

Abstract

The cobalt(II) complex of 3,5-DitBu-IbuPhyrin, [Co(P1)], is an effective catalyst for intramolecular amination of electron-deficient C–H bonds, including those adjacent to electron-withdrawing CO2R, C(O)NR2, C(O)R, and CN groups, in excellent yields with high regio- and stereoselectivity. The [Co(P1)]-catalyzed amination system provides a direct method for the synthesis of α-amino acid derivatives from the corresponding carboxylate precursors.
Nitrogen-containing compounds are of central importance in biology and medicine.1 The development of synthetic methodologies that allow for selective conversion of omnipresent C–H bonds into valuable amine functional groups promises to transform the art and practice of organic synthesis and should lead to many new applications. Among several approaches, catalytic C–H amination via metal-mediated nitrene insertion represents one of the most general and direct methods for installation of amino functionalities with potential control of various selectivities. Research efforts in this direction have been exceedingly prolific and have led to the development of a number of C–H amination systems based on different combinations of metal catalysts and nitrene sources.2 Represented by dirhodium(II) tetracarboxylate catalysts in conjunction with in situ-generated iminoiodane nitrene sources, existing catalytic systems have enabled effective amination of benzylic, allylic and other electron-rich C–H bonds with high regio- and stereoselectivity.2 The potential of catalytic amination, however, has not been fully extended to other types of more challenging C–H bonds, especially the electron-deficient C–H bonds due to their incompatibility with electrophilic metallonitrene intermediates intervened in most current catalytic systems.
The C–H bonds in the α-position of electron-withdrawing groups such as esters, amides, ketones and nitriles are the common electron-deficient C–H bonds that have not been successfully demonstrated for metal-catalyzed amination. Clearly, this mode of transformation would be highly desirable because α-C–H amination of esters and amides may offer a direct method for stereoselective synthesis of biologically important α-amino acid derivatives.3 To the best of our knowledge, the only previous report that briefly touched the subject is the Rh2-catalyzed intramolecular α-C–H amination of N-Boc-protected sulfamide esters.4,5 Evidently, amination of electron-deficient C–H bonds is an unaddressed issue that faces formidable challenges in both reactivity and regioselectivity.
Cobalt(II) porphyrins, a family of stable metalloradicals with well-defined open-shell doublet d7 electronic structure, have recently arisen as a new class of catalysts for selective C–H amination.6 These Co(II)-based metalloradical catalysts have proven to be unusually effective on the activation of various organic azides, including sulfonyl,7 phosphoryl,8 carbonyl9 and aryl10 azides, for amination of broad classes of C–H bonds under neutral and non-oxidative conditions.11 Particularly, Co(II) complexes of D2h-symmetric amidoporphyrins [Co(D2h-Por)] have revealed uncommon catalytic capacity for efficient intramolecular amination of strong primary C–H bonds7b,8 and have also displayed excellent chemoselectivity for intramolecular allylic C–H amination over the competitive C=C aziridination.7c Several lines of experimental and computational evidence back the radical mechanism of Co(II)/azide-based C–H amination that involves an unusual Co(III)-nitrene radical intermediate undergoing a stepwise radical abstraction-substitution pathway.7b,7c,12,13 Considering the non-electrophilic nature of this radical mechanism, which is fundamentally different from the electrophilic metallonitrene mechanism shared by the widely-studied Rh2- and other closed-shell systems, we envisaged the possibility of addressing the aforementioned challenge of intramolecular electron-deficient C–H amination through Co(II)-based metalloradical catalysis.
At the onset of our investigation, we evaluated the catalytic intramolecular C–H amination reaction of N-benzyl sulfamoyl azide 1a,14,15 which contains electron-deficient secondary C–H bonds positioned α- to the ester unit, by Co(II) porphyrins (Scheme 1). Under the typical neutral and nonoxidative conditions of Co(II)-based metalloradical catalysis, we were thrilled to find that even the simple [Co(TPP)] was capable of aminating the electron-deficient secondary α-C–H bonds in 1a to form the corresponding six-membered cyclic sulfamide-based amino acid ester 2a despite the fact that a relatively higher catalyst loading (5 mol %) was employed. Although the yield was moderate (41%), the α-C–H amination was very clean without observation of β-C–H amination, indicative of its slow reaction rate. When the Co(II) complex of D2h-symmetric amidoporphyrin 3,5-DitBuIbuPhyin [Co(P1)] was employed as the catalyst,7b,7c,8 the amination rate was drastically enhanced to afford the desired amino acid derivative 2a in 98% yield in spite of a lower catalyst loading (2 mol %). This ligand-enhanced catalysis is presumably contributed to the cooperative hydrogen bonding interaction between the groups S=O of the substrate and N-H of the catalyst.13a,16
Scheme 1.

Ligand Effect on Intramolecular Amination of Electron-Deficient C–H Bonds by Co(II) Porphyrins.
The [Co(P1)]-catalyzed intramolecular amination was found to be generally effective for various types of electron-deficient C–H bonds (Scheme 2). In addition to the α-C–H bonds of the ester 1a (entry 1), the Co(II)-based system could efficiently catalyze α-C–H amination of amides, ketones, and nitriles as demonstrated with reactions of azides 1b–1d, respectively, affording the desired amination products 2b–2d in excellent yields (entries 2–4). Its complete regioselectivity toward 1,6-amination of electron-deficient C–H bonds was further highlighted by the high-yielding diastereoselective reactions of the β-substituted azides 1e–1f without any interference from 1,5- or 1,6-amination of the electron-rich C–H bonds (entries 5–6). Higher to excellent diastereoselectivities were achieved for regioselective α-C–H amination of enantiopure γ-substituted azide esters 1g–1h, affording optically active bis-α-amino acid derivatives 2g–2h in high yields (entries 7–8), which is in stark contrast to the Rh2-catalyzed amination.4 Furthermore, β,γ-disubstituted substrates such as cyclohexane-based cis- and trans-azide ketones 1i–1j could be diastereoselectively aminated to produce the 6,6-bicyclic structures 2i–2j in high yields (entries 9–10). As expected, the [Co(P1)]-catalyzed amination could be well applied to electron-deficient tertiary C–H bonds as exemplified with azide ester 1k and azide ketone 1l, resulting in high-yielding formation of α,α-disubstituted α-amino acid and ketone 2k and 2l, respectively (entries 11–12). In addition to N-benzyl-substituted substrates, the [Co(P1)]-catalyzed amination has been shown to be compatible with sulfamoyl azides having varied N-substitutions including oxidizable functional groups.7b,7c Relatively slower reaction rates, however, were noticed for azide substrates that contain N-electron-withdrawing groups as shown with azides 1m and 1n (entries 13–14).
 |
(1) |
Scheme 2.

Regio- and Stereoselective Intramolecular Amination of Electron-Deficient C–H Bonds by [Co(P1)]a,b
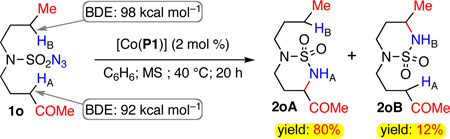
To shed some light on the origin of the remarkable catalytic capacity of the Co(II)-based system toward selective amination of electron-deficient C–H bonds, we performed a direct competition experiment between electron-deficient and -rich secondary C–H bonds with similar steric environments by choosing N-n-butyl sulfamoyl azide ketone 1o as the amination substrate (eq. 1). Under the standard catalysis of [Co(P1)], the reaction of 1o afforded two products 2oA and 2oB in 80% and 12% yields, respectively, indicating that 1,6-amination of electron-deficient C–HA was considerably faster than that of electron-rich C–HB. This seemingly very surprising result can be rationalized on the basis of the stepwise radical abstraction-substitution mechanism of the Co(II)-catalyzed C–H amination.7b,7c,12,13 Since the C–HA bond (92 kcal mol−1) is significantly weaker than the C–HB bond (98 kcal mol−1),17 the H-atom abstraction by the key Co(III)-nitrene radical intermediate is expected to be more facile for HA than HB.
 |
(2) |
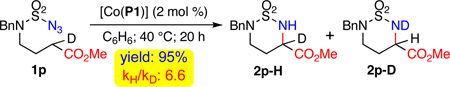
Additional support for the participation of an H-atom transfer (HAT) key step in the radical mechanism of the Co(II)-based metalloradical amination was obtained through the measurement of kinetic isotope effect (KIE) (eq. 2). When sulfamoyl azide ester 1p, in which one of two α-H-atoms is replaced with a deuterium atom, was subjected to the standard catalytic conditions by [Co(P1)], the high-yielding reaction generated a mixture of two α-amino acid esters 2p-H and 2p-D, which resulted from α-C–H and α-C–D amination, respectively, of the monodeuterated substrate. Analysis of the product mixture by 1H-NMR provided an intramolecular KIE of 6.6. This degree of primary KIE, which is substantially greater than the determined value of 1.9 for Rh2-catalyzed intramolecular amination of benzylic C–H bonds,18 is comparable to that measured for Co(II)-based metalloradical allylic C–H amination,7c indicating the involvement of direct C–H bond rupture via HAT process by the key Co(III)-nitrene radical intermediate.7b,7c,12,13
In an effort to trap the resulting carbon-based radical after H-atom abstraction by the Co(III)-nitrene radical intermediate, we carried out catalytic intramolecular C–H amination of azide ester 1a by [Co(P1)] in the presence of stable oxygen-based radical TEMPO (Scheme 3). Addition of substoichiometric amounts of TEMPO was found to have no significant effect on the catalytic reaction as previously observed for Co(II)-catalyzed intramolecular allylic C–H amination.7c This observation, which seems to be abnormal for a catalytic reaction involved with radical intermediates, is actually consistent with the very low barrier (or barrierless) of the subsequent intramolecular homolytic substitution (SHi) step suggested by the DFT calculations of related catalytic systems.12,13 When the amount of added TEMPO was increased to 5 equivalents, however, the conversion of 1a was decreased to 90% with 10% of the starting azide 1a remained under the same catalytic conditions (Scheme 3). While the α-amino acid ester 2a was still formed as the major product in 67% yield, the reaction generated a new product 3a in 18% yield. Detailed characterizations revealed 3a as a TEMPO-containing sulfamide in which the TEMPO unit is attached to the α-position of the ester group via C–O bond formation. As illustrated in Scheme 3, the production of 3a was likely originated from the intermediate 1aC that was formed from TEMPO trapping of the C-based radical intermediate 1aB. Presumably due to steric reasons, the initially generated Co(III)-nitrene radical intermediate 1aA could undergo the HAT process to generate 1aB without being affected by the presence of TEMPO. Together with the competition experiment and kinetic isotope effect, we take these results as new experimental evidence for the stepwise radical abstraction-substitution mechanism of Co(II)-based metalloradical C–H amination.7b,7c,12,13
Scheme 3.

TEMPO Trapping of Carbon-Based Radical Intermediate Involved in Radical Mechanism of Co(II)-Based Metalloradical C–H Amination.
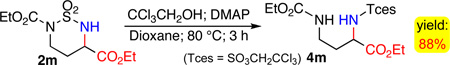
Considering their stereogenic centers that are substituted with important functionalities such as ester, amide, ketone and nitrile groups, the family of six-membered cyclic sulfamides constructed through Co(II)-catalyzed intramolecular α-amination of electron-deficient C–H bonds may find useful applications as intermediates for stereoselective synthesis, in addition to their known biomedical applications as potential inhibitors of several important enzymes.19 In particular, the resulting cyclic sulfamide esters and amides are the direct precursors for the synthesis of α,γ-diamino acid derivatives upon removal of the SO2 unit. As a demonstration of this aspect of application, cyclic sulfamide ester 2m could be effectively converted to α,γ-diamino acid ester 4m in 88% yield when it was treated with CCl3CH2OH in the presence of DMAP (eq. 3).4 Since the α- and γ-amine groups in 4m are differentially protected, its further transformations should provide access to various α,γ-diamino acid derivatives.
 |
(3) |
In summary, we have demonstrated, for the first time, the stereoselective amination of electron-deficient C(sp3)–H bonds by applying Co(II)-based metalloradical catalysis to intramolecular α-C–H amination of esters, amides, ketones, and nitriles. In addition to high-yielding and its neutral and nonoxidative conditions, the Co(II)-catalyzed amination can proceed with high regio-, and diastereoselectivity. Among other potential applications, the catalytic process offers an enabling approach for the stereoselective synthesis of α-amino acid derivatives through direct α-C–H amination of the corresponding esters and amides. We have shown, through the combination of a competition reaction, measurement of kinetic isotope effect and radical trapping experiment, that the unparalleled profile of activity and selectivity exhibited by Co(II)-catalyzed amination is attributed to its unique radical mechanism that involves the Co(III)-nitrene radical intermediate undergoing a stepwise radical abstractionsubstitution pathway. As a further outcome of metalloradical catalysis, C–H bond-dissociation energy (BDE) is recognized as a fundamentally important factor in controlling and differentiating reactivity and selectivity of various C–H bonds.
Supplementary Material
Acknowledgment
We are grateful for financial support by NSF (CHE-1152767) and NIH (R01-GM098777).
Footnotes
Supporting Information Available. Experimental procedures and analytical data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.(a) Nugent TC. Chiral Amine Synthesis: Methods, Developments and Applications. Willey-VCH: Weinheim; 2010. [Google Scholar]; (b) Ricci A. Amino Group Chemistry: From Synthesis to the Life Sciences. Willey-VCH: Weinheim; 2008. [Google Scholar]; (c) Ricci A. Modern amination methods. Willey-VCH: Weinheim; 2000. [Google Scholar]; (d) Salvatore RN, Yoon CH, Jung KW. Tetrahedron. 2001;57:7785. [Google Scholar]; (e) Johannsen M, Jorgensen KA. Chem. Rev. 1998;98:1689. doi: 10.1021/cr970343o. [DOI] [PubMed] [Google Scholar]; (f) Muller TE, Beller M. Chem. Rev. 1998;98:675. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]; (g) Wolfe JP, Wagaw S, Marcoux JF, Buchwald SL. Acc. Chem. Res. 1998;31:805. [Google Scholar]; (h) Hartwig JF. Acc. Chem. Res. 1998;31:852. [Google Scholar]
- 2.(a) Zalatan DN, Du Bois J. Top. Curr. Chem. 2010;292:347. doi: 10.1007/128_2009_19. [DOI] [PubMed] [Google Scholar]; (b) Collet F, Dodd RH, Dauban P. Chem. Commun. 2009:5061. doi: 10.1039/b905820f. [DOI] [PubMed] [Google Scholar]; (c) Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Davies HML. Angew. Chem. Int. Ed. 2006;45:6422. doi: 10.1002/anie.200601814. [DOI] [PubMed] [Google Scholar]; (e) Davies HML, Long MS. Angew. Chem. Int. Ed. 2005;44:3518. doi: 10.1002/anie.200500554. [DOI] [PubMed] [Google Scholar]; (f) Muller P, Fruit C. Chem. Rev. 2003;103:2905. doi: 10.1021/cr020043t. [DOI] [PubMed] [Google Scholar]
- 3.(a) Najera C, Sansano JM. Chem. Rev. 2007;107:4584. doi: 10.1021/cr050580o. [DOI] [PubMed] [Google Scholar]; (b) Maruoka K, Ooi T. Chem. Rev. 2003;103:3013. doi: 10.1021/cr020020e. [DOI] [PubMed] [Google Scholar]; (c) Easton CJ. Chem. Rev. 1997;97:53. doi: 10.1021/cr9402844. [DOI] [PubMed] [Google Scholar]
- 4.Kurokawa T, Kim M, Du Bois J. Angew. Chem. Int. Ed. 2009;48:2777. doi: 10.1002/anie.200806192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For Cu(I)-catalyzed intermolecular α-amination of arylacetate esters and aryl ketones with di-tert-butyldiaziridinone for formation of hydantonis and imidazolinones, respectively, see: Zhao BG, Du HF, Shi Y. J. Am. Chem. Soc. 2008;130:7220. doi: 10.1021/ja802242h. Zhao BG, Du HF, Shi Y. J. Org. Chem. 2009;74:4411. doi: 10.1021/jo9004553.
- 6.(a) Lu HJ, Zhang XP. Chem. Soc. Rev. 2011;40:1899. doi: 10.1039/c0cs00070a. [DOI] [PubMed] [Google Scholar]; (b) Che CM, Lo VKY, Zhou CY, Huang JS. Chem. Soc. Rev. 2011;40:1950. doi: 10.1039/c0cs00142b. [DOI] [PubMed] [Google Scholar]; (c) Driver TG. Org Biomol Chem. 2010;8:3831. doi: 10.1039/c005219c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ruppel JV, Kamble RM, Zhang XP. Org. Lett. 2007;9:4889. doi: 10.1021/ol702265h. [DOI] [PubMed] [Google Scholar]; (b) Lu H, Jiang H, Wojtas L, Zhang XP. Angew. Chem. Int. Ed. 2010;49:10192. doi: 10.1002/anie.201005552. [DOI] [PubMed] [Google Scholar]; (c) Lu H, Jiang H, Yang H, Wojtas L, Zhang XP. Chem. Sci. 2011;2:2361. [Google Scholar]
- 8.Lu HJ, Tao JR, Jones JE, Wojtas L, Zhang XP. Org. Lett. 2010;12:1248. doi: 10.1021/ol100110z. [DOI] [PubMed] [Google Scholar]
- 9.Lu HJ, Subbarayan V, Tao JR, Zhang XP. Organometallics. 2010;29:389. [Google Scholar]
- 10.(a) Cenini S, Gallo E, Penoni A, Ragaini F, Tollari S. Chem. Commun. 2000:2265. [Google Scholar]; (b) Ragaini F, Penoni A, Gallo E, Tollari S, Gotti CL, Lapadula M, Mangioni E, Cenini S. Chem. Eur. J. 2003;9:249. doi: 10.1002/chem.200390018. [DOI] [PubMed] [Google Scholar]
- 11.For reviews on the use of azides as nitrene sources for metal-catalyzed nitrene transfers, see: (a) ref. 6a. (b) ref. 6b. (c) ref. 6c. Cenini S, Gallo E, Caselli A, Ragaini F, Fantauzzi S, Piangiolino C. Coord. Chem. Rev. 2006;250:1234. doi: 10.1002/chem.200801148. Katsuki T. Chem. Lett. 2005;34:1304.
- 12.For detailed studies on the radical mechanism of [Co(Por)]-catalyzed C–H amination, including EPR observation of Co(III)-nitrene radical intermediates, see: Lyaskovskyy V, Suarez AIO, Lu H, Jiang H, Zhang XP, de Bruin B. J. Am. Chem. Soc. 2011;133:12264. doi: 10.1021/ja204800a.
- 13.For related DFT studies on the radical mechanism of Co(Por)]-catalyzed olefin aziridination, see: Suarez AIO, Jiang HL, Zhang XP, de Bruin B. Dalton Trans. 2011;40:5697. doi: 10.1039/c1dt10027k. Hopmann KH, Ghosh A. ACS Catal. 2011;1:597.
- 14.Sulfamoyl azides were reported to be chemically stable, even in strong acidic and basic conditions (see ref. 15). Our DSC experiments indicated that these sulfamoyl azides were thermally stable without decomposition up to at least 100 °C; see Supporting Information for a representative DSC plot of azide 1a.
- 15.(a) Griffith J. J. Chem. Soc. C. 1971:3191. [Google Scholar]; (b) Goddard-Borger ED, Stick RV. Org. Lett. 2007;9:3797. doi: 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- 16.Ruppel JV, Jones JE, Huff CA, Kamble RM, Chen Y, Zhang XP. Org. Lett. 2008;10:1995. doi: 10.1021/ol800588p. [DOI] [PubMed] [Google Scholar]
- 17.Luo YR. Comprehensive handbook of chemical bond energies. Boca Raton, FL: CRC; 2007. [Google Scholar]
- 18.Fiori KW, Espino CG, Brodsky BH, Du Bois J. Tetrahedron. 2009;65:3042. [Google Scholar]
- 19.(a) Reitz AB, Smith GR, Parker MH. Expert Opin. Ther. Patents. 2009;19:1449. doi: 10.1517/13543770903185920. [DOI] [PubMed] [Google Scholar]; (b) Winum JY, Scozzafava A, Montero JL, Supuran CT. Expert Opin. Ther. Patents. 2006;16:27. doi: 10.1517/13543776.16.1.27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.