

Abstract
Because PML-RARA-induced acute promyelocytic leukemia (APL) is a morphologically differentiated leukemia, many groups have speculated about whether its leukemic cell of origin is a committed myeloid precursor (e.g. a promyelocyte) versus an hematopoietic stem/progenitor cell (HSPC). We originally targeted PML-RARA expression with CTSG regulatory elements, based on the early observation that this gene was maximally expressed in cells with promyelocyte morphology. Here, we show that both Ctsg, and PML-RARA targeted to the Ctsg locus (in Ctsg-PML-RARA mice), are expressed in the purified KLS cells of these mice (KLS = Kit+Lin−Sca+, which are highly enriched for HSPCs), and this expression results in biological effects in multi-lineage competitive repopulation assays. Further, we demonstrate the transcriptional consequences of PML-RARA expression in Ctsg-PML-RARA mice in early myeloid development in other myeloid progenitor compartments [common myeloid progenitors (CMPs) and granulocyte/monocyte progenitors (GMPs)], which have a distinct gene expression signature compared to wild-type (WT) mice. Although PML-RARA is indeed expressed at high levels in the promyelocytes of Ctsg-PML-RARA mice and alters the transcriptional signature of these cells, it does not induce their self-renewal. In sum, these results demonstrate that in the Ctsg-PML-RARA mouse model of APL, PML-RARA is expressed in and affects the function of multipotent progenitor cells. Finally, since PML/Pml is normally expressed in the HSPCs of both humans and mice, and since some human APL samples contain TCR rearrangements and express T lineage genes, we suggest that the very early hematopoietic expression of PML-RARA in this mouse model may closely mimic the physiologic expression pattern of PML-RARA in human APL patients.
Introduction
The fusion gene PML-RARA is produced by t(15;17)(q22;q21), and is found only in the hematopoietic cells of patients with acute promyelocytic leukemia (APL). When PML-RARA is expressed in mice using regulatory elements from the human or mouse cathepsin G gene (CTSG/Ctsg) or the human S100A8 (MRP8) promoter/enhancer, it can initiate APL; when RARA or PML-RARA are expressed in mouse bone marrow cells via retroviral transduction, both can decrease myeloid maturation and increase self-renewal [1], [2], [3], [4]. Human APL is associated with differentiation arrest at the promyelocyte stage; in mouse models of the disease, this maturation arrest is less pronounced and varies between models, for reasons that are not yet clear. However, the disease is always myeloid-restricted [5]. Because murine models of APL were designed to target PML-RARA expression to myeloid-restricted cells, we and others have suggested that myeloid-restricted disease might result from targeted expression of PML-RARA to the promyelocyte compartment [6], [7], [8], [9], [10]. However, human PML and murine Pml are expressed in early CD34+ hematopoietic progenitor cells, and human PML-RARA expression may not be limited to committed myeloid progenitors and promyelocytes [11], [12].
Several studies have suggested that in APL, the leukemic cell of origin must be a committed myeloid progenitor [10]. First, Turhan et al. did not observe PML-RARA expression in flow-sorted CD34+/CD38− cells (a cell population enriched for less mature hematopoietic progenitors, including stem cells), but did detect PML-RARA expression in CD34+/CD38+ cells (a population enriched for more mature hematopoietic progenitors, including early myeloid committed progenitors) from two APL patients, using semi-quantitative RT-PCR [7]. Secondly, Bonnet and Dick observed engraftment of CD34+/CD38− AML cells into NOD/SCID mice, but no engraftment of similarly sorted CD34+/CD38− cells from APL patients, suggesting that these were not the initiating cells for this subtype of AML [13]. Many authors have suggested that mouse models of APL support this hypothesis, since expression of PML-RARA under the control of Ctsg or MRP-8 regulatory elements has led to myeloid leukemia [8], [9], [10], [14]. However, Chapiro et al. recently reported expression of T-lineage transcripts and TCR rearrangements in 60% of hypogranular t(15;17) APL cases, suggesting the translocation may affect HSPCs with the capacity to differentiate into both myeloid and lymphoid lineages [14]. In addition, APL cells often do not express CD34 on the cell surface, but do often express atypical lymphoid linage markers (CD56, CD19, or CD2), again suggesting that PML-RARA may initiate disease (in human patients) within a multipotent progenitor compartment [15].
In this study, we use state-of-the-art flow-sorting, mRNA amplification, and expression profiling strategies to carefully define the timing of activation of Ctsg and PML-RARA during early hematopoietic development in Ctsg-PML-RARA mice. We found that Ctsg mRNA is expressed not only in the KLS (Kit+/Lin−/Sca+) compartment, but also in SLAM cells (CD150+/CD41−/CD48− KLS), which are even more primitive. We observed striking changes in the gene expression profile of flow-sorted common myeloid progenitors (CMPs) and granulocyte/monocyte progenitors (GMPs) derived from Ctsg-PML-RARA mice, which suggests that PML-RARA has important transcriptional consequences in early myeloid progenitor cells. We extend these findings with functional validation of PML-RARA effects on lymphoid and erythroid lineages, which confirm that PML-RARA is expressed (and functional) at a very early stage in the hematopoietic development of Ctsg-PML-RARA mice. We observed that in flow-sorted Ctsg-PML-RARA promyelocytes, PML-RARA expression does result in significant gene expression changes but does not result in distinct gene expression signature, nor does it promote self-renewal. These results change our understanding of the cellular compartments in which PML-RARA initiates leukemia in the Ctsg-PML-RARA mouse model of APL.
Results
Expression of PML-RARA and the genes used to direct expression in APL mouse models
We analyzed the expression profiles of flow-sorted SLAM cells (cKit+Lin−Sca+CD150+CD41−CD48−), KLS cells (cKit+Lin−Sca+), CMPs (Lin−Sca-1−cKit+CD34+FcγRII/IIIlo), GMPs (Lin−Sca-1−cKit+CD34+FcγRII/IIIhi), megakaryocyte-erythrocyte progenitors (MEPs; Lin−Sca-1−cKit+CD34−FcγRII/IIIlo), “promyelocytes"/early myeloid cells (Ly6gintSCCintB220−CD115−Ter119−), and neutrophils (Ly6g+SCChighB220−CD115−Ter119−) from 2–6 individual Ctsg-PML-RARA or WT mice (Figure S1).
In our mouse model of APL, PML-RARA is inserted into the 5′ untranslated region of Ctsg, and the Ctsg locus therefore regulates its expression [2]. To begin to define when PML-RARA expression is activated in this model during hematopoietic development, we first examined the expression of the Ctsg gene in all the compartments listed above, using young WT and Ctsg-PML-RARA mice (Figure 1). Ctsg expression is not consistently detected in the SLAM compartment by exon array. However, KLS cells express detectable amounts of Ctsg mRNA by exon array, and this expression is absent in KLS cells derived from Ctsg deficient mice (proving the specificity of the probesets for Ctsg transcripts). Ctsg expression increases massively in the CMP compartment; the high level of expression persists in the GMP compartment and in promyelocytes, and declines in neutrophils. Ctsg is minimally expressed in the MEP compartment.
Figure 1. Expression of Ctsg in flow-sorted bone marrow cells and mouse leukemia samples.
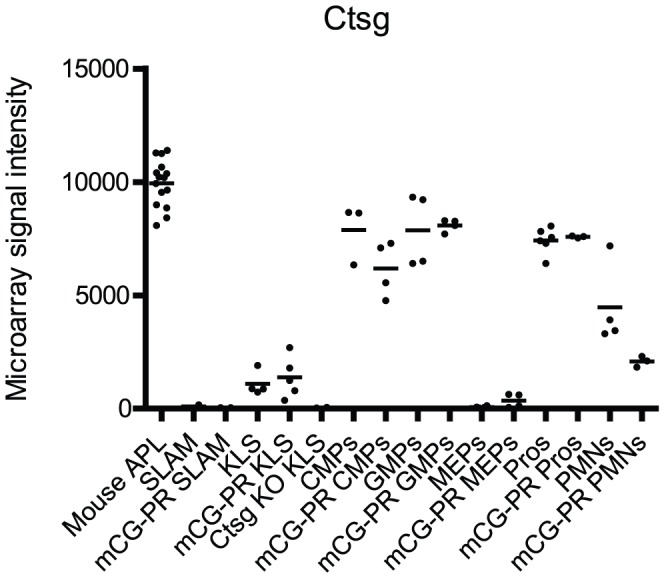
Expression profile in indicated WT and Ctsg-PML-RARA (labeled mCG-PR) flow-sorted bone marrow cells and 15 Ctsg-PML-RARA leukemia samples (labeled Mouse APL) using Nugen amplified mRNA and Affymetrix Mouse Exon 1.0ST arrays. We plotted Ctsg expression for probeset 5542324. Note the absence of Ctsg mRNA in Ctsg deficient cells (labeled Ctsg KO). There were no statistically significant differences in Ctsg expression between the same cell populations, when comparing WT and Ctsg-PML-RARA samples (using two-tailed t test, p-value cutoff 0.05). Comparing the levels of Ctsg expression between the Ctsg-PML-RARA samples using a two-tailed t test, the p-value for SLAM vs. KLS = 0.11, KLS vs. CMPs = 0.00026, CMPs vs. GMPs = 0.022, GMPs vs. MEPs 2.4×10−8, MEPs vs. Pros 2.6×10−7, GMPs vs. Pros = 0.027, Pros vs. PMNs = 2.4×10−6.
Using very sensitive quantitative RT-PCR to further investigate the timing of Ctsg activation, we found that Ctsg mRNA could be detected not only in the KLS compartment but also in SLAM cells (Figure S2A). However, we could not reliably detect PML-RARA expression in SLAM cells, demonstrating that PML-RARA expression is at or below the level of detection by RT-PCR (Figure S2B and S2C). We have previously shown that the expression of the PML-RARA transgene driven from the Ctsg locus is much lower than that of endogenous Ctsg from a WT locus, so this result is consistent with those findings [1], [2]. As expected, PML-RARA mRNA was not detected in bone marrow cells from WT littermate controls [8].
We noted that Ly6g (Gr-1) expression was limited to promyelocytes and neutrophils, and that Kit and Flt3 expression declined during myeloid maturation, as expected (Supplemental Figures S3A, S3B, and S3C). Promyelocytes had relatively low but detectable expression of Cd34 with an average expression intensity of 785.912±72.575 in Ctsg-PML-RARA promyelocytes (n = 3) and 467.255±189.475 in WT promyelocytes (n = 6) with much higher Cd34 expression in the earlier myeloid compartments, CMPs and GMPs, as expected (Figure S3D). Importantly, we detected no expression of Ly6g in SLAM and KLS cells, demonstrating efficient negative selection of differentiated myeloid cells within these populations: all probes had signal intensity <100, whereas signal intensity <200 represents experimental ‘noise’ on this platform (Figure S3A).
We confirmed the purification of the sorted compartments by plotting the expression of myeloid genes that are known to be developmentally-regulated: myeloperoxidase (Mpo), elastase (Ela2), and proteinase3 (Prtn3) are expressed early in myeloid development in primary granules; lactoferrin (Ltf) is expressed later in secondary granules; and matrix metallopeptidase 9 (Mmp9) found in tertiary granules is a marker of mature neutrophils (Figure S4A–E). [9], [16], [17]. In addition, we show that the mature myeloid markers formyl peptide receptor 1 (Fpr1) and lysozyme-2 (Lyz2) are appropriately expressed in our sorted cell populations (Figure S4F and S4G) [18]. The expression data from 11 developmentally-regulated myeloid genes was used to construct a supervised heatmap (by z-score) which clearly illustrates the expected levels of gene expression at the appropriate stage of myeloid development for our sorted cell populations (Figure 2).
Figure 2. A supervised heatmap of 11 developmentally-regulated myeloid genes.

Using the expression data from the Affymetirix Mouse Exon 1.0ST arrays, we created a supervised heatmap with SPOTFIRE using z-score averaging of the probeset with the highest average expression for each of 11 developmentally-regulated myeloid genes across all of our flow-sorted bone marrow cell samples. The legend is shown below the heatmap with downregulated genes in green and upregulated genes in red.
Regulatory elements from other “myeloid-restricted" genes have also been used to generate mouse models of APL. One transgenic strategy (using the S100A8 (MRP8) 5′ flanking region) resulted in myeloid leukemia, while two others (using Fes and Itgam (CD11b) promoters) did not [3], [19], [20]. Using data from the same expression arrays, we found that Fes and Itgam are both expressed at much lower levels in KLS cells than Ctsg, with expression peaking in neutrophils, rather than promyelocytes (Figure 3A and 3B). In contrast, S100A8 is highly expressed in human AML cells and in flow-sorted CD34+ cells, with massive up-regulation at the promyelocyte stage (Figure 3C). High expression of S100a8 was also seen in murine hematopoietic cells, although dynamic regulation during promyelocyte maturation was absent (Figure 3D). The expression pattern of these loci contrasts with that of human PML and murine Pml, which are expressed at much lower levels, and which exhibit little or no dynamic regulation during myeloid maturation (Figure 3E and 3F).
Figure 3. Expression of genes used to target PML-RARA expression in mice.

Expression data for the indicated genes in flow-sorted bone marrow cells using murine Affymetrix Exon 1.0ST arrays or Human Genome U133 Plus 2.0 arrays. Each panel is a highly representative probe on the array. A. cFes (probeset 4758608). B. Itgam (CD11b, probeset 4782002). C. S100A8 (MRP8, probeset 202917). D. S100a8 (Mrp8, probeset 5010279). E. PML (probeset 235508). F. Pml (probeset 4885841). We have previously published the expression profile of PML in AML and human hematopoietic cells [11]. Panel E includes an additional 86 AML cases of AML and data from a second representative probe.
Effects of Ctsg-PML-RARA in multiple hematopoietic compartments
To determine whether PML-RARA expression from the Ctsg locus results in biological effects on multipotent hematopoietic cells, we used four independent approaches. First, we used transcriptional profiling using exon arrays of SLAM, KLS, MEP, CMP, GMP, promyelocyte and mature neutrophil populations from Ctsg-PML-RARA and WT mice. We performed unsupervised hierarchical clustering analyses to assess the global effects of PML-RARA expression on gene expression in KLS and SLAM cells. We observed that KLS (but not SLAM) samples segregated by genotype in an unsupervised clustering analysis, suggesting that expression of PML-RARA within the KLS compartment significantly alters the expression of a specific set of genes (Figure S5A). Although many genes were significantly dysregulated in the comparison between WT and Ctsg-PML-RARA promyelocytes, this cell population did not cluster by genotype in an unsupervised analysis (Figure S5B).
To assess the downstream consequences of PML-RARA expression in early myeloid progenitors, we purified CMPs and GMPs for expression analysis. These populations segregated by Ctsg-PML-RARA genotype in unsupervised hierarchical clustering, whereas MEPs did not cluster by genotype (Figure S6). To ascertain significant differences in specific gene expression between the Ctsg-PML-RARA and WT samples, we performed ANOVA and Significance of Microarrays (SAM) analysis to define significantly dysregulated genes in each subset (Tables S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, S11, S12, 13 and Data S1). In the KLS comparison, one of the most significantly dysregulated genes by ANOVA was Notch1 (Table S2), which is relevant for the recent finding of activated Notch1 signaling in both human and murine APL pathogenesis (Grieselhuber NR et al., submitted). Unexpectedly, one of the most significantly dysregulated genes within the CMP and GMP compartments was Cadherin 1 (Cdh1), a tumor suppressor in epithelial cancers, which was strikingly up-regulated in both the CMP and GMP compartments of Ctsg-PML-RARA mice (Table S7) [21]. We also found that cyclin H (Ccnh) expression is down-regulated in the CMP compartment of Ctsg-PML-RARA mice (Table S3): cyclin H has been previously shown to be part of a complex that phosphorylates the AF-2 domain of RARA, leading to the downstream activation of retinoic acid responsive genes [22].
To highlight the changes in gene expression in the CMP and GMP populations, we performed a supervised clustering analysis of the twenty-two genes that were significantly dysregulated (unadjusted p value<0.001, fold change ≥2) in both the CMP and GMP ANOVA results (the supervised heatmap was created by plotting the relative expression of each of these genes to each other) (Figure 4). Interestingly, this gene expression signature is not present in earlier cell populations (KLS or SLAM), in more mature myeloid cells (promyelocytes and neutrophils), nor the leukemic cells from the Ctsg-PML-RARA mice. Also, there is no concordant overlap between genes dysregulated in the CMP or GMP compartments versus promyelocytes.
Figure 4. A supervised heatmap of the 22 genes significantly dysregulated in both the CMP and GMP compartments.

We created a supervised clustering using SPOTFIRE of the 22 genes that were significantly dysregulated (unadjusted p value<0.001, fold change ≥2) in both the CMP and GMP ANOVA results comparing WT vs. Ctsg-PML-RARA samples. The supervised heatmap was created by plotting the relative expression of each of these genes to each other as a relative percentage, rather than z-score averaging, since the differences in expression levels between some genes was large, and z-score averaging inappropriately highlighted the genes with the highest expression levels. The legend is shown below the heatmap, with minimally expressed genes in green, and highly expressed genes in red.
To further validate the changes in gene expression in the CMP and GMP compartments, we performed supervised clustering analyses of all the significantly dysregulated genes by ANOVA (unadjusted p value<0.001, fold change ≥2) from the GMP and CMP compartments individually (these heatmaps were created by traditional z-score scaling). As expected, the clustering with genes dysregulated in either the CMP or GMP compartment segregated both the GMP and CMP populations, but not the MEP populations, by genotype (Figure S7A and S7B). Thus, the expression of PML-RARA in Ctsg-PML-RARA mice has clear transcriptional consequences that occur early in myeloid development and are specific for the CMP and GMP cell populations. These transcriptional changes, in turn, may influence the gene expression profile of more mature myeloid cells, just as gene expression changes driven by the presence of PML-RARA expression in earlier hematopoietic precursors may shape the distinct expression signature seen in the CMP and GMP compartments.
Secondly, we assessed the function of Ctsg-PML-RARA bone marrow cells after competitive transplantation. We transplanted total bone marrow from healthy 6-week-old (i.e. non-leukemic) Ctsg-PML-RARA mice (CD45.2+) at ratios of 1∶9, 1∶1, and 9∶1 with competitor Ly5.1/Ly5.2 bone marrow (CD45.1+/CD45.2+) into Ly5.1 recipients (CD45.1+). Peripheral blood was assessed at 6 weeks, 3 months, and 6 months post-transplant. Four mice developed leukemia between 3 and 6 months, and could not be analyzed further. As expected, we noted a consistent expansion of Ctsg-PML-RARA + cells within the Ly6g+ (myeloid) compartment at all time points tested (Figure 5A). However, we also observed an expansion of Ctsg-PML-RARA + cells within the CD3+ and B220+ compartments at 3 and 6 months (Figure 5A).
Figure 5. Effects of Ctsg-PML-RARA on multi-lineage hematopoiesis.

A. Effects on myeloid and lymphoid lineage hematopoiesis. Bone marrow cells from indicated mice at 6 weeks of age were mixed at ratios of 1∶1, 9∶1, or 1∶9 with competitor CD45.1+/CD45.2+ bone marrow cells from sex- and age-matched mice. These cells were transplanted into sex-matched, 6-week-old, lethally irradiated CD45.1+ recipients. At the indicated time points, peripheral blood was assessed for ratios of CD45.2+ and CD45.1+/CD45.2+ white blood cells within the B220+, CD3+, or Gr1+ compartments. One sample, two-tailed t-test compared outcomes with the expected values of 10%, 50%, or 90%. Alpha was set at 0.05. Time points with p<0.01 (*) and p<0.05 (†) are indicated. B–C. Effect of Ctsg-PML-RARA on erythroid lineage hematopoiesis. Bone marrow cells from 6-week-old and 8-week-old, healthy mice were plated in methylcellulose containing erythropoietin. B. Total colonies after one week in culture (each data point represents results from an individual mouse in three combined experiments). Results of paired t-test between WT and mCG-PR samples are shown (p-value = 0.002). C. After one week in culture, total colony cells were washed and the number of immature erythrocytes (cKit+CD71dim) was assessed. Results of a paired t-test between WT and mCG-PR samples are shown (p-value = 0.007).
Thirdly, we examined the function of erythroid progenitors derived from Ctsg-PML-RARA mice. Total Ctsg-PML-RARA bone marrow cells contained significantly more BFU-Es than wild-type littermate controls (Figure 5B). There was no difference in the percentage of mature erythroid cells (Ter119+CD71high, 24%±8% vs 37%±18%, p = 0.1; Figure S8A) in these colonies, although Ctsg-PML-RARA BFU-E colonies contained more immature erythroid cells (cKit+CD71dim, 12.7%±2.3% vs 7%±3.5%, p = 0.007; Figure 5C) and more myelomonocytic/monocytic cells (CD11b+, 32%±4.4% vs 21%±9.6%, p = 0.02; Figure S8B). The lack of transcriptional changes in flow sorted MEP cells (Tables S5 and S12) may be accounted for by differences between the immunophenotypically defined MEPs and the physiologically defined colony forming cell (which may be less mature than MEP cells). Evaluation of a large cohort of mice (n = 19 Ctsg-PML-RARA and 15 wild-type littermate controls) revealed normal hemoglobin levels and red cell size in all mice (Figure S8C and S8D), suggesting that PML-RARA-dependent effects on BFU-Es are not associated with a loss of erythroid homeostasis in vivo. These results parallel our previous finding that myeloid CFUs are increased in healthy pre-leukemic Ctsg-PML-RARA mice, but do not lead directly to increased numbers of circulating neutrophils [23], [24].
Finally, we asked whether promyelocyte self-renewal was altered in Ctsg-PML-RARA mice. As expected, we observed that KLS cells from Ctsg-PML-RARA mice contained increased numbers of CFUs compared with purified promyelocytes (Figure 6B and 6C). We also observed a trend toward increased numbers of CFUs in Ctsg-PML-RARA KLS cells compared with wild-type KLS cells. Serial re-plating was detected with Ctsg-PML-RARA KLS cells, as expected, but was not found with Ctsg-PML-RARA promyelocytes (Figure 6C). Transplantation of pools of donor KLS cells (3,000 cells per sub-lethally irradiated Ly5.1 recipient) from either WT or Ctsg-PML-RARA mice led to multi-lineage engraftment, as expected (Figure 6D and Figure S9A–C). In contrast, transplantation of 50,000 purified promyelocytes led to trace engraftment (<1% of bone marrow leukocytes) regardless of genotype, which was not myeloid restricted (Figure 6D–E and Figure S9D–F).
Figure 6. Effects of Ctsg-PML-RARA on flow-sorted promyelocytes.

A. Experimental schema. Bone marrow cells from Ctsg-PML-RARA and littermate WT controls were harvested and KLS and promyelocytes were purified by flow sorting. Aliquots from individual donor mice were plated in methylcellulose containing myeloid cytokines (SCF, IL-3, IL-6, Epo). KLS cells were combined from all donors by genotype and 3,000 cells/recipient were transferred to sub-lethally irradiated Ly5.1 mice (4 recipients per genotype). Purified promyelocytes were separated into two pools, and 50,000 cells/recipient were transferred into sub-lethally irradiated Ly5.1 mice. B. Total CFUs per 10,000 KLS cells plated by week. Every 7 days, total colonies were counted. Total cells were then collected in warm (37°) media and serially replated. C. Total CFUs per 10,000 promyelocytes plated by week. D. Percentage of donor CD45.2+ cells in total bone marrow cells of individual recipient mice 12 weeks after engraftment, with indicated flow sorted cells. All mice that received promyelocytes had <1% CD45.2+ cells. E. Percentage of CD45.2+ cells within the Gr1+, CD3+, and CD19+ bone marrow cells of mice engrafted with promyelocytes.
Discussion
The fusion protein PML-RARA is associated exclusively with myeloid leukemia in both humans and mice (unlike BCR-ABL and MLL fusion proteins, which can lead to myeloid or lymphoid leukemias) [2], [3], [5], [25], [26], [27]. There has therefore been much speculation regarding whether the leukemic cell of origin is a committed progenitor (e.g. a promyelocyte) or multipotent progenitor [5], [6], [7], [8], [10], [14], [28]. The data presented here show that when PML-RARA is inserted into the murine Ctsg locus, it begins to be expressed in KLS cells, where it alters myeloid, lymphoid, and erythroid hematopoiesis (biologically confirming its activity in multipotent cells). It is massively upregulated in CMP and GMP cells, where it significantly alters the expression of a number of genes. In contrast, PML-RARA expression in promyelocytes is not associated with a striking change in gene expression or inappropriate self-renewal. The studies described here define the hematopoietic compartments that are initially exposed to (and perturbed by) PML-RARA in healthy, pre-leukemic Ctsg-PML-RARA mice; the immunophenotype and cytomorphology of leukemia initiating cells within the subsequent leukemias have been defined elsewhere, and are not addressed in this analysis [8], [9].
One of the important arguments used to support a committed progenitor as the leukemic cell of origin has been the successful generation of leukemia using CTSG, Ctsg, and S100A8 loci, which were thought to target PML-RARA to the promyelocyte compartment [10]. However, we and others have now found that in Ctsg-PML-RARA mice, PML-RARA is consistently expressed in cells within the KLS compartment, [8], and is strikingly upregulated in committed myeloid progenitor cells. We have therefore reevaluated the expression patterns of the genes used to direct PML-RARA expression in mice: PML-RARA directed from the CTSG, Ctsg and S100A8 (MRP8) loci cause APL; expression directed by regulatory elements from the cFes and Itgam (CD11b) loci do not [1], [2], [3], [19], [20]. Ctsg, CTSG, S100a8, and S100A8 are all expressed in multipotent progenitor compartments, while cFes and Itgam display low to absent levels of expression in KLS cells, with maximal expression in neutrophils. This suggests that APL development may require PML-RARA expression in an early hematopoietic compartment, and that induction of expression at the promyelocyte stage is insufficient to initiate leukemia, perhaps because PML-RARA cannot reverse the commitment of terminally differentiated neutrophils to die.
Other authors have evaluated the cell-specific effects of PML-RARA and MLL fusion transcripts using viral transduction [4], [29], [30], [31]. We have focused our analysis on transgenic mouse models because interpretation of viral-transduction models is complicated both by the heterogeneity of the bone marrow cells transduced, and by the toxicity of PML-RARA, which is variable among cell types [28], [32], [33].
Importantly, this work addresses a different question than the work of Guibal et al., which defined the immunophenotype of a committed myeloid progenitor (cKit+CD34+FcγRII/III+Ly6gint) as the leukemia-initiating cell in the hMRP8-PML-RARA transgenic mouse model of APL. Here we show the transcriptional and functional consequences of PML-RARA expression in Ctsg-PML-RARA pre-leukemic cells. These findings are not mutually exclusive, since the hematopoietic compartment susceptible to PML-RARA transformation may not be at the same developmental stage as the resultant leukemia [9].
Our work clarifies and expands the findings of Wojiski et al. [8] Using semi-quantitative RT-PCR, they observed PML-RARA expression in the KLS cells of Ctsg-PML-RARA mice. Their functional studies revealed increased myeloid self-renewal in flow-sorted KLS, CMP, and GMP cells from Ctsg-PML-RARA mice. They used a PCR-based protocol to demonstrate long-term engraftment of flow-sorted Ctsg-PML-RARA promyelocytes (Lin−Sca−Ly6g+cKit+CD34+ cells) in sub-lethally irradiated recipient B6 mice. However, this strategy does not determine whether the engrafted cells are myeloid-restricted (i.e. derived from promyelocytes) or whether they are multi-lineage (i.e. arising from stem/progenitor cells that contaminated the purified “promyelocytes"). In contrast, we used Ly5.1 mice as the recipients for our purified promyelocyte populations, and found that even large numbers of promyelocytes (50,000 purified cells per recipient) were insufficient to lead to long-term engraftment of myeloid restricted cells. Our data therefore do not support the idea that the promyelocytes from Ctsg-PML-RARA mice have altered self-renewal properties that contribute to the expansion of these cells in vivo.
There are no standard immunophenotypic markers for defining murine promyelocytes, and our flow-sorting strategy for promyelocytes was different than that used by Wojiski et al.; however, we extensively validated our approach using both expression data from genes known to be developmentally-regulated during myeloid development as well as traditional cytomorphology, in which a trained hematopathologist performed blinded differentials (Figure S1) [8], [34]. Using these techniques, we demonstrate that we have greatly enriched murine promyelocytes using our flow-sorting strategy; however, it remains possible that the different strategy used by Wojiski et al. enriches for an earlier myeloid population that could account for the different results regarding “promyelocyte" self-renewal between our studies.
Nonetheless, our data demonstrate that in healthy, pre-leukemic Ctsg-PML-RARA mice, PML-RARA is first expressed in very early hematopoietic stem/[progenitor cells (KLS cells), and that its expression continues throughout myeloid development (CMPs, GMPs, promyelocytes, and neutrophils). Biological effects of PML-RARA can be seen in pre-leukemic KLS cells, CMPs and GMPs. Thus, in this model system, it is highly likely that PML-RARA initiates leukemia in very early hematopoietic cells, and not in promyelocytes, as originally predicted. Although we cannot formally exclude the possibility that pre-leukemic promyelocytes acquire self-renewal properties, it seems very unlikely, based on recent data that some human APL cells have TCR rearrangements, and express T lineage genes [14]. We suggest that the Ctsg-PML-RARA mouse model may recapitulate the physiologic timing of PML-RARA activation in very primitive human HPSCs that are capable of giving rise to both lymphoid and myeloid cells.
PML-RARA does not appear to alter normal hematopoietic homeostatic feedback or cause increasing cell numbers within an immunophenotypically-defined multipotent progenitor compartment (although healthy Ctsg-PML-RARA mice do have a subtle increase in bone marrow promyelocytes) [2], [8], [11], [24]. Neither we, nor others, have observed increased numbers of KLS, GMP, CMP or MEP cells in mouse models of APL [8], [20], [24], [35]. However, we have identified a competitive advantage in multiple hematopoietic lineages following transplantation [11], and increased numbers of myeloid [26], [36] and erythroid BFUs in the marrow of Ctsg-PML-RARA mice. Since Ctsg-PML-RARA mice virtually never develop lymphocytic leukemia or erythroleukemia, our data suggests that the myeloid restriction of leukemia must be explained by myeloid-restricted genes or proteins that cooperate with PML-RARA in leukemic cells (Figure 7). Although neutrophil elastase is known to be one of the critical interacting proteins, further investigation will be needed to fully define all of the cooperating elements [23], [28].
Figure 7. Model of APL development in Ctsg-PML-RARA mice.

Murine APL requires the expression of PML-RARA in a multipotent progenitor, and cooperation of PML-RARA with myeloid restricted elements; expression of PML-RARA within promyelocytes is insufficient to cause leukemia. Orange ellipses indicate compartments with expression of the indicated genes used to direct expression in transgenic models. Red arrows indicate inappropriate self-renewal resulting from Ctsg-PML-RARA expression. Broken arrows indicate that multiple differentiation steps exist between indicated compartments. SLAM: lineage negative/Kit+/Sca-1+/CD150+/CD41−/CD48−. KLS: c-Kit+/lineage negative/Sca-1+. Pro: promyelocyte. PMN: polymorphic neutrophil. Concept after Lane and Ley [28].
Methods
Mice
Mice expressing PML-RARA from the murine Ctsg locus (Ctsg-PML-RARA) and Ctsg deficient mice (CG-KO) have been previously reported [2], [37]. The Ctsg-PML-RARA transgenic mice used in this study were backcrossed more than 12 generations into the C57Bl/6 Taconic background [38]. The Washington University Animal Studies Committee approved all animal experiments.
Flow Sorting, Expression Array Profiling, and RT-PCR Validation
Bone marrow cells from individual mice were harvested from both femurs and tibia of 6-week-old and 13-week-old mice and prepared previously described [11], [24]. Standard red blood cell lysis was performed with ACK lysis buffer (0.15 M NH4Cl, 10 mM KHC03, 0.1 mM Na2EDTA) on ice for 10 minutes. Non-specific staining was blocked with Miltenyi FcR Blocking Reagent for mouse (Auburn, CA). Isotype-matched antibodies were used as negative controls and the Fluorescence Minus One (FMO) strategy was used to set appropriate gates. Flow sorting was performed on a Reflection high-speed cell sorter (i-Cyt, Champaign, IL). Cells were sorted directly into Trizol (Invitrogen, Carlsbad, CA). Cells were stained by standard protocols with the following antibodies (eBioscience unless otherwise noted): KLS (c-Kit+/lineage negative/Sca-1+) cells were assessed using the following lineage markers: FITC-conjugated αGr-1, αTer119, αCD3, αCD4, αCD8, αB220, αCD19, and αCD127; APC-αc-Kit, and PE-αSca-1. CMPs (Lin−Sca-1−cKit+CD34+FcγRII/IIIlo), GMPs (Lin−Sca-1−cKit+CD34+FcγRII/IIIhi), megakaryocyte-erythrocyte progenitors (MEPs; Lin−Sca-1−cKit+CD34−FcγRII/IIIlo) were done with the same antibody cocktail as KLS with addition of APC-conjugated αFcγRII/III (clone 93). SLAM (CD150+/CD41−/CD48− KLS) cells were assessed using KLS staining as above, except for PerCP-Cy5.5–conjugated αSca-1, with the addition of FITC-αCD41, FITC-αCD48 and PE-αCD150 antibodies. Promyelocyte (Gr1intSCCintB220−CD115−Ter119−) and neutrophil (Gr1+SCChighB220−CD115−Ter119−) sorting was done using APC-αGr-1, PE-αCD115, FITC-αB220 and PE-Cy7-αTer119. Additional cells from both the promyelocyte and neutrophils fraction were sorted into FACS buffer for morphological validation of cytospins. Samples analyzed in expression array profiling were generated during five separate flow-sorting experiments to limit technical bias. 15 mouse Ctsg-PML-RARA leukemia samples were prepared as previously described [38].
For expression array profiling, total cellular RNA was purified using TRIzol reagent (Invitrogen), quantified using UV spectroscopy (Nanodrop Technologies), and qualitatively assessed using an Experion Bioanalyzer. Amplified cDNA was prepared from 20 ng total RNA using the whole transcript WT-Ovation RNA Amplification System and biotin-labeled using the Encore Biotin Module, both from NuGen Technologies, according to the manufacturer's instructions. Labeled targets were then hybridized to Mouse Exon 1.0 ST arrays (Affymetrix), washed, stained, and scanned using standard protocols from the Siteman Cancer Center, Molecular and Genomic Analysis Core Facility (http://pathology.wustl.edu/research/cores/lcg/index.php). Affymetrix Expression Console software was used to process array images, export signal data, and evaluate image and data quality relative to standard Affymetrix quality control metrics. Exon array data for all samples used in this study have been deposited on GEO (http://www.ncbi.nlm.nih.gov/geo/; SuperSeries accession numbers GSE26131 and GSE40022).
For Ctsg qRT-PCR, we performed standard, two-step RT-PCR, including the Genomic DNA Wipeout step, according to the manufacturer's instructions using Ctsg QuantiTect Primers in conjunction with the QuantiTect SYBER Green RT-PCR Kit (both from Qiagen, Germantown, Maryland). For PML-RARA qRT-PCR, we also used the Qiagen RT-PCR kit with the following primers: Forward TCTTCCTGCCCAACAGCAA, Reverse GCTTGTAGATGCGGGGTAGAG. We used GAPDH as the PCR control with the following primers: Forward TGCACCACCAACTGCTTAG, Reverse GGATGCAGGGATGATGTTC.
Bone marrow Transplantation
Cells used for competitive repopulation studies were injected retroorbitally, as previously described [11], [24]. Two separate experiments were performed, and the data was combined for analysis.
Determination of engraftment of flow sorted bone marrow cells is described in Figure S9. Recipient Ly5.1 mice received either lethal (1,100 cGy) or sub-lethal (350 cGy) irradiation 24 hours prior to transplantation with either 1×106 total bone marrow cells, 3,000 KLS cells or 50,000 promyelocytes/mouse. At least 20,000 events (peripheral blood) and 40,000 events (bone marrow) per recipient mouse were collected for analysis.
Hematopoietic Progenitor Assays
Bone marrow cells from 6-week-old and 8-week-old, healthy Ctsg-PML-RARA mice and littermate controls were collected and plated (in duplicate) in 1.1 ml of methylcellulose medium at 41.5×103 cells/ml (MethoCult M3334, StemCell Technologies, Vancouver, Canada) or 8.3×103/ml (MethoCult 3434) [37]. Colonies with >30 cells were counted on day 7. Colonies were collected in 37° DMEM and assessed by flow cytometry using the following antibodies and FACS Scan (Beckman-Dickenson, Franklin Lakes, NJ): FITC-αTer119 (BD Pharmingen, Franklin Lakes, NJ, Ly-76), PE-αCD71 (eBioscience, R17217), PE-αCD11b (eBioscience, M1/70), and APC-αcKit (eBioscience, 2B8), or serially replated.
Statistical analysis
Expression array analysis: exon-level summary was generated using the RMA algorithm in the Affymetrix Expression Console (Affymetrix Inc., USA). Only core probesets were used in order to limit the analysis within well-annotated exons with the exception of Ly6g, whose 2 probesets are within the extended probesets group. Probesets having an expression signal less than 200 in all samples were removed and an unsupervised hierarchical clustering analysis was performed using z-score normalized expression values in SPOTFIRE (Decision Site version 9.1.1, Somerville, USA) or with the Partek Genomics Suite (Saint Louis, MO) with the expression of each gene standardized to a mean of 0 and standard deviation of 1 and using the Pearson dissimilarity as a distance measure. Results did not segregate based on age of the mice used in this analysis (6-week-old vs 13-week-old), and so the data were combined. Supervised clustering analyses were done using SPOTFIRE as described in the text. Comparison of cell populations using ANOVA was done with Partek with fold change ≥2 and unadjusted p-values as noted in the text. Comparisons of cell populations using SAM Version 4.0 (http://www-stat.stanford.edu/~tibs/SAM/, Palo Alto, CA) as an Excel add-in performing two-class unpaired analyses with unlogged data, 100 permutations, standard regression and fold change ≥2 with false discovery rates and q-values noted in the text. Competitive repopulation analysis: a one sample, two-tailed t-test with alpha set at 0.05 compared outcomes with expected values of 10%, 50%, and 90% (Prism, Graphpad 5, La Jolla, CA). CFU-E analysis was done with a paired t-test (Excel, Microsoft, Seattle, WA).
Supporting Information
Murine promyelocyte and neutrophil flow sorting strategy. A. Total bone marrow cells were labeled with B220, Ter119, CD115 and Gr1 and early myeloid cells/promyelocytes (Pros) and neutrophils (PMNs) were identified as B220−, Ter119−, CD115− cells that are either BSCintGr1int (Pros), or BSChighGr1high (PMNs). B. Results of 200 cell differential counts by a blinded hematopathologist from 4 Pros (2 WT and 2 Ctsg-PML-RARA) and 5 PMNs (3 WT and 2 Ctsg-PML-RARA) separate sorted samples. C. Representative cytomorphology of WT and Ctsg-PML-RARA (labeled PR) Pros samples counted in B (1,000×). D. Representative cytomorphology of WT and Ctsg-PML-RARA (labeled PR) PMNs samples counted in B (1,000×).
(EPS)
Validation of Ctsg and PML-RARA expression using quantitative reverse-transcriptase PCR. A. Ctsg expression was validated with quantitative RT-PCR normalized to Gapdh. B. Quantitative RT-PCR using PML-RARA specific primers normalized to Gapdh in the indicated mice and Nugen amplified mRNA. C. Agarose gel of PCR products from Panel B. PML-RARA expected size is 145 bp. Markers are 100 and 200 base-pairs.
(EPS)
Expression of Ly6g , Kit , Flt3 and Cd34 in flow-sorted bone marrow cells and mouse leukemia samples. Expression profile in indicated WT and Ctsg-PML-RARA (labeled mCG-PR) flow-sorted bone marrow cells and 15 Ctsg-PML-RARA leukemia samples (labeled Mouse APL) using Nugen amplified mRNA and Affymetrix Mouse Exon 1.0ST arrays. We plotted Ly6g, Kit, Flt3 and Cd34 expression using representative probesets.
(EPS)
Expression of 7 developmentally-regulated myeloid genes in flow-sorted bone marrow cells. Expression profile in indicated WT and Ctsg-PML-RARA (labeled mCG-PR) flow-sorted bone marrow cells and 15 Ctsg-PML-RARA leukemia samples (labeled Mouse APL) using Nugen amplified mRNA and Affymetrix Mouse Exon 1.0ST arrays. We plotted Elane, Prtn3, Mpo, Ltf, Mmp9, Lyz2, and Fpr1 expression using representative probesets.
(EPS)
Expression profile of flow-sorted Ctsg-PML-RARA and WT bone marrow cells by unsupervised clustering analyses. Bone marrow cells were flow sorted as described in Figure 1, and analyzed using Affymetrix Exon 1.0ST arrays. A: Unsupervised hierarchal clustering using SPOTFIRE of expression array data from flow-sorted SLAM and KLS cells from littermate 6-week-old and 13-week-old healthy WT (−/−) vs. mCG-PR mice (+/−). Clustering occurs by genotype within the KLS samples, but not the SLAM samples. Samples were generated during two independent flow-sorting experiments to minimize technical bias. B. Unsupervised hierarchal clustering using SPOTFIRE of expression array data of flow-sorted promyelocytes cells from littermate 8-week-old healthy WT (−/−) vs. mCG-PR mice (+/−). Clusters do not segregate by genotype. Samples were generated with three independent flow-sorting experiments to minimize technical bias. The legend is shown below the heatmaps with downregulated genes in green and upregulated genes in red.
(EPS)
Unsupervised heatmap clustering analyses for WT vs. Ctsg-PML-RARA CMP, GMP and MEP samples. Bone marrow cells were flow sorted as described in the Methods section, and analyzed using Affymetrix Exon 1.0ST arrays. An unsupervised clustering analysis was performed using Partek with Pearson dissimilarity as the distance measure. Probesets with signal intensity of less than 200 in all samples were removed from the analyses. The legend is shown below each heatmap with downregulated genes in green and upregulated genes in red. A. Unsupervised heatmap clustering analyses for CMP WT vs. Ctsg-PML-RARA samples. B. Unsupervised heatmap clustering analyses for GMP WT vs. Ctsg-PML-RARA samples. C. Unsupervised heatmap clustering analyses for MEP WT vs. Ctsg-PML-RARA samples.
(EPS)
Individual supervised clustering analyses for the significantly dysregulated genes by ANOVA from the GMP and CMP comparisons between WT and Ctsg-PML-RARA samples. These heatmaps were created by traditional z-score scaling using SPOTFIRE. The clustering with genes dysregulated in either the CMP or GMP compartment by ANOVA (with unadjusted p value<0.001, fold change ≥2) segregated both the GMP and CMP populations, but not the MEP populations, by genotype. The legend is shown below the heatmaps with downregulated genes in green and upregulated genes in red. A. Supervised clustering heatmap using genes dysregulated by ANOVA comparison between WT and Ctsg-PML-RARA CMP populations. B. Supervised clustering heatmap using genes dysregulated by ANOVA comparison between WT and Ctsg-PML-RARA GMP populations.
(EPS)
Erythroid colony formation (CFU-E) in Ctsg-PML-RARA vs. WT mice. Bone marrow cells from 6-week-old and 8-week-old, healthy mice were plated in methylcellulose containing erythropoietin (Methocult 3334). Results of paired t-tests are shown in each panel. A. Immunophenotypes of cells in these colonies were assessed as indicated: A. Ter119+CD71high (mature erythrocytes) and B. CD11b+ (myelomonocytic/monocytic cells). C. and D. Hemoglobin and mean corpuscular volume (MCV) values were determined in a cohort of healthy 4-month-old mice with the indicated genotypes.
(EPS)
Examples of bone marrow immunophenotypes following transplantation of flow sorted KLS and promyelocytes. Bone marrow KLS and promyelocytes were sorted from littermate Ctsg-PML-RARA (mCG-PR) and WT mice and transplanted into sub-lethally irradiated Ly5.1 recipients (Figure 6). Twelve weeks after transplantation, bone marrow cells were harvested and assessed for Gr1, CD3, CD19, CD45.2 and CD45.1 expression. At least 40,000 events were collected per recipient. A–C. Representative recipient of KLS transplantation. D–F. Representative recipient of promyelocyte transplantation.
(EPS)
Comparative gene expression analysis between cell populations derived from Ctsg-PML-RARA versus WT mice. The supplementary data includes a summary of gene expression differences (by ANOVA and SAM analysis) for comparisons between Ctsg-PML-RARA and WT SLAM, KLS, CMP, GMP, MEP and promyelocyte cell populations. The analysis includes a comparison of dysregulated genes to previously identified PML-RARA binding sites and to known dysregulated mRNA abundance identified in primary human APL samples [39], [40].
(DOCX)
Ctsg-PML-RARA versus WT SLAM ANOVA Results.
(XLSX)
Ctsg-PML-RARA versus WT KLS ANOVA Results.
(XLSX)
Ctsg-PML-RARA versus WT CMP ANOVA Results.
(XLSX)
Ctsg-PML-RARA versus WT GMP ANOVA Results.
(XLSX)
Ctsg-PML-RARA versus WT MEP ANOVA Results.
(XLSX)
Ctsg-PML-RARA versus WT Promyelocyte ANOVA Results.
(XLSX)
Concordant Dysregulated Genes in both the CMP and GMP Compartments by ANOVA.
(XLSX)
Ctsg-PML-RARA versus WT SLAM SAM Results.
(XLSX)
Ctsg-PML-RARA versus WT KLS SAM Results.
(XLSX)
Ctsg-PML-RARA versus WT CMP SAM Results.
(XLSX)
Ctsg-PML-RARA versus WT GMP SAM Results.
(XLSX)
Ctsg-PML-RARA versus WT MEP SAM Results.
(XLSX)
Ctsg-PML-RARA versus WT Promyelocyte SAM Results.
(XLSX)
Acknowledgments
We thank Christine Pham for the generous gift of Ctsg-KO mice and Mieke Hoock for excellent mouse colony management. We thank Cheng “Cynthia" Li and Jackie Hughes for their assistance with flow-sorting protocols and cell staining. We thank Mark Murakami and Dan Link for assistance with promyelocyte and neutrophil separation protocols. We thank the Alvin J. Siteman Cancer Center for the use of the High Speed Cell Sorter Core, which performed hematopoietic cell sorting, and for use of the Biomedical Informatics Core, which provided informatics support.
Funding Statement
This work was supported by a Leukemia and Lymphoma Society Fellows award 5340-10 (J.S.W.), by United States National Institutes of Health (NIH) K99 HL103975 (J.S.W), by NIH Training Grant T32 HL0078088 (L.D.W.), by NIH Grants CA83962 and CA101937 and the Barnes-Jewish Hospital Foundation (T.J.L.), and UL1 RR024992 from the National Center for Research Resources (NCRR), a component of the NIH, and NIH Roadmap for Medical Research. The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant #P30 CA91842. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Grisolano JL, Wesselschmidt RL, Pelicci PG, Ley TJ (1997) Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood 89: 376–387. [PubMed] [Google Scholar]
- 2. Westervelt P, Lane AA, Pollock JL, Oldfather K, Holt MS, et al. (2003) High-penetrance mouse model of acute promyelocytic leukemia with very low levels of PML-RARalpha expression. Blood 102: 1857–1865. [DOI] [PubMed] [Google Scholar]
- 3. Brown D, Kogan S, Lagasse E, Weissman I, Alcalay M, et al. (1997) A PMLRARalpha transgene initiates murine acute promyelocytic leukemia. Proc Natl Acad Sci U S A 94: 2551–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Du C, Redner RL, Cooke MP, Lavau C (1999) Overexpression of wild-type retinoic acid receptor alpha (RARalpha) recapitulates retinoic acid-sensitive transformation of primary myeloid progenitors by acute promyelocytic leukemia RARalpha-fusion genes. Blood 94: 793–802. [PubMed] [Google Scholar]
- 5. Kogan SC (2007) Mouse models of acute promyelocytic leukemia. Curr Top Microbiol Immunol 313: 3–29. [DOI] [PubMed] [Google Scholar]
- 6. Westervelt P, Ley TJ (1999) Seed versus soil: the importance of the target cell for transgenic models of human leukemias. Blood 93: 2143–2148. [PubMed] [Google Scholar]
- 7. Turhan A, Lemoine F, Debert C, Bonnet M, Baillou C, et al. (1995) Highly purified primitive hematopoietic stem cells are PML-RARA negative and generate nonclonal progenitors in acute promyelocytic leukemia. Blood 85: 2154–2161. [PubMed] [Google Scholar]
- 8. Wojiski S, Guibal FC, Kindler T, Lee BH, Jesneck JL, et al. (2009) PML-RARalpha initiates leukemia by conferring properties of self-renewal to committed promyelocytic progenitors. Leukemia 23: 1462–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guibal FC, Alberich-Jorda M, Hirai H, Ebralidze A, Levantini E, et al. (2009) Identification of a myeloid committed progenitor as the cancer-initiating cell in acute promyelocytic leukemia. Blood 114: 5415–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grimwade D, Enver T (2004) Acute promyelocytic leukemia: where does it stem from? Leukemia 18: 375–384. [DOI] [PubMed] [Google Scholar]
- 11. Welch JS, Yuan W, Ley TJ (2011) PML-RARA can increase hematopoietic self-renewal without causing a myeloproliferative disease in mice. J Clin Invest 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, et al. (2008) PML targeting eradicates quiescent leukaemia-initiating cells. Nature 453: 1072–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bonnet D, Dick JE (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3: 730–737. [DOI] [PubMed] [Google Scholar]
- 14. Chapiro E, Delabesse E, Asnafi V, Millien C, Davi F, et al. (2006) Expression of T-lineage-affiliated transcripts and TCR rearrangements in acute promyelocytic leukemia: implications for the cellular target of t(15;17). Blood 108: 3484–3493. [DOI] [PubMed] [Google Scholar]
- 15. Guglielmi C, Martelli MP, Diverio D, Fenu S, Vegna ML, et al. (1998) Immunophenotype of adult and childhood acute promyelocytic leukaemia: correlation with morphology, type of PML gene breakpoint and clinical outcome. A cooperative Italian study on 196 cases. Br J Haematol 102: 1035–1041. [DOI] [PubMed] [Google Scholar]
- 16. Theilgaard-Monch K, Jacobsen LC, Borup R, Rasmussen T, Bjerregaard MD, et al. (2005) The transcriptional program of terminal granulocytic differentiation. Blood 105: 1785–1796. [DOI] [PubMed] [Google Scholar]
- 17. Borregaard N, Cowland JB (1997) Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 89: 3503–3521. [PubMed] [Google Scholar]
- 18. Yuan W, Payton JE, Holt MS, Link DC, Watson MA, et al. (2007) Commonly dysregulated genes in murine APL cells. Blood 109: 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pandolfi PP (1997) Transgenic models of acute myeloid leukemias. Hematology 1997: Education Program of the American Society for Hematology. San Diego, CA.
- 20. Early E, Moore MA, Kakizuka A, Nason-Burchenal K, Martin P, et al. (1996) Transgenic expression of PML/RARalpha impairs myelopoiesis. Proc Natl Acad Sci U S A 93: 7900–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paredes J, Figueiredo J, Albergaria A, Oliveira P, Carvalho J, et al. (2012) Epithelial E- and P-cadherins: Role and clinical significance in cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1826: 297–311. [DOI] [PubMed] [Google Scholar]
- 22. Bour G, Gaillard E, Bruck N, Lalevee S, Plassat J-L, et al. (2005) Cyclin H binding to the RARA activation function (AF)-2 domain directs phosphorylation of the AF-1 domain by cyclin-dependent kinase 7. Proc Natl Acad Sci U S A 102: 16608–16613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Uy GL, Lane AA, Welch JS, Grieselhuber NR, Payton JE, et al. (2010) A protease-resistant PML-RAR{alpha} has increased leukemogenic potential in a murine model of acute promyelocytic leukemia. Blood 116: 3604–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Welch JS, Klco JM, Varghese N, Nagarajan R, Ley TJ (2011) Rara haploinsufficiency modestly influences the phenotype of acute promyelocytic leukemia in mice. Blood 117: 2460–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou G-b, Li G, Chen S-j, Chen Z (2007) From dissection of disease pathogenesis to elucidation of mechanisms of targeted therapies: leukemia research in the genomic era. Acta Pharmacol Sin 9 1434–1449. [DOI] [PubMed] [Google Scholar]
- 26. Ilaria RL Jr (2004) Animal models of chronic myelogenous leukemia. Hematol Oncol Clin North Am 18: 525–543, vii. [DOI] [PubMed] [Google Scholar]
- 27. McCormack E, Bruserud O, Gjertsen BT (2008) Review: genetic models of acute myeloid leukaemia. Oncogene 27: 3765–3779. [DOI] [PubMed] [Google Scholar]
- 28. Lane AA, Ley TJ (2005) Neutrophil elastase is important for PML-retinoic acid receptor alpha activities in early myeloid cells. Mol Cell Biol 25: 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. So CW, Karsunky H, Passegue E, Cozzio A, Weissman IL, et al. (2003) MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell 3: 161–171. [DOI] [PubMed] [Google Scholar]
- 30. Lavau C, Luo RT, Du C, Thirman MJ (2000) Retrovirus-mediated gene transfer of MLL-ELL transforms primary myeloid progenitors and causes acute myeloid leukemias in mice. Proc Natl Acad Sci U S A 97: 10984–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Minucci S, Monestiroli S, Giavara S, Ronzoni S, Marchesi F, et al. (2002) PML-RAR induces promyelocytic leukemias with high efficiency following retroviral gene transfer into purified murine hematopoietic progenitors. Blood 100: 2989–2995. [DOI] [PubMed] [Google Scholar]
- 32. He LZ, Tribioli C, Rivi R, Peruzzi D, Pelicci PG, et al. (1997) Acute leukemia with promyelocytic features in PML/RARalpha transgenic mice. Proc Natl Acad Sci U S A 94: 5302–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sternsdorf T, Phan VT, Maunakea ML, Ocampo CB, Sohal J, et al. (2006) Forced retinoic acid receptor alpha homodimers prime mice for APL-like leukemia. Cancer Cell 9: 81–94. [DOI] [PubMed] [Google Scholar]
- 34.McGarry MP, Protheroe CA, Lee JJ (2009) Mouse Hematology: A Laboratory Manual. Long Island, NY: Cold Spring Harbor Laboratory Press. 99 p.
- 35. Walter MJ, Park JS, Ries RE, Lau SK, McLellan M, et al. (2005) Reduced PU.1 expression causes myeloid progenitor expansion and increased leukemia penetrance in mice expressing PML-RARalpha. Proc Natl Acad Sci U S A 102: 12513–12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rego EM, Wang ZG, Peruzzi D, He LZ, Cordon-Cardo C, et al. (2001) Role of promyelocytic leukemia (PML) protein in tumor suppression. J Exp Med 193: 521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. MacIvor DM, Shapiro SD, Pham CT, Belaaouaj A, Abraham SN, et al. (1999) Normal neutrophil function in cathepsin G-deficient mice. Blood 94: 4282–4293. [PubMed] [Google Scholar]
- 38. Wartman LD, Larson DE, Xiang Z, Ding L, Chen K, et al. (2011) Conserved progression mutations revealed by sequencing a mouse acute promyelocytic leukemia genome. J Clin Invest 121: 1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martens JH, Brinkman AB, Simmer F, Francoijs KJ, Nebbioso A, et al. (2010) PML-RARalpha/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell 17: 173–185. [DOI] [PubMed] [Google Scholar]
- 40. Payton JE, Grieselhuber NR, Chang LW, Murakami M, Geiss GK, et al. (2009) High throughput digital quantification of mRNA abundance in primary human acute myeloid leukemia samples. J Clin Invest 119: 1714–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Murine promyelocyte and neutrophil flow sorting strategy. A. Total bone marrow cells were labeled with B220, Ter119, CD115 and Gr1 and early myeloid cells/promyelocytes (Pros) and neutrophils (PMNs) were identified as B220−, Ter119−, CD115− cells that are either BSCintGr1int (Pros), or BSChighGr1high (PMNs). B. Results of 200 cell differential counts by a blinded hematopathologist from 4 Pros (2 WT and 2 Ctsg-PML-RARA) and 5 PMNs (3 WT and 2 Ctsg-PML-RARA) separate sorted samples. C. Representative cytomorphology of WT and Ctsg-PML-RARA (labeled PR) Pros samples counted in B (1,000×). D. Representative cytomorphology of WT and Ctsg-PML-RARA (labeled PR) PMNs samples counted in B (1,000×).
(EPS)
Validation of Ctsg and PML-RARA expression using quantitative reverse-transcriptase PCR. A. Ctsg expression was validated with quantitative RT-PCR normalized to Gapdh. B. Quantitative RT-PCR using PML-RARA specific primers normalized to Gapdh in the indicated mice and Nugen amplified mRNA. C. Agarose gel of PCR products from Panel B. PML-RARA expected size is 145 bp. Markers are 100 and 200 base-pairs.
(EPS)
Expression of Ly6g , Kit , Flt3 and Cd34 in flow-sorted bone marrow cells and mouse leukemia samples. Expression profile in indicated WT and Ctsg-PML-RARA (labeled mCG-PR) flow-sorted bone marrow cells and 15 Ctsg-PML-RARA leukemia samples (labeled Mouse APL) using Nugen amplified mRNA and Affymetrix Mouse Exon 1.0ST arrays. We plotted Ly6g, Kit, Flt3 and Cd34 expression using representative probesets.
(EPS)
Expression of 7 developmentally-regulated myeloid genes in flow-sorted bone marrow cells. Expression profile in indicated WT and Ctsg-PML-RARA (labeled mCG-PR) flow-sorted bone marrow cells and 15 Ctsg-PML-RARA leukemia samples (labeled Mouse APL) using Nugen amplified mRNA and Affymetrix Mouse Exon 1.0ST arrays. We plotted Elane, Prtn3, Mpo, Ltf, Mmp9, Lyz2, and Fpr1 expression using representative probesets.
(EPS)
Expression profile of flow-sorted Ctsg-PML-RARA and WT bone marrow cells by unsupervised clustering analyses. Bone marrow cells were flow sorted as described in Figure 1, and analyzed using Affymetrix Exon 1.0ST arrays. A: Unsupervised hierarchal clustering using SPOTFIRE of expression array data from flow-sorted SLAM and KLS cells from littermate 6-week-old and 13-week-old healthy WT (−/−) vs. mCG-PR mice (+/−). Clustering occurs by genotype within the KLS samples, but not the SLAM samples. Samples were generated during two independent flow-sorting experiments to minimize technical bias. B. Unsupervised hierarchal clustering using SPOTFIRE of expression array data of flow-sorted promyelocytes cells from littermate 8-week-old healthy WT (−/−) vs. mCG-PR mice (+/−). Clusters do not segregate by genotype. Samples were generated with three independent flow-sorting experiments to minimize technical bias. The legend is shown below the heatmaps with downregulated genes in green and upregulated genes in red.
(EPS)
Unsupervised heatmap clustering analyses for WT vs. Ctsg-PML-RARA CMP, GMP and MEP samples. Bone marrow cells were flow sorted as described in the Methods section, and analyzed using Affymetrix Exon 1.0ST arrays. An unsupervised clustering analysis was performed using Partek with Pearson dissimilarity as the distance measure. Probesets with signal intensity of less than 200 in all samples were removed from the analyses. The legend is shown below each heatmap with downregulated genes in green and upregulated genes in red. A. Unsupervised heatmap clustering analyses for CMP WT vs. Ctsg-PML-RARA samples. B. Unsupervised heatmap clustering analyses for GMP WT vs. Ctsg-PML-RARA samples. C. Unsupervised heatmap clustering analyses for MEP WT vs. Ctsg-PML-RARA samples.
(EPS)
Individual supervised clustering analyses for the significantly dysregulated genes by ANOVA from the GMP and CMP comparisons between WT and Ctsg-PML-RARA samples. These heatmaps were created by traditional z-score scaling using SPOTFIRE. The clustering with genes dysregulated in either the CMP or GMP compartment by ANOVA (with unadjusted p value<0.001, fold change ≥2) segregated both the GMP and CMP populations, but not the MEP populations, by genotype. The legend is shown below the heatmaps with downregulated genes in green and upregulated genes in red. A. Supervised clustering heatmap using genes dysregulated by ANOVA comparison between WT and Ctsg-PML-RARA CMP populations. B. Supervised clustering heatmap using genes dysregulated by ANOVA comparison between WT and Ctsg-PML-RARA GMP populations.
(EPS)
Erythroid colony formation (CFU-E) in Ctsg-PML-RARA vs. WT mice. Bone marrow cells from 6-week-old and 8-week-old, healthy mice were plated in methylcellulose containing erythropoietin (Methocult 3334). Results of paired t-tests are shown in each panel. A. Immunophenotypes of cells in these colonies were assessed as indicated: A. Ter119+CD71high (mature erythrocytes) and B. CD11b+ (myelomonocytic/monocytic cells). C. and D. Hemoglobin and mean corpuscular volume (MCV) values were determined in a cohort of healthy 4-month-old mice with the indicated genotypes.
(EPS)
Examples of bone marrow immunophenotypes following transplantation of flow sorted KLS and promyelocytes. Bone marrow KLS and promyelocytes were sorted from littermate Ctsg-PML-RARA (mCG-PR) and WT mice and transplanted into sub-lethally irradiated Ly5.1 recipients (Figure 6). Twelve weeks after transplantation, bone marrow cells were harvested and assessed for Gr1, CD3, CD19, CD45.2 and CD45.1 expression. At least 40,000 events were collected per recipient. A–C. Representative recipient of KLS transplantation. D–F. Representative recipient of promyelocyte transplantation.
(EPS)
Comparative gene expression analysis between cell populations derived from Ctsg-PML-RARA versus WT mice. The supplementary data includes a summary of gene expression differences (by ANOVA and SAM analysis) for comparisons between Ctsg-PML-RARA and WT SLAM, KLS, CMP, GMP, MEP and promyelocyte cell populations. The analysis includes a comparison of dysregulated genes to previously identified PML-RARA binding sites and to known dysregulated mRNA abundance identified in primary human APL samples [39], [40].
(DOCX)
Ctsg-PML-RARA versus WT SLAM ANOVA Results.
(XLSX)
Ctsg-PML-RARA versus WT KLS ANOVA Results.
(XLSX)
Ctsg-PML-RARA versus WT CMP ANOVA Results.
(XLSX)
Ctsg-PML-RARA versus WT GMP ANOVA Results.
(XLSX)
Ctsg-PML-RARA versus WT MEP ANOVA Results.
(XLSX)
Ctsg-PML-RARA versus WT Promyelocyte ANOVA Results.
(XLSX)
Concordant Dysregulated Genes in both the CMP and GMP Compartments by ANOVA.
(XLSX)
Ctsg-PML-RARA versus WT SLAM SAM Results.
(XLSX)
Ctsg-PML-RARA versus WT KLS SAM Results.
(XLSX)
Ctsg-PML-RARA versus WT CMP SAM Results.
(XLSX)
Ctsg-PML-RARA versus WT GMP SAM Results.
(XLSX)
Ctsg-PML-RARA versus WT MEP SAM Results.
(XLSX)
Ctsg-PML-RARA versus WT Promyelocyte SAM Results.
(XLSX)