggested two possible dimers with a significant buried area at the interface, namely 1258 Å2 and 1087 Å2 per monomer, respectively. Remarkably, these interfaces are not conserved in the first crystal form, which actually displays less extensive contact surfaces, the largest masking 608 Å2 per monomer, none of them considered as significant by the PISA algorithm.
Each of the two interfaces suggested by PISA results in a possible dimer with pseudo-two-fold symmetry. Based on their appearence, we call them the ‘open’ and the ‘closed’ dimers, respectively (Figure 3a and 3b). In the closed dimer the concave sides of the monomers face each other thereby creating a chamber with a volume of  8650 Å3. In the open dimer the interface involves their convex side. Not only do the open and closed dimers bear similar contact surface areas, but they also display the same number (15) of inter-subunit hydrogen bonds, defined according to Mills & Dean criteria [26]. There are however two global differences between these two dimers. First, the closed dimer exhibits two strong symmetrical salt bridges between Asp211 on one monomer and Arg215 on the other. Furthermore, a third salt bridge is observed between Glu195 in monomer B and His219 in monomer A (Figure 3c). Conversely, only weak ion-pair interactions are found in the open dimer, with the best N-O bridge [22] established between residues Glu29 and His237. Second, shape complementarity of the interface surfaces, as calculated by the SC program [27] is significantly better (0.670) for the closed dimer than for the open dimer (0.563).
8650 Å3. In the open dimer the interface involves their convex side. Not only do the open and closed dimers bear similar contact surface areas, but they also display the same number (15) of inter-subunit hydrogen bonds, defined according to Mills & Dean criteria [26]. There are however two global differences between these two dimers. First, the closed dimer exhibits two strong symmetrical salt bridges between Asp211 on one monomer and Arg215 on the other. Furthermore, a third salt bridge is observed between Glu195 in monomer B and His219 in monomer A (Figure 3c). Conversely, only weak ion-pair interactions are found in the open dimer, with the best N-O bridge [22] established between residues Glu29 and His237. Second, shape complementarity of the interface surfaces, as calculated by the SC program [27] is significantly better (0.670) for the closed dimer than for the open dimer (0.563).
Figure 3. Crystal packing of ATV.

 .
a) Overall view of the open and b) closed dimers found in the second crystal form. In both cases the pseudo-two-fold axis is vertical in the plane of the paper. c) View of the interface between monomer A (colored as in in Figure 1) and monomer B (grey) in the closed dimer. Amino acid residues involved in cross-monomer salt bridges are labelled in one-letter code. The secondary structure elements that support them are also labelled. d) A view of the dimers arranged as a continuous helical fiber in the crystal. The edges of a unit cell box are shown in grey color.
.
a) Overall view of the open and b) closed dimers found in the second crystal form. In both cases the pseudo-two-fold axis is vertical in the plane of the paper. c) View of the interface between monomer A (colored as in in Figure 1) and monomer B (grey) in the closed dimer. Amino acid residues involved in cross-monomer salt bridges are labelled in one-letter code. The secondary structure elements that support them are also labelled. d) A view of the dimers arranged as a continuous helical fiber in the crystal. The edges of a unit cell box are shown in grey color.
Noteworthy is that the dimers observed in the ATV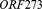 crystals do not combine to form a tetramer but rather generate fibers (see below). This observation is at odds with the fact that these crystals appear under pH conditions (pH
crystals do not combine to form a tetramer but rather generate fibers (see below). This observation is at odds with the fact that these crystals appear under pH conditions (pH  6) closer to those used for the protein purification, where the MALS/SEC data indicate the presence of tetramers, than to pH 3.6, where dimers are detected by this technique. We sought to resolve this apparent discrepancy by using SAXS to characterize the ATV
6) closer to those used for the protein purification, where the MALS/SEC data indicate the presence of tetramers, than to pH 3.6, where dimers are detected by this technique. We sought to resolve this apparent discrepancy by using SAXS to characterize the ATV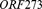 oligomer in solution and examine its shape and dimensions. Attempts to obtain SAXS data at pH
oligomer in solution and examine its shape and dimensions. Attempts to obtain SAXS data at pH  6 were unsuccessful, due to concentration-dependent protein aggregation. Therefore we collected SAXS data under the pH and ionic strength conditions used for the protein purification. Guinier analysis yielded an Rg of 35.8 Å, which is bigger than values calculated for the closed (22.9 Å) and the open (24.4 Å) dimers. The maximum dimension of the particle (D
6 were unsuccessful, due to concentration-dependent protein aggregation. Therefore we collected SAXS data under the pH and ionic strength conditions used for the protein purification. Guinier analysis yielded an Rg of 35.8 Å, which is bigger than values calculated for the closed (22.9 Å) and the open (24.4 Å) dimers. The maximum dimension of the particle (D = 111.0 Å), obtained from the distance distribution function (P(r)), was also larger than those computed from the structures of the closed (67.9 Å) and open (84.0 Å) dimers. Finally, the excluded volume of the hydrated particle (Porod volume) was 221.5 nm3 that, according to the empirical formula
= 111.0 Å), obtained from the distance distribution function (P(r)), was also larger than those computed from the structures of the closed (67.9 Å) and open (84.0 Å) dimers. Finally, the excluded volume of the hydrated particle (Porod volume) was 221.5 nm3 that, according to the empirical formula 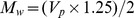 [28], gives a molar mass of 133 kDa, in good agreement with an ATV
[28], gives a molar mass of 133 kDa, in good agreement with an ATV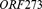 tetramer (132 kDa). The Kratky plot was consistent with a properly folded protein (Figure 4b), whereas the P(r) function was monomodal and suggested a compact, slightly elongated particle (Figure 4c).
tetramer (132 kDa). The Kratky plot was consistent with a properly folded protein (Figure 4b), whereas the P(r) function was monomodal and suggested a compact, slightly elongated particle (Figure 4c).
Figure 4. SAXS analysis of ATV.

 .
a) SAXS intensity as a function of the momentum transfer. This profile corresponds to the measurements taken at 4.7 mg/ml protein cocentration and pH 8.5. Average values are in red and the standard error in grey. b) The Kratky plot (see text for details) corresponds to a folded protein. c) Pair-distance distribution, P(r), function of the data shown in panel a). d) Three orthogonal views of the ab initio envelope calculated imposing orthorhombic symmetry.
.
a) SAXS intensity as a function of the momentum transfer. This profile corresponds to the measurements taken at 4.7 mg/ml protein cocentration and pH 8.5. Average values are in red and the standard error in grey. b) The Kratky plot (see text for details) corresponds to a folded protein. c) Pair-distance distribution, P(r), function of the data shown in panel a). d) Three orthogonal views of the ab initio envelope calculated imposing orthorhombic symmetry.
Next we employed the program DAMMIF to carry out ab initio shape reconstruction of the oligomer. We conducted several series of independent runs with either no forced symmetry or imposing P2, P222, P3 or P4 symmetries (Table 2, Figure S1a). Within each symmetry class, the models were very reproducible with average normalized spatial discrepancy (NSD) values below 1.0, consistent with structurally similar solutions. Furthermore, all the models were similar in terms of agreement with the experimental data, as measured by DAMMIF 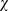 parameter. Slightly better agreement was attained with P1 and P222 (Figure 4d) models, whereas P4 models fitted the data systematically worse than the rest. We note that the averaged models have slightly bigger volumes, ranging from 256.5 Å (P4 model) to 282.5 Å (P1 model), than that obtained by Porod analysis.
parameter. Slightly better agreement was attained with P1 and P222 (Figure 4d) models, whereas P4 models fitted the data systematically worse than the rest. We note that the averaged models have slightly bigger volumes, ranging from 256.5 Å (P4 model) to 282.5 Å (P1 model), than that obtained by Porod analysis.
Table 2. Summary of SAXS results.
| DAMMIFa | |||||
| Symmetry |
|
NSD |
 Vol Vol (nm3) (nm3) |
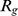 (Å) (Å) |
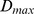 (Å) (Å) |
| P1 | 1.654 0.011 0.011 |
0.583 0.019 0.019 |
282.5 | 34.4 | 115 |
| P2 | 1.659 0.021 0.021 |
0.680 0.106 0.106 |
278.0 | 34.2 | 115 |
| P222 | 1.648 0.008 0.008 |
0.486 0.017 0.017 |
272.2 | 34.7 | 114 |
| P3 | 1.704 0.036 0.036 |
0.654 0.032 0.032 |
278.4 | 33.6 | 102 |
| P4 | 1.770 0.052 0.052 |
0.768 0.127 0.127 |
256.5 | 32.8 | 90 |
ab initio shape reconstruction.
Rigid-body modelling.
In a complementary approach, we used SASREF [29] to generate rigid-body refined models based on the available structures of ATV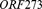 , that is the monomer and the two possible dimers identified in the second crystal form. For the monomer we used the same symmetries as for the ab initio shape reconstruction, whereas for the dimers we calculated models with P1 and P2 symmetry. As expected, better agreement to the experimental curve was achieved by the trials with more degrees of freedom, i.e. those involving the monomer (Table 2), with the notable exception of the P3 symmetry, which gave the worst agreement of all the rigid-body models. Examination of the P3 models showed that the monomers are disconnected since they need to occupy a volume that is better accounted for by four monomers. Amongst the models generated from the monomers, those with P4 symmetry performed only better than the P3 models and worse than the rest, in terms of
, that is the monomer and the two possible dimers identified in the second crystal form. For the monomer we used the same symmetries as for the ab initio shape reconstruction, whereas for the dimers we calculated models with P1 and P2 symmetry. As expected, better agreement to the experimental curve was achieved by the trials with more degrees of freedom, i.e. those involving the monomer (Table 2), with the notable exception of the P3 symmetry, which gave the worst agreement of all the rigid-body models. Examination of the P3 models showed that the monomers are disconnected since they need to occupy a volume that is better accounted for by four monomers. Amongst the models generated from the monomers, those with P4 symmetry performed only better than the P3 models and worse than the rest, in terms of  (Table 2). To further explore the possibility that the protein may form trimers in solution and to exclude a problem with SASREF for the generation of appropriated trigonal models, we used the symmdock software [30] to generate trimers of ATV
(Table 2). To further explore the possibility that the protein may form trimers in solution and to exclude a problem with SASREF for the generation of appropriated trigonal models, we used the symmdock software [30] to generate trimers of ATV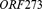 with trigonal symmetry. The best symmdock model has a score of 11592 compared to a score of 8892 for the second best model, with scores monotonously decreasing thereafter. The best symmdock model has an Rg of 25.8 Å, the second best model has Rg = 25.9 Å and the average Rg of the best 10 models is 27.3
with trigonal symmetry. The best symmdock model has a score of 11592 compared to a score of 8892 for the second best model, with scores monotonously decreasing thereafter. The best symmdock model has an Rg of 25.8 Å, the second best model has Rg = 25.9 Å and the average Rg of the best 10 models is 27.3 1.0 Å. Similarly, the first model has D
1.0 Å. Similarly, the first model has D = 84.5 Å, the second model has D
= 84.5 Å, the second model has D = 83.8 Å and the average of the best 10 models gives D
= 83.8 Å and the average of the best 10 models gives D = 84.3
= 84.3 2.8 Å. These values are clearly different from the experimental values obtained by SAXS analysis. Further, the SAXS profiles calculated from these trimers by the program CRYSOL [31], fitted very poorly the experimental profile, with average
2.8 Å. These values are clearly different from the experimental values obtained by SAXS analysis. Further, the SAXS profiles calculated from these trimers by the program CRYSOL [31], fitted very poorly the experimental profile, with average 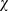 = 39.98
= 39.98 6.73. We conclude that under these experimental conditions ATV
6.73. We conclude that under these experimental conditions ATV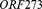 does not form trimers.
does not form trimers.
Interestingly, the rigid-body refined models calculated from the crystallographic open dimer came next to those from the monomer in terms of agreement with the SAXS data. Furthermore, models calculated from the open dimer were all very similar, irrespective of the symmetry used (overall 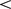 NSD
NSD = 1.128
= 1.128 0.492). We conclude from these results that under the conditions tested (pH 8.5, 100 mM NaCl) ATV
0.492). We conclude from these results that under the conditions tested (pH 8.5, 100 mM NaCl) ATV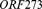 forms a tetramer, possibly with point group 222 symmetry (Figure S1b).
forms a tetramer, possibly with point group 222 symmetry (Figure S1b).
ATV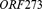 is a hyperthermostable and hyperacidostable protein
is a hyperthermostable and hyperacidostable protein
ATV viruses proliferate at extreme pH ( 1.5) and temperature (
1.5) and temperature ( 85°C). Under these conditions, structural proteins have to bear an extreme stability to preserve fold and function. The ATV
85°C). Under these conditions, structural proteins have to bear an extreme stability to preserve fold and function. The ATV CD spectrum recorded at 20°C and pH 7.2 displays two minima at 215 and 222 nm, characteristic of a folded protein containing mainly
CD spectrum recorded at 20°C and pH 7.2 displays two minima at 215 and 222 nm, characteristic of a folded protein containing mainly  -helices (Figure 5). The CD spectra recorded at 20°C and 80°C, both at neutral and acidic (pH 0) conditions are all nearly superimposable, establishing the extreme protein resistance to high temperature, low pH and a combination of both factors. Deconvolution of the CD spectra using the CDSSTR program [32], as implemented in the Dichroweb server [33], was consistent with the secondary structure derived from the crystal structure (42% helices and 18% strands, see Figure 5) within the uncertainties of this approach [32]. These analyses suggest that, at the two pH values studied, increasing the temperature to 80°C slightly destabilises the protein helices without affecting its content in
-helices (Figure 5). The CD spectra recorded at 20°C and 80°C, both at neutral and acidic (pH 0) conditions are all nearly superimposable, establishing the extreme protein resistance to high temperature, low pH and a combination of both factors. Deconvolution of the CD spectra using the CDSSTR program [32], as implemented in the Dichroweb server [33], was consistent with the secondary structure derived from the crystal structure (42% helices and 18% strands, see Figure 5) within the uncertainties of this approach [32]. These analyses suggest that, at the two pH values studied, increasing the temperature to 80°C slightly destabilises the protein helices without affecting its content in 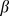 -strands.
-strands.
Figure 5. Circular dichroism spectra of ATV.

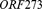 . Mean residue ellipticity spectra recorded at pH 7.2 (black lines) and pH 0 (red lines), either at 20°C (full lines) or at 80°C (dashed lines). The content in helices and strands, as determined by deconvolution of the spectra, is shown (see text for details).
. Mean residue ellipticity spectra recorded at pH 7.2 (black lines) and pH 0 (red lines), either at 20°C (full lines) or at 80°C (dashed lines). The content in helices and strands, as determined by deconvolution of the spectra, is shown (see text for details).
In contrast, at pH 11 and 80°C, ATV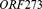 unfolds: the signal at 200 nm is negative and the signal in the range 215–260 nm is closer to zero (not shown). Importantly, the fact that ATV
unfolds: the signal at 200 nm is negative and the signal in the range 215–260 nm is closer to zero (not shown). Importantly, the fact that ATV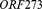 withstands a combination of low pH and high temperature agrees well with the environment of the ATV virus in the thermal spring where it was discovered.
withstands a combination of low pH and high temperature agrees well with the environment of the ATV virus in the thermal spring where it was discovered.
Final remarks
Apart from an increased presence of salt-bridges and other ionic pair interactions, ATV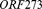 bears some rare traits for a hyperthermostable protein (see [21] for a thorough discussion of these properties). First, its fold is less compact than expected and displays several cavities. Second, its C′-terminus and especially its N′-terminus are not well structured. Finally, it carries disordered loops. The presence of cavities and of disordered loops would seem to suggest an enzymatic function. However, the novelty of the fold and the fact that the protein sequence does not retrieve any significant hit in public databases hinder the assignment of a biological function to ATV
bears some rare traits for a hyperthermostable protein (see [21] for a thorough discussion of these properties). First, its fold is less compact than expected and displays several cavities. Second, its C′-terminus and especially its N′-terminus are not well structured. Finally, it carries disordered loops. The presence of cavities and of disordered loops would seem to suggest an enzymatic function. However, the novelty of the fold and the fact that the protein sequence does not retrieve any significant hit in public databases hinder the assignment of a biological function to ATV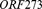 . Furthermore, although the protein has a number of surface pockets we could not assign them to any bona fide ligand/substrate. Significantly, ATV
. Furthermore, although the protein has a number of surface pockets we could not assign them to any bona fide ligand/substrate. Significantly, ATV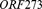 is the fourth most abundant protein in virion preparations [15] and, moreover, no homologue of ATV
is the fourth most abundant protein in virion preparations [15] and, moreover, no homologue of ATV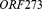 is present in the STSV1 virus [16]. In contrast, the ATV homologue (ATV
is present in the STSV1 virus [16]. In contrast, the ATV homologue (ATV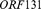 ) of the STSV1 major capsid protein (STSV1
) of the STSV1 major capsid protein (STSV1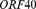 ) is also the most abundant protein in the ATV virion [15].
) is also the most abundant protein in the ATV virion [15].
The two viruses ATV and STSV1 both exhibit large fusiform bodies but while the former generates long bipolar tails extracellularly, the latter generates one long tail intracellularly [14], [16]. Both genomes encode several pairs of homologous proteins, albeit distantly related, but they differ markedly in their virion protein contents. Whereas ATV virions contain several major protein components, STSV1 carries only one major component, the coat protein STSV1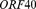 . It has been hypothesised that some of the virion components of ATV actively contribute to the extracellular tail development [15] and in a detailed study of one of the major components, a MoxR-type ATPase ATV
. It has been hypothesised that some of the virion components of ATV actively contribute to the extracellular tail development [15] and in a detailed study of one of the major components, a MoxR-type ATPase ATV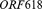 , evidence was provided for a co-chaperone activity together with a Von Willebrand domain A protein ATV
, evidence was provided for a co-chaperone activity together with a Von Willebrand domain A protein ATV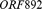 [34]. Moreover, a model was presented whereby this co-chaperone facilitated tail development together with another virion protein ATV
[34]. Moreover, a model was presented whereby this co-chaperone facilitated tail development together with another virion protein ATV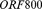 , which exhibits intermediate filament-like properties. Further, it was proposed that novel ATV DNA binding proteins, also present in the virions, were involved in drawing DNA along the tails [34]. In this context, a structural role for ATV
, which exhibits intermediate filament-like properties. Further, it was proposed that novel ATV DNA binding proteins, also present in the virions, were involved in drawing DNA along the tails [34]. In this context, a structural role for ATV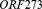 cannot be ruled out in spite of its enzyme-like features. In this regard, the stability of ATV
cannot be ruled out in spite of its enzyme-like features. In this regard, the stability of ATV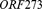 to extreme acidic pH suggests that it may be in contact with the external environment of the virion and thus participate in its coat and contribute to the development of ATV tails. Finally, the two interfaces identified in the second-form crystals of ATV
to extreme acidic pH suggests that it may be in contact with the external environment of the virion and thus participate in its coat and contribute to the development of ATV tails. Finally, the two interfaces identified in the second-form crystals of ATV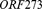 are assembled head-to-tail, resulting in helicoidal fibers that extend indefinitely along the crystallographic c-axis (Figure 3d). Although we have not observed fiber formation when handling the protein, such a process could be dependent on pH. In this respect, the crystal pH (
are assembled head-to-tail, resulting in helicoidal fibers that extend indefinitely along the crystallographic c-axis (Figure 3d). Although we have not observed fiber formation when handling the protein, such a process could be dependent on pH. In this respect, the crystal pH ( 6) might reflect the protein natural environment better than the pH (8.5) of the protein solutions. Our results should help guiding further experiments necessary to understand the biological function of ATV
6) might reflect the protein natural environment better than the pH (8.5) of the protein solutions. Our results should help guiding further experiments necessary to understand the biological function of ATV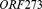 and its possible involvement in the development of ATV tails.
and its possible involvement in the development of ATV tails.
Materials and Methods
ATV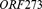 construction and purification procedures
construction and purification procedures
The ATV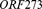 gene was cloned into the pDEST14 expression vector according to standard Gateway protocols. The final construct included a coding sequence for a C–terminal hexa–histidine tag. A variant of ATV
gene was cloned into the pDEST14 expression vector according to standard Gateway protocols. The final construct included a coding sequence for a C–terminal hexa–histidine tag. A variant of ATV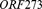 (ATV
(ATV ), carrying the Leu31Met, Leu117Met and Leu240Met mutations, was generated by syntetic production of the mutated gene (Geneart).
), carrying the Leu31Met, Leu117Met and Leu240Met mutations, was generated by syntetic production of the mutated gene (Geneart).
Plasmids were transformed into the Escherichia coli T7 Iq pLysS expression strain (New England Biolabs). Cells were grown at 37°C in Lysogeny Broth (LB) until the OD reached 0.6. Protein expression was induced then with 0.5 mM isopropyl-
reached 0.6. Protein expression was induced then with 0.5 mM isopropyl-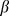 -thio-galactoside (IPTG) and the cultures maintained at 25°C. After
-thio-galactoside (IPTG) and the cultures maintained at 25°C. After  16 hours, cells were harvested and lysed by sonication in 50 mM sodium phosphate buffer (pH 8), 300 mM NaCl, 10 mM imidazole and a protease inhibitor cocktail (Complete EDTA-free, Roche). Soluble protein was separated from inclusion bodies and cell debris by centrifuging for 30 min at 20,000 g. We used an ÄKTA FPLC system for a two-step purification. First, the lysates were applied onto a Ni
16 hours, cells were harvested and lysed by sonication in 50 mM sodium phosphate buffer (pH 8), 300 mM NaCl, 10 mM imidazole and a protease inhibitor cocktail (Complete EDTA-free, Roche). Soluble protein was separated from inclusion bodies and cell debris by centrifuging for 30 min at 20,000 g. We used an ÄKTA FPLC system for a two-step purification. First, the lysates were applied onto a Ni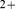 affinity chromatography column (HisTrap 5 ml, GE Healthcare) and eluted with 250 mM imidazole in 50 mM sodium phosphate buffer (pH 8) and 300 mM NaCl. A preparative Superdex 200 (GE Healthcare) gel filtration column was then run in 10 mM Bicine pH 8.5, 100 mM NaCl to remove aggregated material.
affinity chromatography column (HisTrap 5 ml, GE Healthcare) and eluted with 250 mM imidazole in 50 mM sodium phosphate buffer (pH 8) and 300 mM NaCl. A preparative Superdex 200 (GE Healthcare) gel filtration column was then run in 10 mM Bicine pH 8.5, 100 mM NaCl to remove aggregated material.
Seleno-Methionine-labeled ATV was prepared following standard procedures in the minimum medium M9 by blocking the methionine biosynthesis pathway [35]. Expression, purification, and characterisation of the SeMet-labeled ATV
was prepared following standard procedures in the minimum medium M9 by blocking the methionine biosynthesis pathway [35]. Expression, purification, and characterisation of the SeMet-labeled ATV protein were carried out using the same protocols as for the native protein.
protein were carried out using the same protocols as for the native protein.
Size-Exclusion Chromatography-coupled Multi-Angle Light Scattering
Size-Exclusion chromatography (SEC) was performed on an Alliance 2695 HPLC system (Waters). A Shodex KW803 column, operated at 0.5 mL/min, was used in either 20 mM Hepes (pH 7.4) with 100 mM NaCl or in 20 mM citrate buffer (pH 3.6) with 150 mM NaCl. Multi-Angle Light Scattering (MALS), Ultra-Violet (UV) spectrophotometry, Quasi-Elastic Light Scattering (QELS) and Refractive index (RI) measurements were achieved with MiniDawn Treos (Wyatt Technology), Photo Diode Array 2996 (Waters), DynaPro (Wyatt Technology) and Optilab rEX (Wyatt Technology) detectors, respectively. Weight-averaged molar mass (Mw) and hydrodynamic radius calculations were performed with the ASTRA software (Wyatt Technology) using a dn/dc value of 0.185 mL/g.
Circular Dichroism
Temperature and pH stability studies were carried out by far-UV Circular Dichroism (CD) spectroscopy. CD spectra were recorded with a JASCO J-810 spectropolarimeter (JASCO Corporation, Japan) equipped with a Peltier temperature control system. Far–UV measurements (195–260 nm) were performed using a 0.1 cm path quartz cuvette, with a scanning speed of 20 nm/min, spectral bandwidth of 1 nm, and were averaged over three scans. The solvent spectra were subtracted in all experiments to eliminate background effects. CD measurements in millidegrees were performed at a protein concentration of 0.15 mg/mL in 10 mM sodium phosphate buffer at pH 7.2. Stability tests at pH 0 and pH 11 were carried out in 1 M HCl and 1 mM NaOH, respectively. Thermal denaturation was monitored by increasing the temperature from 20°C to 80°C.
The CD spectra were deconvoluted by using the CDSSTR program [32], with reference database 7, as implemented in the Dichroweb server [33]. The normalised root-mean square deviations were in the 0.004–0.013 range, consistent with excellent agreement between the experimental data and the fitting from the deconvolution.
Small-Angle X-ray Scattering measurements and analysis
All Small-Angle X-ray Scattering (SAXS) measurements were carried out at the ID14eh13 beamline (ESRF, Grenoble, France) at a working energy of 13.32 keV corresponding to 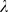 = 0.931 Å. Data were collected on a Pilatus 1 M detector placed at a sample-detector distance of 2.43 m.
= 0.931 Å. Data were collected on a Pilatus 1 M detector placed at a sample-detector distance of 2.43 m.
SAXS data were collected using 30 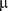 l of protein solution at 2.3, 4.7 and 9.2 mg/ml in 10 mM Bicine (pH 8.5) buffer with 100 mM NaCl, loaded by a robotic system into a 2-mm quartz capillary mounted in a vacuum. This procedure enables the sample to move across the beam during exposure thus minimizing the effect of radiation damage. Ten exposures each of 10 s were made in this way for each condition. Individual frames were processed automatically and independently at the beamline by the data collection software (BsxCUBE), yielding radially averaged normalized intensities as a function of the momentum transfer q, with
l of protein solution at 2.3, 4.7 and 9.2 mg/ml in 10 mM Bicine (pH 8.5) buffer with 100 mM NaCl, loaded by a robotic system into a 2-mm quartz capillary mounted in a vacuum. This procedure enables the sample to move across the beam during exposure thus minimizing the effect of radiation damage. Ten exposures each of 10 s were made in this way for each condition. Individual frames were processed automatically and independently at the beamline by the data collection software (BsxCUBE), yielding radially averaged normalized intensities as a function of the momentum transfer q, with 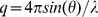 , where
, where 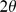 is the total scattering angle and
is the total scattering angle and 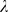 is the X-ray wavelength. Data were collected in the range q = 0.04–6 nm−1. The ten frames were combined to give the average scattering curve for each measurement and any data points affected by aggregation, possibly induced by radiation damage, were excluded. Scattering from the buffer alone was also measured before and after each sample measurement and the average of these two buffer measurements was used for background subtraction using the program PRIMUS [36] from the ATSAS package [37]. PRIMUS was also used to perform Guinier analysis [38] of the low q data, which provides an estimate of the radius of gyration (Rg). Regularized indirect transforms of the scattering data were carried out with the program GNOM [39] to obtain P(r) functions of interatomic distances. The P(r) function has a maximum at the most probable intermolecular distance and goes to zero at D
is the X-ray wavelength. Data were collected in the range q = 0.04–6 nm−1. The ten frames were combined to give the average scattering curve for each measurement and any data points affected by aggregation, possibly induced by radiation damage, were excluded. Scattering from the buffer alone was also measured before and after each sample measurement and the average of these two buffer measurements was used for background subtraction using the program PRIMUS [36] from the ATSAS package [37]. PRIMUS was also used to perform Guinier analysis [38] of the low q data, which provides an estimate of the radius of gyration (Rg). Regularized indirect transforms of the scattering data were carried out with the program GNOM [39] to obtain P(r) functions of interatomic distances. The P(r) function has a maximum at the most probable intermolecular distance and goes to zero at D , the maximum intramolecular distance. Values of D
, the maximum intramolecular distance. Values of D were chosen that yielded solutions that fit the experimental data well and have a smooth and strictly positive P(r) function. This approach also allows the calculation of Rg values that agreed with the values found by the Guinier analysis.
were chosen that yielded solutions that fit the experimental data well and have a smooth and strictly positive P(r) function. This approach also allows the calculation of Rg values that agreed with the values found by the Guinier analysis.
Ab initio 3D shape reconstructions
We built 3D bead models fitting the scattering data with the program DAMMIF [40]. Ten independent DAMMIF runs were performed for each scattering profile, with data extending up to 0.25 Å−1, using slow mode settings, assuming either P1, P2, P222, P3 or P4 symmetry and allowing for a maximum 500 steps to grant convergence. The models resulting from independent runs at each symmetry were superimposed using the DAMAVER suite [41]. This yielded an initial alignment of structures based on their axes of inertia followed by minimisation of the normalized spatial discrepancy (NSD), which is zero for identical objects and larger than 1 for systematically different objects [42]. The aligned structures were then averaged, giving an effective occupancy to each voxel in the model, and filtered at half-maximal occupancy to produce models of the appropriate volume that were used for all subsequent analyses. To provide a clearer representation of the 3D shape reconstructions, bead models were converted to density maps by the program pdb2vol from the Situs package [43].
Rigid body modelling of the SAXS data
We used the program CRYSOL [31] to generate theoretical scattering curves from monomer A of the best resolution model of ATV , as well as from the two putative dimers observed in this crystal form. Rigid body modeling was performed with the program SASREF [29], which uses a simulated annealing protocol to build an interconnected ensemble of subunits without steric clashes, while minimizing the discrepancy between the experimental scattering data and the curves calculated from the appropriate subunits by CRYSOL.
, as well as from the two putative dimers observed in this crystal form. Rigid body modeling was performed with the program SASREF [29], which uses a simulated annealing protocol to build an interconnected ensemble of subunits without steric clashes, while minimizing the discrepancy between the experimental scattering data and the curves calculated from the appropriate subunits by CRYSOL.
Crystallisation and Structure Determination
ATV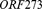 crystallisation trials were carried out in sitting-drop vapour diffusion method at 20°C in 96-well Greiner crystallisation plates using a nanodrop-dispensing robot (Cartesian Inc.). The first crystals, belonging to space group
crystallisation trials were carried out in sitting-drop vapour diffusion method at 20°C in 96-well Greiner crystallisation plates using a nanodrop-dispensing robot (Cartesian Inc.). The first crystals, belonging to space group 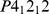 (Table 1), were obtained in 5%–15% PEG 8000, 0.2 M MgCl2, 0.1 M Tris (pH 7–8). A native data set (
(Table 1), were obtained in 5%–15% PEG 8000, 0.2 M MgCl2, 0.1 M Tris (pH 7–8). A native data set (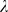 = 0.91839 Å) to 3.85 Å resolution was collected at the ID14eh4 beamline (ESRF, Grenoble, France).
= 0.91839 Å) to 3.85 Å resolution was collected at the ID14eh4 beamline (ESRF, Grenoble, France).
Crystals of a second form grew in a few days by mixing 1.5 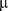 L protein at 5 mg/mL with 0.5
L protein at 5 mg/mL with 0.5  L 3.6% isopropanol, 1.9 M (NH4)2SO4, 5 mM MgCl2, 2 mM AMP. The mother liquor was pH 6. Crystals were cryoprotected with mother-liquor supplemented with 25% glycerol and 2.3 M (NH4)2SO4 and flash vitrified in liquid nitrogen. Two data sets were collected: a native data set (
L 3.6% isopropanol, 1.9 M (NH4)2SO4, 5 mM MgCl2, 2 mM AMP. The mother liquor was pH 6. Crystals were cryoprotected with mother-liquor supplemented with 25% glycerol and 2.3 M (NH4)2SO4 and flash vitrified in liquid nitrogen. Two data sets were collected: a native data set ( = 0.91839 Å) to 2.15 Å resolution at the ID29 beamline (ESRF, Grenoble, France) and a Se-SAD data set (
= 0.91839 Å) to 2.15 Å resolution at the ID29 beamline (ESRF, Grenoble, France) and a Se-SAD data set ( = 0.97911 Å) to 2.77 Å at the Proxima 1 beamline (SOLEIL, Gif-sur-Yvette, France).
= 0.97911 Å) to 2.77 Å at the Proxima 1 beamline (SOLEIL, Gif-sur-Yvette, France).
Data integration and scaling were done using the XDS package [44] and POINTLESS [45] was used to help establishing the space group. The structure of ATV was solved by the single-wavelength anomalous diffraction (SAD) method using the autoSHARP program [46] with SHELXD [47] to locate the selenium substructure. Initial automatic building was performed with Buccaneer [48]. Alternative cycles of manual model building with Coot [49] and refinement with either autoBuster-TNT [50] or refmac5 [51] were carried out to improve the initial model.
was solved by the single-wavelength anomalous diffraction (SAD) method using the autoSHARP program [46] with SHELXD [47] to locate the selenium substructure. Initial automatic building was performed with Buccaneer [48]. Alternative cycles of manual model building with Coot [49] and refinement with either autoBuster-TNT [50] or refmac5 [51] were carried out to improve the initial model.
We solved the structure of the first crystal form by molecular replacement with the program PHASER [52], using as template the final model from the second crystal form. Refinement was performed with autoBuster-TNT [50] with the “target” option [53] that uses local similarity restraints to a separate already determined structure, typically at higher resolution, that remains fixed during the refinement of the structure being refined. This procedure facilitates the refinement of low resolution structures. Our final model from the second crystal form was used as “target” structure. Temperature factors were refined with the translation-libration-screw (TLS) approach with a single TLS group.
We used the DSSP program [54] to define the secondary structure elements of the higher resolution crystal structure. Figures were generated using Chimera [55].
Supporting Information
SAXS analysis of ATV
 .
a) Three orthogonal views of each of the five ab initio envelopes calculated imposing (from left to right) no, binary, orthorhombic, trigonal or tetragonal symmetry. b) Fitting of the SASREF model obtained from the open dimer using P2 symmetry into the P222 DAMMIF model. The fitting was performed by the program Chimera [55].
.
a) Three orthogonal views of each of the five ab initio envelopes calculated imposing (from left to right) no, binary, orthorhombic, trigonal or tetragonal symmetry. b) Fitting of the SASREF model obtained from the open dimer using P2 symmetry into the P222 DAMMIF model. The fitting was performed by the program Chimera [55].
(TIF)
Acknowledgments
We gratefully acknowledge the help of Pierre Legrand, at the Proxima 1 beamline, with data collection. We are also grateful to the staff of the ID14eh4 and ID29 beamlines for their support. We thank the Soleil synchrotron and the European Synchrotron Radiation Facility (ESRF; Grenoble, France) for beam time allocation. We are grateful to the Cambridge Crystallographic Data Centre for granting access to Relibase+ to the SILVER consortium laboratories, and to Global Phasing Ltd for the implementation of a Relibase+ common server for the SILVER partners.
Funding Statement
C.F.-R. was funded by a doctoral fellowship from the Fundação para a Ciência e a Tecnologia (http://www.fct.pt/), Portugal (SFRH/BD/44380/2008). The Ville de Marseille (http://www.marseille.fr) provided a financial installation aid to M.O.-L. The Archaea Centre, Copenhagen was supported by a grant from the Danish Natural Science Research Council. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bergh O, Borsheim KY, Bratbak G, Heldal M (1989) High abundance of viruses found in aquatic environments. Nature 340: 467–468. [DOI] [PubMed] [Google Scholar]
- 2. Suttle CA (2007) Marine viruses–major players in the global ecosystem. Nat Rev Microbiol 5: 801–812. [DOI] [PubMed] [Google Scholar]
- 3. Torsvik T, Dundas ID (1974) Bacteriophage of Halobacterium salinarium. Nature 248: 680–681. [DOI] [PubMed] [Google Scholar]
- 4. Zillig W, Kletzin A, Schleper C, Holz I, Janekovic D, et al. (1994) Screening for Sulfolobales, their plasmids and their viruses in Icelandic solfataras. Syst Appl Microbiol 16: 609–628. [Google Scholar]
- 5. Mochizuki T, Krupovic M, Pehau-Arnaudet G, Sako Y, Forterre P, et al. (2012) Archaeal virus with exceptional virion architecture and the largest single-stranded DNA genome. Proc Natl Acad Sci USA 109: 13386–13391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Prangishvili D, Garrett RA, Koonin EV (2006) Evolutionary genomics of archaeal viruses: unique viral genomes in the third domain of life. Virus Res 117: 52–67. [DOI] [PubMed] [Google Scholar]
- 7. Abrescia NGA, Bamford DH, Grimes JM, Stuart DI (2012) Structure unifies the viral universe. Annu Rev Biochem 81: 795–822. [DOI] [PubMed] [Google Scholar]
- 8. Prangishvili D, Krupovic M (2012) A new proposed taxon for double-stranded DNA viruses, the order “Ligamenvirales”. Arch Virol 157: 791–795. [DOI] [PubMed] [Google Scholar]
- 9. Goulet A, Blangy S, Redder P, Prangishvili D, Felisberto-Rodrigues C, et al. (2009) Acidianus filamentous virus 1 coat proteins display a helical fold spanning the filamentous archaeal viruses lineage. Proc Natl Acad Sci USA 106: 21155–21160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goulet A, Vestergaard G, Felisberto-Rodrigues C, Campanacci V, Garrett RA, et al. (2010) Getting the best out of long-wavelength X-rays: de novo chlorine/sulfur SAD phasing of a structural protein from ATV. Acta Crystallogr D Biol Crystallogr 66: 304–308. [DOI] [PubMed] [Google Scholar]
- 11. Goulet A, Pina M, Redder P, Prangishvili D, Vera L, et al. (2010) Orf157 from the archaeal virus Acidianus filamentous virus 1 defines a new class of nuclease. J Virol 84: 5025–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goulet A, Spinelli S, Blangy S, van Tilbeurgh H, Leulliot N, et al. (2009) The crystal structure of ORF14 from Sulfolobus islandicus filamentous virus. Proteins 76: 1020–1022. [DOI] [PubMed] [Google Scholar]
- 13. Goulet A, Spinelli S, Blangy S, van Tilbeurgh H, Leulliot N, et al. (2009) The thermo- and acidostable ORF-99 from the archaeal virus AFV1. Protein Sci 18: 1316–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Häring M, Vestergaard G, Rachel R, Chen L, Garrett RA, et al. (2005) Independent virus development outside a host. Nature 436: 1101–1102. [DOI] [PubMed] [Google Scholar]
- 15. Prangishvili D, Vestergaard G, Häring M, Aramayo R, Basta T, et al. (2006) Structural and genomic properties of the hyperthermophilic archaeal virus ATV with an extracellular stage of the reproductive cycle. J Mol Biol 359: 1203–1216. [DOI] [PubMed] [Google Scholar]
- 16. Xiang X, Chen L, Huang X, Luo Y, She Q, et al. (2005) Sulfolobus tengchongensis spindle-shaped virus STSV1: virus-host interactions and genomic features. J Virol 79: 8677–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Buchan DWA, Ward SM, Lobley AE, Nugent TCO, Bryson K, et al. (2010) Protein annotation and modelling servers at University College London. Nucleic Acids Res 38: W563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hendlich M, Bergner A, Günther J, Klebe G (2003) Relibase: design and development of a database for comprehensive analysis of protein-ligand interactions. J Mol Biol 326: 607–620. [DOI] [PubMed] [Google Scholar]
- 19. Hendlich M, Rippmann F, Barnickel G (1997) LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model 15: 359–363, 389. [DOI] [PubMed] [Google Scholar]
- 20. Jambon M, Imberty A, Deléage G, Geourjon C (2003) A new bioinformatic approach to detect common 3D sites in protein structures. Proteins 52: 137–145. [DOI] [PubMed] [Google Scholar]
- 21. Petsko GA (2001) Structural basis of thermostability in hyperthermophilic proteins, or “there's more than one way to skin a cat”. Methods Enzymol 334: 469–478. [DOI] [PubMed] [Google Scholar]
- 22. Kumar S, Nussinov R (2002) Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys J 83: 1595–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holm L, Rosenström P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38: W545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krissinel E, Henrick K (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60: 2256–2268. [DOI] [PubMed] [Google Scholar]
- 25.Krissinel E, Henrick K (2005) Detection of protein assemblies in crystals. In: R Berthold M, Glen R, Diederichs K, Kohlbacher O, Fischer I, editors, Computational Life Sciences, Springer Berlin/Heidelberg, volume 3695 of Lecture Notes in Computer Science. pp. 163–174.
- 26. Mills JE, Dean PM (1996) Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J Comput Aided Mol Des 10: 607–622. [DOI] [PubMed] [Google Scholar]
- 27. Lawrence MC, Colman PM (1993) Shape complementarity at protein/protein interfaces. J Mol Biol 234: 946–950. [DOI] [PubMed] [Google Scholar]
- 28. Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, et al. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr 45: 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petoukhov MV, Svergun DI (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J 89: 1237–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ (2005) Geometry-based exible and symmetric protein docking. Proteins 60: 224–231. [DOI] [PubMed] [Google Scholar]
- 31. Svergun D, Barberato C, Koch MHJ (1995) CRYSOL – a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28: 768–773. [Google Scholar]
- 32. Compton LA, Johnson WC Jr (1986) Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal Biochem 155: 155–167. [DOI] [PubMed] [Google Scholar]
- 33. Whitmore L, Wallace BA (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89: 392–400. [DOI] [PubMed] [Google Scholar]
- 34. Scheele U, Erdmann S, Ungewickell EJ, Felisberto-Rodrigues C, Ortiz-Lombardía M, et al. (2011) Chaperone role for proteins P618 and P892 in the extracellular tail development of Acidianus two-tailed virus. J Virol 85: 4812–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Studier FW (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41: 207–234. [DOI] [PubMed] [Google Scholar]
- 36. Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36: 1277–1282. [Google Scholar]
- 37. Konarev PV, Petoukhov MV, Volkov VV, Svergun DI (2006) ATSAS 2.1, a program package for small-angle scattering data analysis. J Appl Crystallogr 39: 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guinier A (1939) La diffraction des rayons x aux très pétits angles: application à l'etude de phénomènes ultramicroscopiques. Ann Phys (Paris) 12: 161–237. [Google Scholar]
- 39. Svergun DI (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 25: 495–503. [Google Scholar]
- 40. Franke D, Svergun DI (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr 42: 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Volkov VV, Svergun DI (2003) Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr 36: 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kozin MB, Svergun DI (2001) Automated matching of high- and low-resolution structural models. J Appl Crystallogr 34: 33–41. [Google Scholar]
- 43. Wriggers W (2010) Using Situs for the integration of multi-resolution structures. Biophys Rev 2: 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Evans PR (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr 67: 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vonrhein C, Blanc E, Roversi P, Bricogne G (2007) Automated structure solution with autoSHARP. Methods Mol Biol 364: 215–230. [DOI] [PubMed] [Google Scholar]
- 47. Sheldrick GM (2010) Experimental phasing with shelxc/d/e: combining chain tracing with density modification. Acta Crystallogr D Biol Crystallogr 66: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cowtan K (2006) The buccaneer software for automated model building. 1. tracing protein chains. Acta Crystallogr D Biol Crystallogr 62: 1002–1011. [DOI] [PubMed] [Google Scholar]
- 49. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, et al. (2011) BUSTER version 2.11.2. Cambridge, UK: Global Phasing Ltd. [Google Scholar]
- 51. Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, et al. (2011) Refmac5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, et al. (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, et al. (2012) Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr 68: 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22: 2577–2637. [DOI] [PubMed] [Google Scholar]
- 55. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. (2004) UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612. [DOI] [PubMed] [Google Scholar]
- 56. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, et al. (2007) Molprobity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35: W375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SAXS analysis of ATV
.
a) Three orthogonal views of each of the five ab initio envelopes calculated imposing (from left to right) no, binary, orthorhombic, trigonal or tetragonal symmetry. b) Fitting of the SASREF model obtained from the open dimer using P2 symmetry into the P222 DAMMIF model. The fitting was performed by the program Chimera [55].
(TIF)