

Abstract
It is well established that the activity of chromatin-modifying enzymes is crucial for regulating gene expression associated with hippocampal-dependent memories. However, very little is known about how these epigenetic mechanisms influence the formation of cortically dependent memory, particularly when there is competition between opposing memory traces, such as that which occurs during the acquisition and extinction of conditioned fear. Here we demonstrate, in C57BL/6 mice, that the activity of p300/CBP-associated factor (PCAF) within the infralimbic prefrontal cortex is required for long-term potentiation and is necessary for the formation of memory associated with fear extinction, but not for fear acquisition. Further, systemic administration of the PCAF activator SPV106 enhances memory for fear extinction and prevents fear renewal. The selective influence of PCAF on fear extinction is mediated, in part, by a transient recruitment of the repressive transcription factor ATF4 to the promoter of the immediate early gene zif268, which competitively inhibits its expression. Thus, within the context of fear extinction, PCAF functions as a transcriptional coactivator, which may facilitate the formation of memory for fear extinction by interfering with reconsolidation of the original memory trace.
Introduction
Fear extinction is the gradual reduction in the fear response by repeated presentation of a non-reinforced conditioned stimulus, which generates a new memory that competes with the original fear memory trace. The role of the medial prefrontal cortex in this process has received increased attention because it is simultaneously involved in regulating memory for both fear and its extinction. Thus, inactivation of the prelimbic prefrontal cortex (PLPFC) impairs fear acquisition (Laurent and Westbrook, 2009; Mamiya et al., 2009), whereas lesions or infusions of protein synthesis inhibitors into the infralimbic prefrontal cortex (ILPFC) slow fear extinction (Vidal-Gonzalez et al., 2006; Sierra-Mercado et al., 2011). Although current evidence indicates that there is a regional bias with respect to the regulation of opposing memories, it is likely that specific neurons throughout the medial prefrontal cortex contribute to each kind of memory, similar to the observation of fear- and extinction-specific neurons within the basolateral amygdala (Herry et al., 2008). The activity of these neurons would be presumed to be contingent on afferent projections from their connections with regions that contribute to both fear and extinction, as well as local reciprocal connections between the PLPFC and ILPFC (Hoover and Vertes, 2007). The existence of this regional heterogeneity prompted us to consider whether there are distinctive mechanisms associated with competing memory processes.
Fear extinction depends on coordinated gene expression and the synthesis of new synaptic proteins, which involves epigenetic mechanisms (Bredy et al., 2007; Bruel-Jungerman et al., 2007; Barrett and Wood, 2008; Bredy and Barad, 2008; Alberini, 2009; Stafford et al., 2012). Indeed, since the discovery that histone acetyltransferase (HAT) contributes to novel taste learning, our understanding of how the epigenome influences learning and memory has advanced considerably (Swank and Sweatt, 2001; Levenson and Sweatt, 2006; Koshibu et al., 2009; Day and Sweatt, 2010). For example, p300 and CREB-binding protein (CBP) are essential for contextual fear, object recognition, and spatial memories (Alarcón et al., 2004, Wood et al., 2005; Vecsey et al., 2007, Stefanko et al., 2009). The HAT p300/CBP-associated factor (PCAF) has also been implicated in memory formation, as PCAF knock-out mice are impaired in spatial learning and the reversal of an operant conditioning task, but show enhanced contextual fear memory (Maurice et al., 2008; Duclot et al., 2010). Furthermore, spatial learning leads to an increase in PCAF gene expression in the dorsal hippocampus (Bousiges et al., 2010). While HAT activity is clearly involved in different types of memory formation, how these epigenetic mechanisms exert their influence when there is competition between opposing memories for control over behavior, such as that which occurs during the acquisition and extinction of conditioned fear, is not understood. In the current series of experiments, we have discovered that PCAF functions as a coactivator within the ILPFC to selectively regulate fear extinction, a finding that makes this epigenetic mechanism an attractive target for the treatment of fear-related anxiety disorders.
Materials and Methods
Mice.
Naive 10–12-week-old C57BL/6 male mice were housed four per cage, maintained on a 12 h light/dark schedule, and allowed ad libitum access to food and water. All testing was conducted during the light phase in red-light-illuminated testing rooms following protocols approved by the Animal Ethics Committee of the University of Queensland.
Nuclear protein extraction.
Preparation of nuclear protein extracts derived from 1 mm3 tissue punches encompassing the ILPFC [anterior–posterior (AP) +1.34 mm to +1.98 mm] of naive, fear-conditioned, and extinction-trained mice (n = 5/group) were performed using the CelLytic NuCLEAR extraction kit (Sigma Aldrich) as per the manufacturer's instructions. Briefly, the extracted tissue was washed twice in 1× PBS and resuspended in 1000 μl of hypotonic 1× lysis buffer containing 10 μl of 0.1 m DTT and 10 μl of protease inhibitor. The tissue was then homogenized, centrifuged in suspension for 20 min (10,000–11,000 × g) and the supernatant removed. The remaining crude pellet was resuspended in 140 μl of extraction buffer containing 1.5 μl of 0.1 m DTT and 1.5 μl of protease inhibitor, and shaken gently for 30 min. Finally, the solution was centrifuged for 5 min at 20,000 × g and the supernatant transferred to a clean, chilled tube where it was snap frozen in aliquots with liquid nitrogen and stored at −70°C.
Protein antibody microarray.
Epigenetic regulatory protein expression was examined using the Panorama Antibody Microarray Gene Regulation kit (Sigma Aldrich) as per the manufacturer's instructions. Briefly, the sample was dialyzed at 4°C for 2 h in dialysis buffer (1000× volume of sample). The dialysis buffer was subsequently replaced with freshly prepared carbonate buffer (0.1 m, pH 9.5–9.6, carbonate-bicarbonate buffer) for an additional 2 h at 4°C. The sample was labeled by adding 1 ml of extract (1 mg/ml) to a vial containing either Cy3 or Cy5 labeling dye and mixed thoroughly. The reaction was incubated at room temperature for 30 min, mixing the solution every 10 min, after which excess Cy3/Cy5 was removed by using SigmaSpin columns (Sigma Aldrich). Protein concentration was determined using the Bradford protein assay, and the samples were subsequently stored at 2–8°C. Cy3- and Cy5-labeled samples (50–150 μg) of equal concentrations (10–30 μg/ml each) were mixed in a tube with 5 ml of array incubation buffer (Sigma Aldrich) and added to an incubation tray. PBS-washed slides were immersed in the incubation tray where they were covered with aluminum foil and incubated for 30 min at a shaking frequency of ∼30 rpm. Subsequently, the slides were washed three times for 5 min in three separate wells containing 5 ml of washing buffer each. Finally, excess liquid was decanted and the slides were incubated for 2 min in PBS before being air-dried for 20 min, protected from light. Microarray data acquisition was performed via a PerkinElmer ScanArray Express laser microarray scanner with subsequent quantification using ImaGene 8.0 software (BioDiscovery). The signal background pixel intensity median was subtracted from the signal pixel intensity median values for all spots (background correction). Experimental values were then divided by control values for each corresponding spot. The (overall) median for all spot ratios was calculated. This process was repeated for the slides in which the dyes were exchanged (experimental vs control) to account for dye bias (dye swap method). Normalization of values was achieved by dividing the ratio for each spot by the overall ratio median for both dye configurations. The normalized ratios for both dye configurations were then averaged and standard deviations calculated. The ratios for replicates were averaged, and this final fold change value was used to infer relative protein abundance in extinction-trained relative to fear-conditioned mice.
Western blot.
Protein concentration of nuclear preparations was determined according to the method described by Bradford (1976). Individual samples, each encompassing one ILPFC (n = 4–5 per group) as described above, were run on a single 15-well gel. Briefly, samples were prepared on ice (to a final volume of 20 μl) and then vortexed and denatured for 10 min at 70°C. Gels were run with 1× Tris buffered saline-Tween (TBS-T) and proteins transferred onto nitrocellulose membrane (Hybond-ECL; GE Healthcare). The membrane was blocked with 5 ml of 5% skim milk powder (Carnation) in TBS-T for 1 h at room temperature, washed with TBS-T for 5 min (3×), and incubated with 5 ml of H3K9me2 (1:1000; Abcam), CBP (1:1000; Abcam), MeCP2 (1:1000; Abcam), PCAF (1:1000; Millipore), HDAC2 (1:1000; Cell Signaling Technology), and p300 (1:1000; Millipore) antibodies in TBS-T in 0.5% skim milk powder for 24 h at 4°C. The membrane was washed with TBS-T (3×), incubated for 1 h with anti-mouse IRDye 680 goat anti-mouse secondary antibody (1:2500; LI-COR) or IRDye 800 goat anti-rabbit secondary antibody (1:2500; LI-COR) in TBS-T, and washed in TBS-T for 10 min (5×) and 20 min (1×). Optical density readings of the film were taken using a LI-COR analysis system. A one-way ANOVA with Tukey's post hoc tests was used for analysis, and the ratio of the relative optical density for target over the relative optical density for total H3 was calculated, thus providing an internal control for each sample.
Immunohistochemistry.
Mice were killed with 100 mg/kg ketamine, after which 50 ml of 1:100,000 sodium nitrite in PBS was pumped through the circulatory system, serving as a vasodilator. To fix the tissue, 4% paraformaldehyde in PBS was used. Following extraction, the brains were stored for 24 h in formalin. Subsequently, they were washed three times in PBS for 20 min before being stored in 0.05% sodium azide. The brains were placed in 30% sucrose for a minimum of 24 h before cryostat slicing. Sectioning at 14 μm was performed using a Zeiss Microm HM560 cryostat, and sections were mounted on Menzel-Glaser Superfrost Plus microscope slides. Briefly, the sections were incubated 1–2 h in blocking buffer, after which primary antibodies (PCAF and ATF4, 1:1000) were added and the slides incubated at 4°C overnight. The slides were then washed three times with PBS containing 0.02% Tween 20 (PBS-T), after which secondary antibodies were added (DyLight 488-conjugated AffiniPure sheep anti-mouse IgG or DyLight 549-conjugated AffiniPure goat anti-rabbit IgG; Jackson ImmunoResearch Laboratories). The slides were then incubated at room temperature for 2 h, washed three times with PBS or PBS-T, and coverslipped.
Behavioral training (for tissue collection).
Naive animals remained in their home cages until death. For the other two groups, fear conditioning consisted of three pairings [2 min intertrial interval (ITI)] of a 2 min, 80 dB, white-noise conditioned stimulus (CS) coterminating with a 2 s, 0.7 mA footshock in Context A. Mice were matched into equivalent treatment groups based on freezing during the third training CS. One day later, the fear-conditioned mice were brought to Context B, where the extinction group (EXT, n = 3–5/group) was presented with 60 CS presentations (2 min., 5 s ITI), based on our previous work indicating that this number of CS exposures induces near complete extinction (Cain et al., 2005). The fear-conditioned without extinction (FC-No EXT, n = 3–5/group) group spent an equivalent amount of time in Context B without any CS presentations. Tissue punches encompassing the ILPFC were collected from both of these groups 2 h after the end of their Context B session.
Drugs.
For local infusion studies, a PCAF inhibitor (50 μm H3-CoA-20-Tat, infusion rate 1 μl/2 min), control compound (50 μm Ac-DDDD-Tat, 1 μl/2 min), or a PCAF activator (30 μm SPV106, infusion rate 1 μl/2 min) was used. These doses were selected based on effective cell permeability, inhibition and activation of PCAF activity in vitro (Lau et al., 2000; Cole, 2008; Sbardella et al., 2008), and our own pilot experiments demonstrating a null effect at lower doses. For the systemic studies looking at the effects of PCAF activation, SPV106 (2.5, 25, or 250 mg/kg, delivered i.p.) or vehicle (1% DMSO, delivered i.p.) was used. This dose–response curve was based on our previous in vivo work demonstrating effective activation of histone acetylation with 25 mg/kg SPV106 (Sbardella et al., 2008; Colussi et al., 2011; Milite et al., 2011). For the in vitro assays, the small molecule p300/CBP inhibitor C646 (25 μm) (Bowers et al., 2010) was also used.
Surgery.
Double cannulae (Plastics One) were implanted into the ILPFC or PLPFC along the midline in the anterior posterior plane a minimum of 3 d before behavioral training. ILPFC cannulae were centered at AP +1.78 mm, and dorsal–ventral (DV) −2.9 mm. For the PLPFC infusion experiments, cannulae were centered at AP +1.78 mm, DV −1.9 mm. After behavioral testing, all mice were transcardially perfused and their brains dissected, sectioned, and Nissl stained (1:1000) to confirm placement of the cannulae.
Behavioral training and testing.
Experiments investigating the effects of PCAF on the extinction of conditioned fear consisted of three phases: fear acquisition (Context A), fear extinction (Context B), and testing (Context B). Testing occurred 1 d after extinction training to allow for complete memory consolidation. In all experiments, cued fear was induced in untreated, naive mice with three pairings of a 2 min, 80 dB, white-noise CS coterminating with a 2 s, 0.7 mA footshock (2 min ITI). Mice were matched into equivalent treatment groups based on freezing during the third training CS. One day later, the mice were placed in Context B and allowed to acclimate for 2 min. Extinction training comprised either 30 non-reinforced 2 min CS presentations (5 s ITI) for testing the effect of PCAF inhibition or five CS exposures for testing the effect of PCAF activation on the formation of memory for fear extinction. Immediately following the last CS exposure, the mice were infused either systemically or directly into the ILPFC with a PCAF inhibitor, PCAF activator, or control compound (1 μl injection volume, over 2 min). As controls, fear-conditioned mice without extinction (FC-No EXT) were placed in Context B for a time equal to that spent there by extinguished mice but were not exposed to any CS presentations. For the behavioral tests, all mice were returned to Context B in the drug-free state 24 h after extinction training. After a 2 min acclimation, freezing was assessed during two 2 min CS presentations (2 min ITI). In the final study, where we administered a PCAF activator (25 mg/kg) 30 min before extinction training, 24 h after testing in Context B, mice were tested for renewal in Context A. For the studies on fear acquisition, training consisted of three pairings (2 min ITI) of a 2 min, 80 dB, white-noise CS coterminating with a 2 s, 0.7 mA footshock in Context A. Immediately following the last CS–unconditioned stimulus pairing, mice were infused directly into the PLPFC with either the PCAF inhibitor or control compound (3 μg or 1 μl, respectively; 2 min). Twenty-four hours after training, mice were tested for auditory-cued fear memory in Context B. Behavioral freezing—the absence of all nonrespiratory movements—was rated during all phases by an experienced investigator blind to subject treatment, using a 5 s instantaneous time sampling technique. The percentage of observations with freezing was calculated for each mouse, and data represented as mean ± SEM freezing percentages for groups of mice during specified time bins. Total session means were analyzed with one-way ANOVA with Tukey's post hoc tests with FC-No EXT control (for extinction) or No-shock control (for fear acquisition) as reference.
Electrophysiology.
Young adult C57BL/6 mice (28- to 40-d-old) were anesthetized with isoflurane and killed by cervical dislocation. Brains were rapidly removed and placed in ice-cold artificial CSF (aCSF) containing the following (in mm): NaCl 118, KCl 2.5, NaHCO3 25, glucose 10, MgCl2 1.3, CaCl2 2.5, and NaH2PO4 1.2. Coronal brain slices (300 μm) containing the ILPFC were prepared with a vibratome (VT 1000S; Leica). Slices were allowed to recover in oxygenated (95% O2/5% CO2) aCSF at 35°C for at least 30 min, after which they were kept at room temperature for at least another 30 min before experiments were commenced. Slices were transferred to the recording chamber as required and were continuously perfused with oxygenated aCSF through a gravity-fed system while being maintained at 30–32°C. The perfusate (45 ml of aCSF, 45 ml of 25 μm Lys-CoA-20-Tat, or 45 ml of 100 μm CPTH2 in 25 ml of aCSF) was recycled using a peristaltic pump to reduce drug usage (Econo Pump EP-1). Experiments were performed in the presence of picrotoxin (100 mm) and CGP-35348 (1 mm) to block GABAergic transmission. Field responses were recorded from layer 5 of the ILPFC using glass electrodes filled with 3 m NaCl (pipette resistance, 4–7 mW) and a bipolar stimulating electrode was placed in layer 2/3. Synaptic responses were evoked at 0.1 Hz, and long-term potentiation (LTP) was induced using high-frequency stimulation (HFS) (5 pulses at 100 Hz, repeated 20 times with an interpulse interval of 200 ms, repeated 5 times with 6 s between trains). The effects of HFS were calculated by averaging 10 min (60 sweeps) of recordings immediately before and 60 min after LTP induction. Signals were recorded using a patch-clamp amplifier (Multiclamp 700B; Molecular Devices). Responses were filtered at 4–8 kHz and digitized at 10 kHz (ITC-18; Instrutech). All data were acquired, stored, and analyzed on a Macintosh computer using Axograph X (version 1.2.). t tests were used for statistical comparisons between groups. Results are expressed as mean ± SEM.
Cell culture, transfection, and dual luciferase assay.
HEK293T cells were cultured in DMEM containing 10% fetal bovine serum in 96-well plates and maintained in 5% CO2. Transfection was performed at 50% confluency. The cells were transfected with 80 ng of pCRE-LUC, and 200 ng of pCMV-ATF4 or pCMV plasmids for each well. The TK plasmids carrying Renilla luciferase was also cotransfected as an internal control. Cells were harvested 24 h after transfection, and relative luciferase activities were analyzed using the Dual-Luciferase assay kit (Promega) as per the manufacturer's recommendation, on a Fluostar OPTIMA plate reader. Luciferase activity values were determined relative to the control Renilla luciferase activity for monitoring the transfection efficiency.
ATF4 overexpression construct design and luciferase assay.
Total RNA was isolated from cortical tissue samples by phenol-chloroform extraction, after which the RNA was reverse transcribed into a cDNA template using a QuantiTect Reverse Transcription kit (Qiagen). PCR was performed to obtain the ATF4 full-length fragment using ATF4 (PCR primers: forward, 5′-AAGGATCCGC CACCATGACCGAGATGAGCT-3′; reverse, 5′-CCGCTCGAGCGGAACTCTC TTCTTCCC-3′). The purified ATF4 fragment was then double digested with BamHI and XhoI, and ligated to the pCMV-Tag1 vecter (Stratagene) to create the ATF4 overexpression construct pCMV-ATF4. For the luciferase assay, a 233 bp fragment of the −242 to −10 bp sequence in the zif268 promoter containing two proximal CRE elements was amplified using primer pairs for the mouse zif268 promoter (forward, 5′-CGGAGCTCCCACTGCTGCTGTTCCAATA-3′; reverse, 5′-CGGCTAGCGAATCGGCCTCTATT TCAAG-3′) using mouse genomic DNA as a template. The fragment was inserted between the SacI and NheI sites of the pISO vector, upstream of the firefly luciferase reporter gene, to generate pCRE-LUC.
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) was performed following modification of the Invitrogen ChIP kit protocol. Tissue samples encompassing ILPFC were fixed in 1% formaldehyde and cross-linked cell lysates were sheared by sonication in 1% SDS lysis buffer to generate chromatin fragments with an average length of 100–200 bp. The chromatin was then immunoprecipitated using an antibody specific to ATF4 (Santa Cruz Biotechnology) or an equivalent amount of control IgG (anti-rabbit; Santa Cruz Biotechnology) overnight at 4°C. Protein–DNA–antibody complexes were precipitated with protein G-magnetic beads for 1 h at 4°C, followed by two washes in low salt buffer, two washes in high salt buffer, and three washes with 1× tris-EDTA buffer. The precipitated protein–DNA complexes were eluted from the antibody with 1% SDS and 0.1 m NaHCO3, then incubated overnight at 65°C in 200 mm NaCl to reverse formaldehyde cross-links. Following proteinase K digestion, phenol-chloroform extraction, and ethanol precipitation, samples were subjected to qPCR using primer pairs specific for 200 bp segments corresponding to the upstream promoter region of the mouse zif268 gene carrying a CRE.
Coimmunoprecipitation.
Tissue was lyzed with 500 μl of modified RIPA buffer for 15 min on ice. The cell lysates were then immunoprecipitated with PCAF (2 μg; Millipore), ATF4 (2 μg; Santa Cruz Biotechnology), CBP (2 μg; Santa Cruz Biotechnology), p300 (2 μg; Millipore), or control rabbit IgG (2 μg; Santa Cruz Biotechnology) antibody following the protocol of Dynabeads Protein G (Invitrogen). Proteins were resolved with SDS-PAGE and separately analyzed with PCAF (1:2000), ATF4 (1:1000), CBP (1:1000), or p300 (1:1000) antibodies by standard Western blot.
qRT-PCR.
RNA from samples encompassing ILPFC was prepared using the Trizol extraction method (Invitrogen). Total RNA (1 μg) was used for cDNA synthesis using the Omniscript RT kit (Qiagen). PCR was then performed using primers for zif268 (forward, 5′-CCACTGCTGCTGTTCCAATA-3′; reverse, 5′-GAATCGGCCTCTATTTCAAGG-3′) and for PGK as an internal control (forward: 5′-TGCACGCTTCAAAAGCGCACG-3′, reverse: 5′-AAGTCCACCCTCATCACGACCC-3′). Quantitative PCR was performed using a RotorGeneQ (Qiagen) cycler using SYBR-green (Qiagen). The threshold cycle for each sample was chosen from the linear range and converted to a starting quantity by interpolation from a standard curve run on the same plate for each set of primers. zif268 mRNA levels were normalized for each well to phosphoglycerate kinase (PGK) mRNA using the ΔΔCT method, and each PCR was run in duplicate for each sample and repeated at least two times. Normalized mRNA levels were expressed as a ratio over PGK relative to FC-No EXT controls. mRNA levels were analyzed by one-way ANOVA followed by a Tukey's post hoc test where appropriate.
Results
Epigenetic regulatory proteins associated with fear extinction
Epigenetic mechanisms influence memory formation through a host of proteins that regulate learning-induced gene expression (Day and Sweatt, 2011). To begin to address how the epigenome contributes to the opposing memory processes, we performed a protein antibody microarray broadly enriched for epigenetic machinery on ILPFC tissue derived from EXT relative to FC-No EXT mice. For fear conditioning, mice were trained on a cued-fear conditioning task and 24 h later were exposed to a novel context for the equivalent duration of extinction training, but without CS exposure, before being killed 2 h later. For fear extinction, mice were fear conditioned as described, then exposed to an extinction training protocol 24 h later, before being killed a further 2 h after extinction training. Comparison between fear-trained and extinction-trained mice revealed many distinct epigenetic regulatory proteins that were preferentially associated with fear extinction (Table 1; Fig. 1). We observed a significant increase in methyl-CpG binding protein, MECP2 (microarray +1.7-fold change, Western blot validation, F(2,11) = 5.55, p < 0.05, Tukey's post hoc test, FC-No EXT vs EXT, p < 0.05; Fig. 1a). MECP2 regulates anxiety-related behavior and influences the function of brain-derived neurotrophic factor (BDNF) (McGill et al., 2006; Adachi et al., 2009). Given that DNA methylation has been implicated in regulating the persistence of remote memories (Miller et al., 2010), an increase in nuclear MECP2 levels may reflect a role for active DNA methylation during the formation of fear extinction memory. With respect to other epigenetic regulatory proteins associated with histone modification, we observed a significant decrease in the expression of the histone deacetylase HDAC2 after fear extinction (microarray −0.67-fold change Western blot validation, F(2,11) = 6.31, p < 0.05, Tukey's post hoc test, FC-No EXT vs EXT, p < 0.05; Fig. 1b). This finding is in accordance with a recent study demonstrating that HDAC2 functions as a negative regulator in memory formation and synaptic plasticity (Guan et al., 2009). Thus, it is possible that downregulation of HDAC2 after fear extinction training, like fear acquisition, is permissive for the memory consolidation, suggesting a general role for this histone deacetylase in regulating memory. The histone modification dimethyl histone 3 lysine 9 (H3K9me2) was also decreased after extinction training (microarray −0.43-fold change, Western blot validation, F(2,11) = 15.21, p < 0.01, Tukey's post hoc test, FC-No EXT vs EXT; p < 0.05, Fig. 1c). This posttranslational modification, associated with transcriptional silencing, has been shown to increase after contextual fear learning (Gupta et al., 2010). Moreover, recent findings suggest that both active and repressive histone modifications occur in a gene-specific manner in the hippocampus and the entorhinal cortex and both are required for the formation of contextual fear memory (Gupta-Agarwal et al., 2012). Furthermore, Gupta-Agarwal et al. (2012) found that inhibition of the histone methyltransferase G9a can influence fear extinction; thus, a gene-specific approach may be necessary to more precisely elucidate the role of H3K9me2 in the formation of memory for fear extinction.
Table 1.
Protein antibody microarray
| Increased EXT versus FC-No EXT | Fold change | Decreased EXT versus FC-No EXT | Fold change |
|---|---|---|---|
| PRMT4 | 2.03 | p300/CBP | 0.679 |
| PRMT6 | 2.00 | HDAC2 | 0.673 |
| DNMT1 | 1.98 | NTF2 | 0.668 |
| Sp1 | 1.92 | HDAC5 | 0.649 |
| HAT1 | 1.88 | hBrm/hsnf2a | 0.630 |
| AP-1/c-Jun | 1.86 | Importin alpha5/7 | 0.618 |
| AP-2a | 1.85 | LAP2 (TMPO) | 0.595 |
| GATA-1 | 1.83 | Phospho-histone H3 (pSer10) | 0.581 |
| Phospho-histone H3 (pSer28) | 1.79 | Actin | 0.580 |
| ATF-2 | 1.75 | TRF-1 | 0.555 |
| p63 | 1.74 | H3K9me2 | 0.431 |
| MBD4 | 1.74 | PCNA | 0.311 |
| MeCP2 | 1.71 | Coilin | 0.279 |
| HDAC3 | 1.67 | ||
| bCATENIN (pSer33) | 1.66 | ||
| PCAF | 1.65 | ||
| HDAC10 | 1.64 | ||
| H3K9me | 1.60 | ||
| HDAC4 | 1.50 |
A direct comparison between fear-trained and extinction-trained mice revealed many epigenetic regulatory proteins that are preferentially associated with fear extinction.
Figure 1.

Nuclear expression of epigenetic regulatory proteins associated with fear extinction. a, There was a significant increase in the expression of methyl-CpG binding protein, MECP2 (microarray +1.7-fold change, Western blot validation, F(2,11) = 5.55, p < 0.05, Tukey's post hoc test, FC-No EXT vs EXT, p < 0.05). b, There was also a significant decrease in the expression of the histone deacetylase HDAC2 after fear extinction training (microarray −0.67-fold change, Western blot validation, F(2,11) = 6.31, p < 0.05, Tukey's post hoc test, FC-No EXT vs EXT, p < 0.05). c, Expression of the histone modification H3K9me2 was also decreased after extinction training (microarray −0.43-fold change, Western blot validation, F(2,11) = 15.21, p < 0.01, Tukey's post hoc test, FC-No EXT vs EXT, p < 0.05). d, We found a significant increase in the expression of PCAF (microarray +1.65-fold change, Western blot validation, F(2,11) = 5.86, p < 0.05, Tukey's post hoc test, FC-No EXT vs EXT, p < 0.05) and a corresponding decrease in both CBP and p300 after extinction training. e, CBP microarray (−0.68-fold change, Western blot validation, F(2,11) = 15.21, p < 0.01, Tukey's post hoc test, FC-No EXT vs EXT, p < 0.05). f, p300 microarray (−0.68-fold change, Western blot validation F(2,11) = 11.09, p < 0.01, Tukey's post hoc test, FC-No EXT vs EXT p < 0.01). *p < 0.05; **p < 0.01.
Finally, and perhaps the most intriguing observation in terms of the contrast between fear extinction and fear acquisition, we found a significant increase in the expression of PCAF (microarray +1.65-fold change, Western blot validation, F(2,11) = 5.86, p < 0.05, Tukey's post hoc test, FC-No EXT vs EXT, p < 0.05; Fig. 1d) and a corresponding decrease in both CBP and p300 after extinction training (CBP microarray −0.68-fold change, Western blot validation, F(2,11) = 15.21, p < 0.01, Tukey's post hoc test, FC-No EXT vs EXT, p < 0.05; Fig. 1e; and p300 microarray −0.68-fold change, Western blot validation, F(2,11) = 11.09, p < 0.01, Tukey's post hoc test, FC-No EXT vs EXT, p < 0.01; Fig. 1f). These data indicate that there are distinct epigenetic regulatory mechanisms within the ILPFC that are associated with either the acquisition of fear or the formation of fear extinction memories.
PCAF activity is required for fear extinction but not for fear acquisition
Next, we examined the functional relevance of the fear extinction learning-induced increase in nuclear PCAF levels observed in our protein antibody microarray. Mice were trained on a cued-fear conditioning task in Context A, then matched for overall freezing scores and randomly assigned to treatment groups. Twenty-four hours later, they were extinction trained in a new context (Context B; 5 non-reinforced CS exposures for the PCAF activator experiment and 30 CS exposures for the PCAF inhibitor experiment) and, immediately after extinction training, microinfused directly into the ILPFC with either an activator of PCAF (30 μm SPV106), an inhibitor of PCAF (50 μm H3-CoA-20-Tat), or a control compound (50 μm, Ac-DDDD-Tat). One group of mice from each experiment was trained on day 1 and exposed to Context B on day 2 without CS exposures (FC-No EXT). These groups were included to control for any nonspecific effects of infusion stress or drug exposure on the original fear memory. On day 3, all mice were returned to Context B and tested. There was a significant reduction in freezing in SPV106-treated EXT mice relative to vehicle-treated FC-No EXT mice (F(3,20) = 5.43, p < 0.01, Tukey's post hoc test, FC-No EXT vehicle vs EXT SPV106, p < 0.05; vs EXT vehicle, n.s.; Fig. 2a). These data support the notion that PCAF activity in the ILPFC enhances the formation of fear extinction memory. In contrast to the effect of the PCAF activator on memory for fear extinction after five CS exposures, a 30-CS extinction training protocol produced a significant decrease in freezing in vehicle-treated EXT mice relative to FC-No EXT mice (Fig. 2b). Importantly, this effect was completely eliminated in mice infused with the PCAF inhibitor H3-CoA-20-Tat (F(3,32) = 5.76, p < 0.01, Tukey's post hoc test, FC-No EXT control vs EXT control, p < 0.05; vs EXT PCAF inhibitor, n.s.), indicating that PCAF activity in the ILPFC is required for the consolidation of fear extinction memory. Notably, the PCAF inhibitor had no effect on the expression of the original fear (Fig. 2b). In a separate experiment, we microinfused either H3-CoA-20-Tat (50 μm) or Ac-DDDD-Tat (50 μm) into the PLPFC immediately after fear training, as described above. Contrary to the effect of PCAF inhibition on fear extinction memory, there was a significant increase in freezing in H3-CoA-20-Tat-treated fear-conditioned mice relative to control drug-treated fear-conditioned mice (Fig. 2c), an effect that was not observed in no-shock controls (F(3,31) = 35.82, p < 0.0001, Tukey's post hoc test, FC control vs FC H3-CoA-20-Tat, p < 0.01; No-shock control vs No-shock H3-CoA-20-Tat, n.s.). These data further support the idea that individual epigenetic regulatory mechanisms are associated with specific memory processes.
Figure 2.
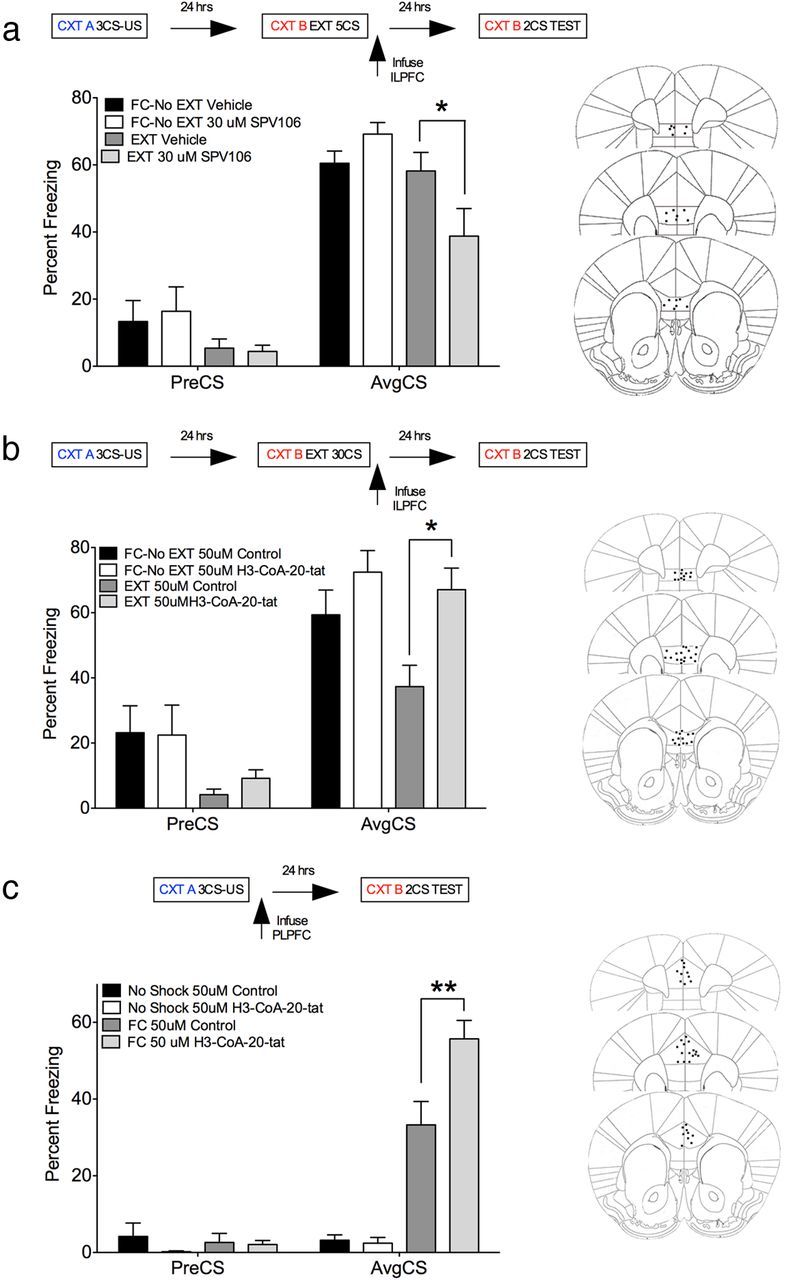
PCAF activity within the ILPFC is required for the consolidation of memory for fear extinction but not for cued fear. a, There was a significant decrease in freezing in SPV106-treated EXT mice relative to vehicle-treated FC-No EXT mice (F(3,20) = 5.43, p < 0.01, Tukey's post hoc test, FC-No EXT vehicle vs EXT SPV106, p < 0.05; vs EXT vehicle, n.s.). b, A 30-CS extinction training protocol produced a significant decrease in freezing in vehicle-treated EXT mice relative to FC-No EXT mice. This effect was eliminated in mice infused with the PCAF inhibitor H3-CoA-20-Tat (F(3,32) = 5.76, p < 0.01, Tukey's post hoc test, FC-No EXT control vs EXT control, p < 0.05; vs EXT PCAF inhibitor, n.s.). c, In a separate experiment, we infused either H3-CoA-20-Tat (3 μg, 1 μl) or Ac-DDDD-Tat (3 μg, 1 μl) directly into the PLPFC immediately after fear training. There was a significant increase in freezing in H3-CoA-20-Tat-treated FC relative to control drug-treated FC mice, an effect that was not observed in no-shock controls (F(5,31) = 35.82, p < 0.0001, Tukey's post hoc test, FC control vs FC H3-CoA-20-Tat, p < 0.01; No-shock control vs No-shock H3-CoA-20-Tat, n.s.). US, Unconditioned stimulus; CTX, context. *p < 0.05; **p < 0.01.
PCAF activity influences synaptic plasticity within the infralimbic prefrontal cortex
Given that LTP is a putative mechanism for plasticity associated with memory formation (Bliss and Lomo, 1973) and that plasticity within the ILPFC is required for the formation of fear extinction memory (Milad and Quirk, 2002), we next investigated the effects of PCAF inhibition on synaptic plasticity in acute brain slices containing ILPFC neurons. LTP was induced by tetanic electric stimulation (100 Hz protocol) of ILPFC layer 2/3 neurons in 300 μm coronal slices, and field potentials were recorded from layer 5 neurons in the presence of GABA receptor inhibitors. When added to the perfusate, a PCAF inhibitor (H3-CoA-20-Tat, 25 μm) eliminated the induction of LTP (one-tailed unpaired t test for the comparison of both groups after LTP induction; Control: 121.3% ± 5.2; H3-CoA-20-Tat: 98.2% ± 6.6, p < 0.05, n = 6/group; Fig. 3). This finding was then confirmed by using a structurally distinct PCAF inhibitor, CPTH2, which again eliminated LTP induction (n = 7; CPTH2: 97.2%, one-tailed unpaired t test compared with control: p < 0.001; data not shown). These data support previous findings demonstrating a role for HATs in regulating LTP (Vecsey et al., 2007), and suggest that LTP within the ILPFC is dependent, in part, on PCAF activity.
Figure 3.

PCAF activity is necessary for synaptic plasticity within the ILPFC. a, LTP was induced by tetanic electric stimulation (100 Hz protocol) of ILPFC layer 2/3 neurons in 300 μm coronal slices, and field potentials were recorded from layer 5 neurons in the presence of GABA receptor inhibitors. b, When added to the perfusate, the PCAF inhibitor (H3-CoA-20-Tat, 25 μm) eliminated the induction of LTP (one-tailed unpaired t test for the comparison of both groups after LTP induction; Control: 121.3% ± 5.2; H3-CoA-20-Tat: 98.2% ± 6.6, *p < 0.05, n = 6/group).
PCAF regulates fear extinction by transiently recruiting ATF4 to the promoter of the immediate early gene zif268, which downregulates its expression
To elucidate a potential mechanism by which PCAF preferentially contributes to the formation of fear extinction memory, we focused on the relationship between PCAF, ATF4, and the immediate early gene zif268. ATF4, also known as CREB2, is a member of the CREB family and is universally expressed in the brain. Importantly, ATF4 is a direct binding partner of PCAF and, in addition to its role as a HAT, PCAF functions as a transcriptional coactivator for ATF4 (Chérasse et al., 2007). Substantial evidence indicates that ATF4 increases the threshold for LTP and impairs long-term memory by competitive inhibition of CREB-mediated transcription in the hippocampus (Bartsch et al., 1995, 2000; Chen et al., 2003; Guan et al., 2003; Ameri and Harris, 2008). The immediate early gene zif268 (also known as Egr1, NGFI-A, and Krox-24) is crucial for the acquisition, retrieval, and reconsolidation of conditioned fear, and zif268 is downregulated in the medial prefrontal cortex during extinction learning (Hall et al., 2001; Bozon et al., 2003; Herry and Mons, 2004; Lee et al., 2004; Kirtley and Thomas, 2010; Lee, 2010; Cheval et al., 2012). Thus, any interaction between ATF4 and zif268 would have significant consequences on the ability of zif268 to regulate memory. Indeed, luciferase assay revealed that overexpression of ATF4 led to decreased activity of the firefly luciferase gene fused to the zif268 promoter (F(2,12) = 5.43, p < 0.05; Tukey's post hoc test, pISO vs pCRE-Luc+ATF4Ox, p < 0.05; Fig. 4a), thereby demonstrating a functional interaction between ATF4 and the CRE within the zif268 gene promoter.
Figure 4.

PCAF recruits ATF4 to the promoter of the immediate early gene zif268, which downregulates its expression. a, ATF4 functionally interacts with the zif268 promoter. b, ATF4 coimmunoprecipitates (IP) with p300 and PCAF in cortical neurons in vitro. c, In the presence of a PCAF inhibitor, H3-CoA-20-Tat, ATF4 binding to the CRE within the zif268 promoter is decreased (two-tailed t test, t = 14.60, df = 6, p < 0.001). d, ATF4 binding to the zif268 promoter is not affected by inhibition of p300/CBP by C646. e, Relative to nonstimulated control samples, 50 mm KCl-induced depolarization (used to mimic neural activity) led to significantly less ATF4 binding to the zif268 promoter in the presence of H3-CoA-20-Tat (F(3,21) = 4.28, p < 0.05; Tukey's post hoc test, Control KCl− vs H3-CoA-20-Tat KCl+, p < 0.05). f, Activity-dependent ATF4 binding to the zif268 promoter is not affected by inhibition of p300/CBP. *p < 0.05.
Next we sought to determine whether HAT activity influences ATF4 binding to the zif268 gene promoter. Although ATF4 interacts with p300 and PCAF in cortical neurons in vitro (Fig. 4b), chromatin immunoprecipitation revealed that in the presence of the PCAF inhibitor, H3-CoA-20-Tat, ATF4 binding to the CRE within the zif268 promoter is inhibited (two-tailed t test, t = 14.60 df = 6, p < 0.001; Fig. 4c); however, this effect was not observed in the presence of a small molecule p300/CBP inhibitor, C646 (Fig. 4d). In a primary cortical neuron preparation used to mimic the effects of neural activity on gene function, we also found that relative to nonstimulated control samples, 50 mm KCl-induced depolarization led to significantly less ATF4 binding to the zif268 promoter in the presence of H3-CoA-20-Tat (F(3,21) = 4.28, p < 0.05; Tukey's post hoc test, Control KCl− vs H3-CoA-20-Tat KCl+, p < 0.05; Fig. 4e). This effect was not observed under KCl-induced depolarization conditions in the presence of C646 (Fig. 4f). Thus, our data suggest that the activity of PCAF, but not p300/CBP, is necessary for ATF4 to be recruited to the zif268 gene promoter and imply that, together with its well established role as a HAT, PCAF also functions as a transcriptional coactivator for ATF4 in cortical neurons.
Immunohistochemistry revealed that PCAF and ATF4 are coexpressed throughout the medial prefrontal cortex (Fig. 5), which further implies a role for this interaction in medial prefrontal cortex-dependent memory processes. To more clearly elucidate the role of ATF4 in fear extinction, we next measured fear-related and extinction learning-induced ATF4 binding to the zif268 promoter and zif268 mRNA expression within the ILPFC in vivo. Relative to fear-conditioned mice, fear extinction learning led to increased ATF4 binding to the zif2568 gene promoter [F(3,15) = 5.40, p < 0.05; Tukey's post hoc test, FC-No EXT (30 min) vs EXT (30 min), p < 0.05; Fig. 6a], which was associated with decreased zif268 mRNA expression at the same time point [F(3,30) = 18.90, p < 0.0001; Tukey's post hoc test, FC-No EXT (30 min) vs EXT (30 min), p < 0.05; Fig. 6b]. Importantly, these effects were transient and were not present 2 h after extinction training, and did not occur in the presence of the PCAF inhibitor, H3-CoA-20-Tat (Fig. 6c,d). These observations emphasize the important relationship between fear extinction learning, PCAF activity, and ATF4-mediated inhibition of a retrieval-induced immediate early gene known to be critical for reconsolidation of fear memory, and suggest a potential mechanism by which PCAF activity contributes to the formation of fear extinction memory.
Figure 5.

Confocal microscopic image of colabeled ATF4 and PCAF labeling within the ILPFC. Top left, ATF4 (red); top right, PCAF (green); bottom left, DAPI (blue); bottom right, merge. Dotted lines show divisions between layers (L); large inset box is magnification (3×) of small boxed area. Arrows indicate examples of colabeling. Scale bar, 100 μm.
Figure 6.

Fear extinction learning induces a transient increase in ATF4 binding to the zif268 promoter and decreases zif268 mRNA expression in vivo. a, Relative to fear-conditioned mice, fear-extinction learning led to increased ATF4 binding to the zif2568 gene promoter [F(3,15) = 5.40, p < 0.05; Tukey's post hoc test, FC-No EXT (30 min) vs EXT (30 min), p < 0.05]. b, This was associated with decreased zif268 mRNA expression at the same time point [F(3,30) = 18.90, p < 0.01; Tukey's post hoc test, FC-No EXT (30 min) vs EXT (30 min), p < 0.05]. c, ATF4 binding to the zif268 promoter after fear-extinction training was inhibited when training occurs in the presence of the PCAF inhibitor H3-CoA-20-Tat (50 μm). d, In the presence of the PCAF inhibitor, fear-extinction learning did not lead to decreased zif268 mRNA expression. *p < 0.05; **p < 0.01.
Systemic administration of a PCAF activator enhances the formation of fear extinction memory and inhibits the return of conditioned fear
To assess the translational potential of targeting PCAF in the treatment of fear-related anxiety disorders, we administered a PCAF activator (SPV106; 2.5, 25, 250 mg/kg, i.p.) or vehicle (1% DMSO/PBS) systemically, immediately after a weak extinction training protocol that does not normally lead to persistent extinction memory (5 non-reinforced white noise exposures, 2 min duration, 5 s ITI). There was a significant reduction in freezing in EXT mice treated with SPV106 (25 mg/kg) relative to vehicle-treated FC-No EXT animals (Fig. 7, top left), an effect that was not observed in vehicle-treated EXT mice (F(5,63) = 3.91, p < 0.01, Tukey's post hoc test, FC-No EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.01; FC-No EXT vehicle vs EXT vehicle, n.s.). Importantly, systemic administration of the PCAF activator SPV106 (25 mg/kg), which enhances the formation of fear extinction memory, had no effect on memory when administered 6 h after training (Fig. 7, top right) nor did it have any effect on the acquisition of cued-fear when administered immediately after conditioning (Fig. 7, middle). We then examined whether PCAF activator-mediated enhancement of fear extinction memory would result in a reduction in the renewal of conditioned fear (Fig. 7, bottom). There was a significant decrease in freezing in EXT mice treated with SPV106 (25 mg/kg) relative to vehicle-treated FC-No EXT animals when they were tested in Context B, as well as significantly reduced freezing when tested for the return of fear (renewal) in Context A [F(3,37) = 3.91, p < 0.05; Tukey's post hoc tests; Context B: average CS (AvgCS): EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.05; Context A: pre-CS; EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.05; AvgCS: EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.01; pre-CS: EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.05; AvgCS: EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.01]. These data demonstrate the selective role of PCAF activity in the formation of fear extinction memory and highlight the strength of targeting this epigenetic mechanism to reduce fear responding after exposure therapy in the treatment of fear-related anxiety disorders.
Figure 7.
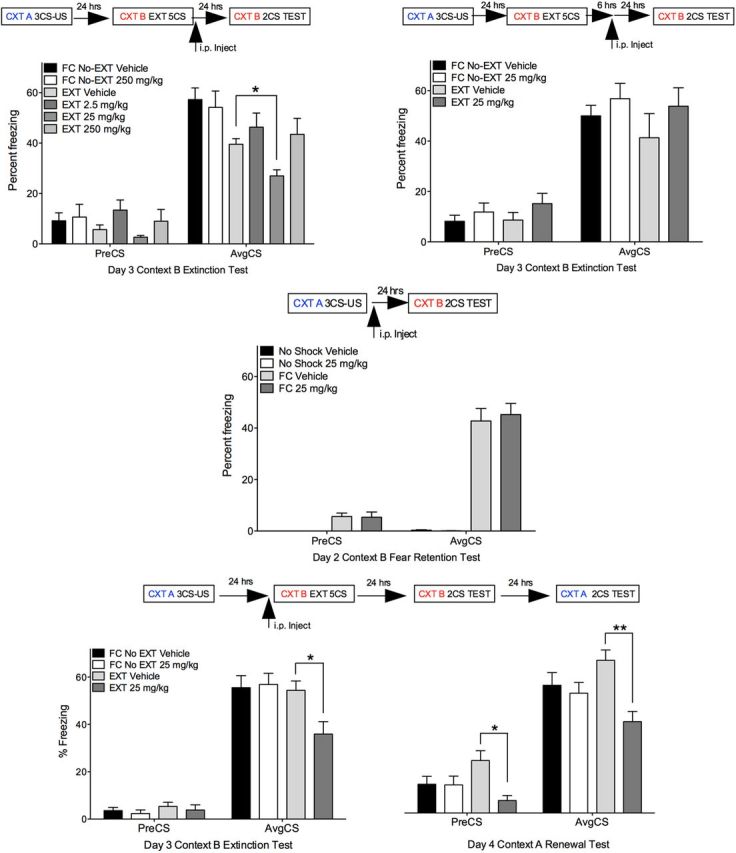
Systemic administration of a PCAF activator enhances the formation of memory for fear extinction. Top left, There was a significant decrease in freezing in EXT mice treated with SPV106 (25 mg/kg) relative to vehicle-treated FC-No EXT mice, an effect that was not observed in vehicle-treated EXT mice (Tukey's post hoc test, EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.05). Top right, Systemic administration of the PCAF activator SPV106 had no effect on memory when administered 6 h after training. Middle, SPV106 did not affect cued-fear memory. Bottom, SPV106 (25 mg/kg) administered before fear-extinction training prevented the renewal of conditioned fear [Context (CTX) B: Tukey's post hoc test, AvgCS: EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.05; Context A: pre-CS; EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.05; AvgCS: EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.01; Tukey's post hoc test, preCS: EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.05; AvgCS: EXT vehicle vs EXT SPV106 25 mg/kg, p < 0.01). *p < 0.05; **p < 0.01.
Discussion
Fear conditioning and its extinction are two distinct forms of learning that engage different molecular pathways. We previously identified fear memory-specific patterns of histone modification around individual promoters of BDNF within the ILPFC (Bredy et al., 2007). Cued-fear learning led to a general increase in H3 acetylation around the P1 and P4 BDNF gene promoters, which correlated with exon-specific BDNF mRNA expression. Conversely, fear-extinction learning led to an increase in H4 acetylation around the P4 BDNF gene promoter, again with a correlated increase in BDNF exon IV mRNA expression (Bredy et al., 2007). Memory for both fear and fear extinction could be strengthened by systemic administration of drugs that nonspecifically target the epigenome to enhance learning-induced histone acetylation (Bredy et al., 2008). Although the HATs p300 and CBP have consistently been implicated in fear learning (Alarcón et al., 2004, Wood et al., 2005; Vecsey et al., 2007, Stefanko et al., 2009; Bousiges et al., 2010), p300 activity within the ILPFC appears to be dispensable for fear extinction (Marek et al., 2011). In contrast to the important role of p300 and CBP in fear learning, we have now discovered that nuclear expression of PCAF is selectively increased in the ILPFC after fear extinction training, that PCAF activity in the ILPFC is required for LTP in this brain region, and that this epigenetic regulatory mechanism is necessary for the formation of fear extinction memory but not for the acquisition of conditioned fear. The influence of PCAF on extinction is mediated, in part, by transiently recruiting the repressive transcription factor ATF4 to the promoter of the immediate early gene zif268. A consequent downregulation of zif268 mRNA may serve to temporarily constrain the reconsolidation of the original fear after retrieval, thereby promoting the formation of a new fear extinction memory.
Fear extinction is generally thought to be a new learning process where the subject forms a new “safe” association with the conditioned stimulus that then acts to inhibit, or oppose, the original fear memory. Pavlov first proposed that extinction of conditional responding is an active learning process and not just passive forgetting, or erasure, of the conditioned association,with the evidence for this including observations of a return of fear after extinction training by changing the test context (renewal) (Bouton and Bolles, 1979), presenting a noncontingent unconditioned stimulus or stress (reinstatement) (Rescorla and Heth, 1975), or simply by allowing time to pass (spontaneous recovery) (Baum, 1988). However, recently the idea that extinction learning leads to erasure, at least in part, of the original fear memory trace has gained significant traction. For example, extinction training in young rodents leads to complete erasure of cued-fear memory (Gogolla et al., 2009; Kim and Richardson, 2010). Further, by taking advantage of the labile nature of memory at the time of retrieval (Przybyslawski and Sara, 1997; Nader et al., 2000), several laboratories have shown that a reactivated memory can either be updated or biased toward extinction by a variety of factors, including stimulus intensity, training to test interval, or the duration of the reminder cue (Eisenberg et al., 2003; Pedreira et al., 2004; Lee et al., 2006; Tronson et al., 2006). Monfils et al. (2009) demonstrated that a single non-reinforced conditioned stimulus before extinction training diminishes the return of fear after fear extinction training, a finding subsequently replicated in mice (Clem and Huganir, 2010), although others have observed variable effects using a similar protocol (Chan et al., 2010). Interestingly, chronic fluoxetine treatment leads to a permanent erasure of fear when combined with fear extinction training, which occurs via direct effects on BDNF (Karpova et al., 2011). These findings are in accordance with an important role for BDNF in regulating the extinction of conditioned fear (Bredy et al., 2007; Peters et al., 2010; Andero et al., 2011) and suggest that a transient increase in BDNF levels at the time of extinction learning might promote memory erasure. The precise mechanism by which fear memories can be disrupted to allow fear extinction to proceed is not fully understood, but appears to involve dendritic spine remodeling in the cortex (Lai et al., 2012) and the engagement of protein degradation pathways (Lee et al., 2008). Our data, including the observation that systemic administration of the PCAF activator SPV106 leads to a reduction in the renewal of conditioned fear (Fig. 7, bottom), support these findings and suggest that there are critical, learning-specific, epigenetic mechanisms engaged at the time of fear memory retrieval, which may contribute to the process of memory destabilization and promote the formation of fear extinction memory.
PCAF is involved in anxiety-related behavior and spatial and reversal learning (Maurice et al., 2008; Duclot et al., 2010), whereas both p300 and CBP are necessary for the formation of contextual fear, object recognition, and spatial memory (Alarcón et al., 2004; Korzus et al., 2004; Wood et al., 2005; Oliveira et al., 2007; Vecsey et al., 2007; Stefanko et al., 2009; Oliveira et al., 2011), but not for fear extinction (Marek et al., 2011). Together with our previous observations (Marek et al., 2011), the current data strongly suggest that p300/CBP and PCAF play opposing roles in the regulation of fear extinction. Indeed, there are fundamental differences in how chromatin complexes incorporating PCAF or p300/CBP are targeted toward genomic loci. One such multi-subunit complex, ATAC, contains PCAF as its catalytic subunit and is preferentially targeted to enhancers in a cell-type-specific manner independent of p300 (Krebs et al., 2011). HATs also directly interact with HDACs to regulate gene expression. For example, HDAC3 is necessary for the acquisition of hippocampal-dependent memory (McQuown et al., 2011) and it was very recently demonstrated that HDAC1 activity within the hippocampus selectively regulates the extinction of contextual fear (Bahari-Javan et al., 2012). Given that PCAF forms a complex with p300, together with HDAC1 (Yamagoe et al., 2003) as well as HDAC3 (Yao et al., 2001), it is possible that there are critical, yet undetermined, synergistic interactions between individual HATs and HDACs that serve to rapidly regulate gene expression depending on the physiological state of the neuron, and which are preferentially engaged depending on the memory process.
ATF4, also known as CREB2, is a member of the CREB family and has been shown to increase the threshold for LTP and impair long-term memory by competitive inhibition of CREB-mediated transcription (Bartsch et al., 1995, 2000; Chen et al., 2003; Guan et al., 2003; Ameri and Harris, 2008). Like PCAF, CBP and p300 have been shown to interact with ATF4 (Liang and Hai, 1997; Lassot et al., 2005) and, similarly, we have found that p300 and PCAF interact with ATF4 in primary cortical neurons (Fig. 4). However, in contrast to the requirement of PCAF activity, we observed no effect of p300/CBP inhibition on ATF4 occupancy at the zif268 promoter. Although there are no assays currently available to directly test for PCAF activity in vivo, and thus we cannot exclude HAT activity as an underlying mechanism for the effect of PCAF, our data suggest that within the context of fear extinction, it is more likely that PCAF functions as a transcriptional coactivator to transiently regulate zif268 mRNA expression. Thus, an important question remains as to how PCAF-specific recruitment of ATF4 regulates the expression of zif268 specifically during fear extinction training. The promoter region of the zif268 gene contains two proximal CRE elements and CREB interacts with these CRE sites within the zif268 promoter (Al Sarraj et al., 2005; Pradines et al., 2008; Mayer and Thiel, 2009). In addition, it is generally accepted that members of the CREB family can form homodimers or heterodimers with transcription factors such as AP-1 and the basic helix-loop-helix family (Hai and Hartman, 2001; Saxlund et al., 2004; Kang et al., 2007). For example, Luo et al. (2003) found that binding of ATF4 to the CRE site of the Grp78 promoter requires the formation of heterodimers with other transcription cofactors, including CREB. Interestingly, ATF4 can also exert opposite effects on gene regulation depending on the context. ATF4/c-Jun heterodimers interact with an AP-1/CREB binding site in the promoter region of the Ape1 gene, which functions permissively to increase Ape1 mRNA expression (Fung et al., 2007). Conversely, an interaction between ATF4 and the pseudokinase TRB3 leads to increased ATF4 transcriptional activity and competitive binding of ATF4 to a CRE element within the Snap25 promoter, which inhibits CREB-mediated Snap25 mRNA expression in β cells (Liew et al., 2010). Considering these findings, it is conceivable that, in the initial phase of extinction learning, ATF4 occupies one or both of the proximal CRE binding sites within the zif268 promoter and competes with CREB binding that would normally be recruited by other coactivators such as CBP or p300, thereby inhibiting zif268 gene expression. Indeed, our results show that 30 min after extinction training, ATF4 binding to the zi268 gene promoter is increased (Fig. 6a), and there is a concomitant decrease in zif268 mRNA expression at the same time-point (Fig. 6b), effects that do not occur in the presence of a PCAF inhibitor (Fig. 6c,d). Importantly, the influence of extinction training on ATF binding is specific to this form of learning and was not observed in animals that had been fear-conditioned or fear-conditioned and exposed to a neutral context without extinction training.
These findings suggest an intimate relationship between specific epigenetic regulatory mechanisms and fear extinction that is sensitive to both the external stimulus at the time of learning and the precise timing of gene regulation associated with the formation of competing memory traces. It is evident that the epigenetic regulation of gene expression and its relationship with memory formation, particularly with respect to fear extinction, is far more complex than currently understood and is not simply a generalized effect of chromatin-modifying enzymes on histone acetylation.
In summary, a PCAF-dependent and fear-extinction learning-induced increase in ATF4 occupancy within the zif268 gene promoter disrupts zif268 mRNA expression around the time of retrieval, which may promote the formation of a fear extinction memory by transiently interfering with the reconsolidation of the original memory trace. Thus, rather than its traditionally recognized role as a HAT, PCAF appears to exert its influence on fear extinction by functioning as transcriptional coactivator. Given that an increase in PCAF activity at the time of extinction learning leads to a reduction in the renewal of conditioned fear, targeted activation of PCAF represents a novel approach to selectively enhance memory for fear extinction, which may prove useful as a pharmacological adjunct to behavior therapy in the treatment of fear-related anxiety disorders.
Footnotes
This work was supported by grants from the Michael Smith Fund for Health Research to T.W.B. and M.S.K., the National Health and Medical Research Council Grants APP1023127 to T.W.B. and APP569575 to P.S., and the Australian Research Council Grant DP1096148 to T.W.B. We thank Rowan Tweedale for helpful editing of the manuscript, Dee McGrath for graphics support, and Dr. Kevin Dudley for technical assistance.
References
- Adachi M, Autry AE, Covington HE, 3rd, Monteggia LM. MECP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of rett syndrome. J Neurosci. 2009;29:4218–4227. doi: 10.1523/JNEUROSCI.4225-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and ltp are impaired in cbp+/− mice: a model for the cognitive deficit in rubinstein-taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Sarraj J, Vinson C, Han J, Thiel G. Regulation of GTP cyclohydrolase i gene transcription by basic region leucine zipper transcription factors. J Cell Biochem. 2005;96:1003–1020. doi: 10.1002/jcb.20580. [DOI] [PubMed] [Google Scholar]
- Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40:14–21. doi: 10.1016/j.biocel.2007.01.020. [DOI] [PubMed] [Google Scholar]
- Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ. Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry. 2011;168:163–172. doi: 10.1176/appi.ajp.2010.10030326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahari-Javan S, Maddalena A, Kerimoglu C, Wittnam J, Held T, Bähr M, Burkhardt S, Delalle I, Kügler S, Fischer A, Sananbenesi F. HDAC1 regulates fear extinction in mice. J Neurosci. 2012;32:5062–5073. doi: 10.1523/JNEUROSCI.0079-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RM, Wood MA. Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem. 2008;15:460–467. doi: 10.1101/lm.917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 1995;83:979–992. doi: 10.1016/0092-8674(95)90213-9. [DOI] [PubMed] [Google Scholar]
- Bartsch D, Ghirardi M, Casadio A, Giustetto M, Karl KA, Zhu H, Kandel ER. Enhancement of memory-related long-term facilitation by apaf, a novel transcription factor that acts downstream from both CREB1 and CREB2. Cell. 2000;103:595–608. doi: 10.1016/s0092-8674(00)00163-x. [DOI] [PubMed] [Google Scholar]
- Baum M. Spontaneous recovery from the effects of flooding (exposure) in animals. Behav Res Ther. 1988;26:185–186. doi: 10.1016/0005-7967(88)90118-0. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the unanaestetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousiges O, Vasconcelos AP, Neidl R, Cosquer B, Herbeaux K, Panteleeva I, Loeffler JP, Cassel JC, Boutillier AL. Spatial memory consolidation is associated with induction of several lysine-acetyltransferase (histone acetyltransferase) expression levels and H2B/H4 acetylation-dependent transcriptional events in the rat hippocampus. Neuropsychopharmacology. 2010;35:2521–2537. doi: 10.1038/npp.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouton ME, Bolles RC. Role of conditioned contextual stimuli in reinstatement of extinguished fear. J Exp Psychol Anim Behav Process. 1979;5:368–378. doi: 10.1037//0097-7403.5.4.368. [DOI] [PubMed] [Google Scholar]
- Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, Larocca C, Saldanha SA, Abagyan R, Sun Y, Meyers DJ, Marmorstein R, Mahadevan LC, Alani RM, Cole PA. Virtual ligand screening of the p300/CBP histone acetyltransferase:identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozon B, Davis S, Laroche S. A requirement for the immediate early gene zif268 in reconsolidation of recognition memory after retrieval. Neuron. 2003;40:695–701. doi: 10.1016/s0896-6273(03)00674-3. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Bredy TW, Barad M. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn Mem. 2008;15:39–45. doi: 10.1101/lm.801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual bdnf gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14:268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel-Jungerman E, Davis S, Laroche S. Brain plasticity mechanisms and memory: a party of four. Neuroscientist. 2007;13:492–505. doi: 10.1177/1073858407302725. [DOI] [PubMed] [Google Scholar]
- Cain CK, Godsil BP, Jami S, Barad M. The L-type calcium channel blocker nifedipine impairs extinction, but not reduced contingency effects, in mice. Learn Mem. 2005;12:277–284. doi: 10.1101/lm.88805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WY, Leung HT, Westbrook RF, McNally GP. Effects of recent exposure to a conditioned stimulus on extinction of Pavlovian fear conditioning. Learn Mem. 2010;17:512–521. doi: 10.1101/lm.1912510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Muzzio IA, Malleret G, Bartsch D, Verbitsky M, Pavlidis P, Yonan AL, Vronskaya S, Grody MB, Cepeda I, Gilliam TC, Kandel ER. Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB-2) and C/EBP proteins. Neuron. 2003;39:655–669. doi: 10.1016/s0896-6273(03)00501-4. [DOI] [PubMed] [Google Scholar]
- Chérasse Y, Maurin AC, Chaveroux C, Jousse C, Carraro V, Parry L, Deval C, Chambon C, Fafournoux P, Bruhat A. The p300/CBP-associated factor (PCAF) is a cofactor of atf4 for amino acid-regulated transcription of CHOP. Nucleic Acids Res. 2007;35:5954–5965. doi: 10.1093/nar/gkm642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheval H, Chagneau C, Levasseur G, Veyrac A, Faucon-Biguet N, Laroche S, Davis S. Distinctive features of EGR transcription factor regulation and DNA binding activity in CA1 of the hippocampus in synaptic plasticity and consolidation and reconsolidation of fear memory. Hippocampus. 2012;22:631–642. doi: 10.1002/hipo.20926. [DOI] [PubMed] [Google Scholar]
- Clem RL, Huganir RL. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science. 2010;330:1108–1112. doi: 10.1126/science.1195298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–597. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colussi C, Rosati J, Straino S, Spallotta F, Berni R, Stilli D, Rossi S, Musso E, Macchi E, Mai A, Sbardella G, Castellano S, Chimenti C, Frustaci A, Nebbioso A, Altucci L, Capogrossi MC, Gaetano C. Nepsilon-lysine acetylation determines dissociation from gap junctions and lateralization of connexin 43 in normal and dystrophic heart. Proc Natl Acad Sci U S A. 2011;108:2795–2800. doi: 10.1073/pnas.1013124108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD. DNA methylation and memory formation. Nat Neurosci. 2010;13:1319–1323. doi: 10.1038/nn.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD. Epigenetic mechanisms in cognition. Neuron. 2011;70:813–829. doi: 10.1016/j.neuron.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duclot F, Jacquet C, Gongora C, Maurice T. Alteration of working memory but not in anxiety or stress response in p300/CBP associated factor (PCAF) histone acetylase knockout mice bred on a C57BL/6 background. Neurosci Lett. 2010;475:179–183. doi: 10.1016/j.neulet.2010.03.077. [DOI] [PubMed] [Google Scholar]
- Eisenberg M, Kobilo T, Berman DE, Dudai Y. Stability of retrieved memory: inverse correlation with trace dominance. Science. 2003;301:1102–1104. doi: 10.1126/science.1086881. [DOI] [PubMed] [Google Scholar]
- Fung H, Liu P, Demple B. ATF4-dependent oxidative induction of the DNA repair enzyme APE1 counteracts arsenite cytotoxicity and suppresses arsenite-mediated mutagenesis. Mol Cell Biol. 2007;27:8834–8847. doi: 10.1128/MCB.00974-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogolla N, Caroni P, Lüthi A, Herry C. Perineuronal nets protect fear memories from erasure. Science. 2009;325:1258–1261. doi: 10.1126/science.1174146. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Kim JH, Lomvardas S, Holick K, Xu S, Kandel ER, Schwartz JH. P38 map kinase mediates both short-term and long-term synaptic depression in aplysia. J Neurosci. 2003;23:7317–7325. doi: 10.1523/JNEUROSCI.23-19-07317.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD. Histone methylation regulates memory formation. J Neurosci. 2010;30:3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta-Agarwal S, Franklin AV, Deramus T, Wheelock M, Davis RL, McMahon LL, Lubin FD. G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J Neurosci. 2012;32:5440–5453. doi: 10.1523/JNEUROSCI.0147-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/camp responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1–11. doi: 10.1016/s0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ. Cellular imaging of zif268 expression in the hippocampus and amygdala during contextual and cued fear memory retrieval: selective activation of hippocampal CA1 neurons during the recall of contextual memories. J Neurosci. 2001;21:2186–2193. doi: 10.1523/JNEUROSCI.21-06-02186.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Mons N. Resistance to extinction is associated with impaired immediate early gene induction in medial prefrontal cortex and amygdala. Eur J Neurosci. 2004;20:781–790. doi: 10.1111/j.1460-9568.2004.03542.x. [DOI] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct. 2007;212:149–179. doi: 10.1007/s00429-007-0150-4. [DOI] [PubMed] [Google Scholar]
- Kang JH, Kim MJ, Jang HI, Koh KH, Yum KS, Rhie DJ, Yoon SH, Hahn SJ, Kim MS, Jo YH. Proximal cyclic amp response element is essential for exendin-4 induction of rat egr-1 gene. Am J Physiol Endocrinol Metab. 2007;292:E215–E222. doi: 10.1152/ajpendo.00181.2006. [DOI] [PubMed] [Google Scholar]
- Karpova NN, Pickenhagen A, Lindholm J, Tiraboschi E, Kulesskaya N, Agústsdóttir A, Antila H, Popova D, Akamine Y, Bahi A, Sullivan R, Hen R, Drew LJ, Castrén E. Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science. 2011;334:1731–1734. doi: 10.1126/science.1214592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Richardson R. New findings on extinction of conditioned fear early in development: theoretical and clinical implications. Biol Psychiatry. 2010;67:297–303. doi: 10.1016/j.biopsych.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Kirtley A, Thomas KL. The exclusive induction of extinction is gated by bdnf. Learn Mem. 2010;17:612–619. doi: 10.1101/lm.1877010. [DOI] [PubMed] [Google Scholar]
- Koshibu K, Gräff J, Beullens M, Heitz FD, Berchtold D, Russig H, Farinelli M, Bollen M, Mansuy IM. Protein phosphatase 1 regulates the histone code for long-term memory. J Neurosci. 2009;29:13079–13089. doi: 10.1523/JNEUROSCI.3610-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs AR, Karmodiya K, Lindahl-Allen M, Struhl K, Tora L. SAGA and ATAC histone acetyl transferase complexes regulate distinct sets of genes and ATAC defines a class of p300-independent enhancers. Mol Cell. 2011;44:410–423. doi: 10.1016/j.molcel.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CS, Franke TF, Gan W. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature. 2012;483:87–91. doi: 10.1038/nature10792. [DOI] [PubMed] [Google Scholar]
- Lassot I, Estrabaud E, Emiliani S, Benkirane M, Benarous R, Margottin-Goguet F. P300 modulates atf4 stability and transcriptional activity independently of its acetyltransferase domain. J Biol Chem. 2005;280:41537–41545. doi: 10.1074/jbc.M505294200. [DOI] [PubMed] [Google Scholar]
- Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A, Wolffe AP, Nakatani Y, Roeder RG, Cole PA. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- Laurent V, Westbrook RF. Inactivation of the infralimbic but not the prelimbic cortex impairs consolidation and retrieval of fear extinction. Learn Mem. 2009;16:520–529. doi: 10.1101/lm.1474609. [DOI] [PubMed] [Google Scholar]
- Lee JL. Memory reconsolidation mediates the updating of hippocampal memory content. Front Behav Neurosci. 2010;4:168. doi: 10.3389/fnbeh.2010.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Everitt BJ, Thomas KL. Independent cellular processes for hippocampal memory consolidation and reconsolidation. Science. 2004;304:839–843. doi: 10.1126/science.1095760. [DOI] [PubMed] [Google Scholar]
- Lee JL, Milton AL, Everitt BJ. Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J Neurosci. 2006;26:10051–10056. doi: 10.1523/JNEUROSCI.2466-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Lee SH, Kim H, Kaang BK. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science. 2008;319:1253–1256. doi: 10.1126/science.1150541. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms: a common theme in vertebrate and invertebrate memory formation. Cell Mol Life Sci. 2006;63:1009–1016. doi: 10.1007/s00018-006-6026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Hai T. Characterization of human activating transcription factor 4, a transcriptional activator that interacts with multiple domains of cAMP-responsive element-binding protein (CREB)-binding protein. J Biol Chem. 1997;272:24088–24095. doi: 10.1074/jbc.272.38.24088. [DOI] [PubMed] [Google Scholar]
- Liew CW, Bochenski J, Kawamori D, Hu J, Leech CA, Wanic K, Malecki M, Warram JH, Qi L, Krolewski AS, Kulkarni RN. The pseudokinase tribbles homolog 3 interacts with ATF4 to negatively regulate insulin exocytosis in human and mouse beta cells. J Clin Invest. 2010;120:2876–2888. doi: 10.1172/JCI36849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Baumeister P, Yang S, Abcouwer SF, Lee AS. Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J Biol Chem. 2003;278:37375–37385. doi: 10.1074/jbc.M303619200. [DOI] [PubMed] [Google Scholar]
- Mamiya N, Fukushima H, Suzuki A, Matsuyama Z, Homma S, Frankland PW, Kida S. Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J Neurosci. 2009;29:402–413. doi: 10.1523/JNEUROSCI.4639-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek R, Coelho CM, Sullivan RK, Baker-Andresen D, Li X, Ratnu V, Dudley KJ, Meyers D, Mukherjee C, Cole PA, Sah P, Bredy TW. Paradoxical enhancement of fear extinction memory and synaptic plasticity by inhibition of the histone acetyltransferase p300. J Neurosci. 2011;31:7486–7491. doi: 10.1523/JNEUROSCI.0133-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice T, Duclot F, Meunier J, Naert G, Givalois L, Meffre J, Célérier A, Jacquet C, Copois V, Mechti N, Ozato K, Gongora C. Altered memory capacities and response to stress in p300/cbp-associated factor (PCAF) histone acetylase knockout mice. Neuropsychopharmacology. 2008;33:1584–1602. doi: 10.1038/sj.npp.1301551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer SI, Thiel G. Calcium influx into min6 insulinoma cells induces expression of EGR-1 involving extracellular signal-regulated protein kinase and the transcription factors Elk-1 and CREB. Eur J Cell Biol. 2009;88:19–33. doi: 10.1016/j.ejcb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- McGill BE, Bundle SF, Yaylaoglu MB, Carson JP, Thaller C, Zoghbi HY. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2006;103:18267–18272. doi: 10.1073/pnas.0608702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, Mullican SE, Jones S, Rusche JR, Lazar MA, Wood MA. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci. 2011;31:764–774. doi: 10.1523/JNEUROSCI.5052-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Milite C, Castellano S, Benedetti R, Tosco A, Ciliberti C, Vicidomini C, Boully L, Franci G, Altucci L, Mai A, Sbardella G. Modulation of the activity of histone acetyltransferases by long chain alkylidenemalonates (locams) Bioorg Med Chem. 2011;19:3690–3701. doi: 10.1016/j.bmc.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Miller CA, Gavin CF, White JA, Parrish RR, Honasoge A, Yancey CR, Rivera IM, Rubio MD, Rumbaugh G, Sweatt JD. Cortical DNA methylation maintains remote memory. Nat Neurosci. 2010;13:664–666. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–955. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Oliveira AM, Wood MA, McDonough CB, Abel T. Transgenic mice expressing an inhibitory truncated form of p300 exhibit long-term memory deficits. Learn Mem. 2007;14:564–572. doi: 10.1101/lm.656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AM, Estévez MA, Hawk JD, Grimes S, Brindle PK, Abel T. Subregion-specific p300 conditional knock-out mice exhibit long-term memory impairments. Learn Mem. 2011;18:161–169. doi: 10.1101/lm.1939811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedreira ME, Pérez-Cuesta LM, Maldonado H. Mismatch between what is expected and what actually occurs triggers memory reconsolidation or extinction. Learn Mem. 2004;11:579–585. doi: 10.1101/lm.76904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–1290. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradines E, Loubet D, Schneider B, Launay JM, Kellermann O, Mouillet-Richard S. CREB-dependent gene regulation by prion protein: impact on MMP-9 and betadystroglycan. Cell Signal. 2008;20:2050–2058. doi: 10.1016/j.cellsig.2008.07.016. [DOI] [PubMed] [Google Scholar]
- Przybyslawski J, Sara SJ. Reconsolidation of memory after its reactivation. Behav Brain Res. 1997;84:241–246. doi: 10.1016/s0166-4328(96)00153-2. [DOI] [PubMed] [Google Scholar]
- Rescorla RA, Heth CD. Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behav Process. 1975;1:88–96. [PubMed] [Google Scholar]
- Saxlund MA, Sadler-Riggleman I, Skinner MK. Role of basic helix-loop-helix (BHLH) and CREB transcription factors in the regulation of Sertoli cell androgen-binding protein expression. Mol Reprod Dev. 2004;68:269–278. doi: 10.1002/mrd.20080. [DOI] [PubMed] [Google Scholar]
- Sbardella G, Castellano S, Vicidomini C, Rotili D, Nebbioso A, Miceli M, Altucci L, Mai A. Identification of long chain alkylidenemalonates as novel small molecule modulators of histone acetyltransferases. Bioorg Med Chem Lett. 2008;18:2788–2792. doi: 10.1016/j.bmcl.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–538. doi: 10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Raybuck JD, Ryabinin AE, Lattal KM. Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction. Biol Psychiatry. 2012;72:25–33. doi: 10.1016/j.biopsych.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanko DP, Barrett RM, Ly AR, Reolon GK, Wood MA. Modulation of long-term memory for object recognition via hdac inhibition. Proc Natl Acad Sci U S A. 2009;106:9447–9452. doi: 10.1073/pnas.0903964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swank MW, Sweatt JD. Increased histone acetyltransferase and lysine acetyltransferase activity and biphasic activation of the ERK/RSK cascade in insular cortex during novel taste learning. J Neurosci. 2001;21:3383–3391. doi: 10.1523/JNEUROSCI.21-10-03383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Wiseman SL, Olausson P, Taylor JR. Bidirectional behavioral plasticity of memory reconsolidation depends on amygdalar protein kinase A. Nat Neurosci. 2006;9:167–169. doi: 10.1038/nn1628. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via creb: CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, Quirk GJ. Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learn Mem. 2006;13:728–733. doi: 10.1101/lm.306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagoe S, Kanno T, Kanno Y, Sasaki S, Siegel RM, Lenardo MJ, Humphrey G, Wang Y, Nakatani Y, Howard BH, Ozato K. Interaction of histone acetylases and deacetylases in vivo. Mol Cell Biol. 2003;23:1025–1033. doi: 10.1128/MCB.23.3.1025-1033.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao YL, Yang WM, Seto E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol Cell Biol. 2001;21:5979–5991. doi: 10.1128/MCB.21.17.5979-5991.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]