

Abstract
The impact of acute brain injury and delayed neurological deficits due to cerebral vasospasm (CVS) are major determinants of outcomes after subarachnoid hemorrhage (SAH). Although hyperbaric oxygen (HBO) had been used to treat patients with SAH, the supporting evidence and underlying mechanisms have not been systematically reviewed. In the present paper, the overview of studies of HBO for cerebral vasospasm is followed by a discussion of HBO molecular mechanisms involved in the protection against SAH-induced brain injury and even, as hypothesized, in attenuating vascular spasm alone. Faced with the paucity of information as to what degree HBO is capable of antagonizing vasospasm after SAH, the authors postulate that the major beneficial effects of HBO in SAH include a reduction of acute brain injury and combating brain damage caused by CVS. Consequently, authors reviewed the effects of HBO on SAH-induced hypoxic signaling and other mechanisms of neurovascular injury. Moreover, authors hypothesize that HBO administered after SAH may “precondition” the brain against the detrimental sequelae of vasospasm. In conclusion, the existing evidence speaks in favor of administering HBO in both acute and delayed phase after SAH; however, further studies are needed to understand the underlying mechanisms and to establish the optimal regimen of treatment.
Keywords: Intracranial aneurysm, Subarachnoid hemorrhage, Cerebral vasospasm, Neurological deficits, Hyperbaric oxygen, Neuroprotection
Introduction
Cerebral vasospasm (CVS) has been defined as a narrowing of major cerebral arteries that occurs on average between 5 and 15 days (peak 5–7 days) after subarachnoid hemorrhage (SAH) [1]. The occurrence of cerebral vasospasm is associated with high morbidity and mortality after SAH [2]. However, studies have found that the selective endothelin A-receptor (ETA) antagonist clazosentan does not significantly improve morbidity and mortality, even though it reduces angiographic vasospasm [3, 4]. Thorough explanation as to why clazosentan showed only limited clinical benefits in patients has been reviewed by earlier authors [2]. For one thing, treatment with clazosentan might be ineffective against the early brain injury, which greatly determines neurological outcomes after SAH [5] This early stage of injury is constituted by the change of pathophysiological factors (including raised intracranial pressure (ICP), decreases in cerebral blood flow (CBF) and cerebral perfusion pressure (CPP), blood–brain barrier (BBB) disruption, brain swelling, brain edema, acute vasospasm, and dysfunction of autoregulation) within the first 48 h after SAH [6, 7]. Considering the results of clazosentan studies, more integrative treatment strategies for vasospasm need to be explored under close hemodynamic and ICP monitoring [8]. The current clinical management of CVS includes preventive measures (e.g., nimodipine for 21 days following SAH) and several interventions for symptomatic patients, including hypervolemia, hypertension, and hemodilution (triple-H) therapy or mechanical therapies [9, 10]. Despite all these developments, CVS morbidity and mortality continue to be a significant clinical problem. However, a sizable window of opportunity for other therapies seems to exist, consistent with the fact that proximal vasospasm often starts around day 4 while clinical symptoms only develop days later [11]. In addition, therapies that produce vasodilatation of small arteries and ameliorate microcirculation may greatly alleviate neurological deficit even without reducing radiological vasospasm [9, 12].
Cerebral tissue ischemia triggered by the initial bleeding or secondary to vasospasm is a major cause of SAH-induced brain injury. HBO treatment, which results in a relief of cerebral hypoxia, appears to be a well-suited modality for combating acute ischemia following SAH [13]. It would be reasonable to expect that HBO applied within the therapeutic window of opportunity may also protect against delayed ischemia caused by cerebral vasospasm.
This paper provides an overview of clinical and experimental studies investigating HBO treatment of SAH and the ensuing cerebral vasospasm. The number of relevant clinical studies remains limited, which is partly a consequence of insufficient preclinical testing resulting in limited knowledge of basic mechanisms underlying HBO treatment. This lack of knowledge is a major limitation for selecting optimal treatment regimen and successful translation from bench to bedside. In order to identify the areas that need further investigation, we have also reviewed the molecular pathways that can be modulated by HBO in the brain vasculature and in the cerebral tissues after SAH.
The results used for the primary analysis were extracted from studies of HBO in SAH published in PubMed and ISI up to January of 2011. There were eight clinical reports and four laboratory investigations on rats, two of which used blood injection model and the other two used a perforation model of SAH.
In the clinical studies, the main analyzed endpoints included amelioration of neurological scores and reduction of mortality as well as reversal of radiological vasospasm and alleviation of the raised ICP. The number of patients, number of HBO sessions, and the level of hyperbaria were recorded in each study. Laboratory investigations were analyzed with emphasis on the reversal of angiographic vasospasm and the reduction of acute brain injury. The impact of HBO on the functional performance of animals was also evaluated. The inclusion of masked evaluation of outcomes and randomization were considered as indices of methodological quality.
As delineated later in more detail, the results of this survey suggest that HBO can favorably modify the molecular mechanisms of early brain injury and sequelae of cerebral vasospasm; therefore, it has a potential to become a valuable treatment option for SAH patients.
Clinical HBO Treatment of Neurological Impairment and Cerebral Vasospasm After SAH
The clinical evidence in favor of HBO therapy for vasospasm is limited. A few clinical single-center studies without randomization found therapeutic benefits of HBO in SAH. Six different centers successfully used HBO for treating SAH in a total of 319 patients [14–21]. Positive neurological outcomes after HBO treatment were found by two studies of surgically treated aneurysm cases [16, 20]. In the study conducted by Isakov et al. [16], 47 HBO-treated patients (6–15 sessions at 1.6–2.0 atm absolute (ATA)) showed a shortened duration of critical condition and relief of neurological symptoms after operations on the cerebral vessels because of ruptured aneurysms. The majority of 56 neurosurgical patients treated by Ugruimov etal. [20] showed amelioration of neurological deficits, whereas patients enrolled by Kitaoka et al. [21] improved mentally with HBO treatment after surgeries on aneurysms of the anterior communicating arteries This particular result is of paramount clinical importance as cognitive dysfunction affects up to 60% of SAH survivors [22]. Koshi et al. [15] found good outcomes in 12 out of 24 patients with symptomatic cerebral vasospasm treated with HBO (2–21 sessions for 60 min at 2.5 ATA) in addition to mild hypertensive hypervolemia. Ohta et al. [18] reported that HBO (2 ATA) reduced ICP in 19 patients with SAH. The same group found an improvement of somatosensory-evoked potentials in HBO-treated (2 ATA) mild SAH cases, although in patients with moderate to severe neurological symptoms, the improvement was found less frequently [19]. Levina et al. [17] studied the effect of HBO (6–15 sessions at pressure of 1.2–1.6 ATA) on 110 cases of aneurysm clipping surgery after SAH and found a reduced ischemic lesion size and the alleviation of edema on CT scans in the majority of patients. The authors of a recent review suggested that HBO exerts a beneficial effect on the penumbra in the ischemic brain regions and advocated the addition of HBO to triple-H therapy [23, 24].
Although the triple-H therapy improves CBF after SAH, it reduces the capability of blood to carry oxygen due to hemodilution. Moreover, in the ischemic regions of the brain, the occurrence of plasma flow without erythrocytes through the cerebral capillary vessels is already increased [25]. HBO may overcome this drawback of triple-H therapy by increasing the quantity of oxygen physically dissolved in the plasma. It has been suggested that maximizing oxygen delivery carries a potential to prevent neurological injury from delayed cerebral ischemia after SAH in patients with anemia [26]. HBO could be considered as the alternative of red blood cell transfusion administered to increase oxygen delivery in anemic patients after SAH. In conclusion, the level of evidence from the existing clinical studies of HBO in SAH can be classed as insufficient. The prospective randomized clinical trials on the subject are not available. It would be necessary to perform meaningful randomized clinical studies before HBO could be applied in standard care of SAH.
Another important question—whether normobaric oxygen (NBO) can be as effective as HBO in the treatment of CVS—has been addressed experimentally. Kocaoagullar et al. [27] compared NBO versus HBO treatment of rats with cerebral vasospasm after SAH and found that NBO was less effective in ameliorating neurological deficits associated with CVS. Interestingly though, despite improved neurological severity score, the authors did not find significant changes in the diameter of basilar artery in response to HBO treatment. However, the authors adopted a relatively mild treatment regimen of only one HBO session at 3 ATA and 1-h duration. Thus, the effect of HBO on the severity of angiographic spasm needs to be further investigated with the use of repeated HBO sessions and different levels of hyperbaria. In different experimental systems, HBO was shown to ameliorate both the vasoconstrictive and vasodilatory capabilities of the blood vessels, which stresses the potential of HBO for vascular protection in SAH considering that both the vasoconstrictive and vasodilatory capabilities of cerebral blood vessels are impaired after hemorrhage. For example, in the rat multiple organ failure syndrome, HBO (2 ATA at the 4th and 11th hours after study onset) improved both contraction of arteries in response to endothelin-1 (ET-1) and vasodilation induced by acetylcholine. These findings may suggest that HBO reduces vascular injury, which in turn results in an improved functionality of cerebral vessels [28]. This may point toward the potential of HBO for vascular protection in SAH considering that both the vasoconstrictive and vasodilatory capabilities of cerebral blood vessels are impaired in this type of hemorrhage.
HBO and Mechanisms of Prolonged Vasoconstriction
Several mechanisms underlying prolonged vascular spasm may be considered as targets for HBO treatment and are outlined below. ET-1 is a major vasoconstrictor that induces cerebral vasospasm following SAH. HBO has been shown to reduce brain edema, decrease infarct volume, and contribute to neurological functional recovery in the focal brain ischemia caused by cerebral infusion of ET-1. The impact on ET-1-induced arterial narrowing, however, has not been determined [29]. Therefore, it cannot be excluded that the beneficial effect of HBOT on ET-1-induced ischemia is due to effects other than an antagonistic action on vasospasm. In addition, the concentrations of ET-1 that induce ischemia in experiments are by magnitudes higher than those measured in SAH patients [30, 31]. It may suggest that ET-1 is potentiated by other factors playing a consistent pathophysiological role in the development of vasospasm [31]. However, another plausible explanation is that not only the level of ET-1 but also an increase in the vascular density of ET and 5HT1B receptors underlie vascular narrowing after SAH [32]. The levels of ET-1 nearly equivalent to physiological levels are sufficient to induce submaximal contraction of cerebral arteries excised from rats after SAH [33]. It is also known that ischemia upregulates endothelin receptors in the brain [34]. Thus, it may be hypothesized that hyperbaric oxygen can antagonize the vasoconstrictive propensity of cerebral vessels after SAH by alleviation of ischemic condition with subsequent reduction in the ET receptor upregulation, which, however, requires experimental verification.
Protein Kinase Pathways
In the acute phase, cerebral vascular spasm occurs through a calcium-dependent mechanism. In response to hemoglobin exposure, the intracellular level of calcium increases and drives myosin light chain kinase (MLCK) to phosphorylate the myosin light chains, which produces transient contraction. However, the mechanism of the prolonged phase of vasospasm is largely calcium-independent. Near resting levels of calcium were detected during protracted vasospasm [5]. This phenomenon can be caused by calcium sensitization [35] mediated by several kinases acting in concert with ET-1 released by endothelial cells, astrocytes, and leukocytes in response to hemoglobin and acute cerebral ischemia. ET-1 in general produces lasting receptor stimulation. Hirata et al. [36] first demonstrated that ET-1 can occupy its receptors for up to 48 h.
The major involvement of Rho A kinase in the process of prolonged contraction has been also postulated [37]. ET-1 activates Rho A, which in turn activates Rho kinase. The activated Rho kinase interacts with a MLCK via the stimulation of protein kinase C (PKC) that inhibits myosin phosphatase [37]. This leads to the increased phosphorylation of myosin light chains, resulting in prolonged contraction without elevation in intracellular calcium [38]. Interestingly, HBO has been found to reduce levels of Rho A in the brain [39]. Normobaric oxygen may lower the effect on the PKC, as shown in cultured alveolar cells [40] and in the mouse lung endothelial cells [41]. The pivotal role of PKC in the mechanism of vasospasm has been reviewed [42].
Protein tyrosine kinase (PTK) also plays an important role in the mechanism of vasospasm [43]. Studies have shown that the major causative mechanism of CVS shifts from PKC to PTK during the prolonged phase of arterial narrowing, which corresponds to a progression from the active myogenic tone to the non-myogenic tone [43]. Interestingly, the low level of oxidative stress has been shown to inactivate the Src family tyrosine kinases in human umbilical vein and aortic endothelial cells and in fibroblasts [44]. Src kinase, with its downstream effector mitogen-activated protein kinases (MAPK), has been implicated in CVS after SAH [45]. It should be stressed, however, that protein kinases play a role of enhancers or mediators of vasospasm rather than agents that can maintain vasospasm through perpetual activation. Several studies showed that the inhibition of kinases may attenuate but not prevent vasospasm [46, 47]. In contrast, the surgical evacuation of subarachnoid hematoma, especially within 24 h of SAH, was able to prevent cerebral vasospasm that otherwise develops in response to spasmogenic compounds released by lysed blood [48, 49]. This suggests that subarachnoid clot lysis and evacuation may be required for the full therapeutic benefits of HBO to develop (Fig. 1).
Fig. 1.
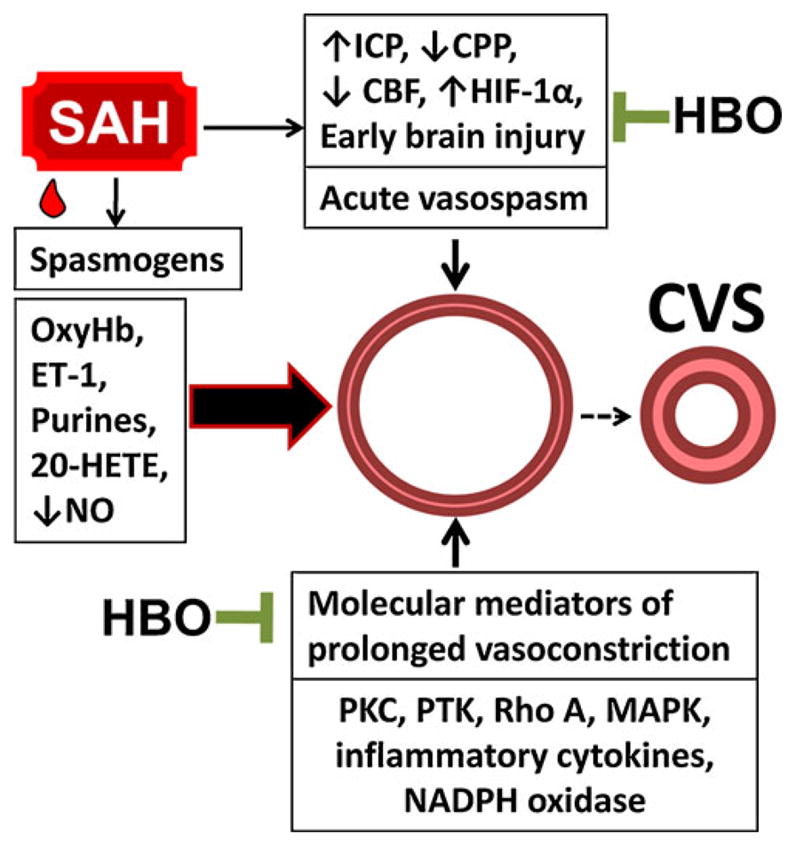
Molecular mechanisms of HBO treatment in SAH and cerebral vasospasm. Effects of HBO include countering activation of protein kinases, suppressing inflammatory mediators, and reducing oxidative stress. By targeting these mechanisms, HBO may ameliorate SAH-induced early brain injury and antagonize CVS
Oxidative Stress After SAH
The reactive oxygen species (ROS) produced from enzymatic sources and formed during hemoglobin autoxidation constitute a major etiologic factor underlying the development of cerebral vasospasm after SAH. Concordantly, antioxidants have been shown to attenuate arterial narrowing in experimental SAH produced by autologous blood injection [50, 51]. There have been several mechanisms proposed to explain how ROS may contribute to cerebral vasospasm after SAH. ROS oxidize bilirubin to bilirubin oxidation products (BOXes) [49]. BOXes inhibit eNOS and, due to the reduced availability of NO, impair vasodilation mechanism. Nitric oxide is a major vasodilator produced principally by endothelial cells and acts through the stimulation of guanyl cyclase that produces cyclic guanosine monophosphate with subsequent dephosphorylation of myosin light chains [52]. Therefore, the endothelial dysfunction and, more so, injury resulting in the reduction of NO level may contribute to vascular constriction after SAH. In addition, reactive oxygen species, together with thrombin and clotting cascade components, stimulate the production of 20-hydroxy-eicosatetraenoic acid, a vasoconstrictor metabolite of arachidonic acid which blocks calcium-activated potassium channels, thereby leading to a decrease in cerebral blood flow after SAH [53].
One of the major sources of free radicals after subarachnoid bleeding is NADPH oxidase-producing superoxide. The superoxide radical can combine with nitric oxide to form peroxynitrite, which targets nitric oxide synthase (NOS) [54]. HBO (2.8 ATA for 2 h) has been shown to inhibit NADPH oxidase activity/expression and reduce the level of lipid peroxidation products in the cerebral tissues after SAH [55]. The pharmacological inhibitors of NADPH oxidase, diphenyleneiodonium and apocynin, potently reduced cerebral vasospasm in the rat model of SAH [56, 57]. However, the specific effect of HBO on NADPH oxidase in vascular tissues awaits investigation.
The reduced bioavailability of NO can be also caused by the negative regulation of NOS by PKC activated after SAH [58]. Furthermore, the depletion of nitric oxide occurs due to scavenging by hemoglobin (sink effect) and the damage of nitric oxide-secreting cells in the vascular adventitia [11]. As shown by the studies of pulmonary circulation, HBO can attenuate vascular constriction by inducing extra-endothelial nitric oxide production [59]. Microdialysis analysis revealed that HBO induced nitric oxide production in brains of experimental animals, which may result in the elevated CBF upon prolonged HBO exposure [60–62].
Although HBO appears to have a beneficial impact on oxidative stress after SAH, it has been shown to amplify iron-induced brain edema in the setting of experimental intracerebral hemorrhage [63]. These finding may suggest that HBO enhances oxidative stress in the presence of lysed blood clot in the cerebral tissues. Therefore, the use of HBO should be considered cautiously in patients with SAH extending into brain parenchyma.
Anti-apoptosis for Cerebral Vasospasm
SAH triggers a cascade of molecular events leading to endothelial apoptosis [64]. The expression of active caspase-3 and the presence of DNA strand breaks have been detected in endothelial cells of spastic cerebral arteries after experimental SAH [64]. Electron microscopic investigations revealed apoptotic changes in endothelial cells of cerebral arteries collected from a patient who died after suffering severe CVS caused by aneurysm rupture [65]. The loss of endothelial cells producing nitric oxide can affect the fragile balance between vasoconstrictors and vasodilators acting on the vascular wall. Studies have shown that the endothelial injury after SAH is associated with inflammatory mediators. Tumor necrosis factor α (TNF-α) and interleukin-1β (IL-1β) have been shown to exert a pro-apoptotic effect on cultured cerebral microvascular endothelial cells [66]. In the experimental settings, caspase inhibitors have been shown to attenuate CVS after SAH and to reduce levels of inflammatory mediators including IL-1β [67]. Other studies further corroborated these results by showing that treatment with a broad caspase inhibitor decreased TNF-α expression in arterial wall and attenuated CVS [64]. The beneficial effect of HBO on apoptosis in the brain after SAH has been demonstrated by one group of investigators. HBO (2.8 ATA for 2 h) given at 1 h after SAH reduced neuronal apoptosis and diminished BBB disruption, which suggests both neuroprotection and reduced damage to the endothelial cells [13]. Even though this study did not investigate the specific effect of HBO on endothelial apoptosis after SAH, the results from different experimental systems seem to be favorable. In the ischemic wound model, the cleaved caspase-3 expressed predominantly in the endothelial cells within injury significantly decreased after HBOT (2.4 ATA for 90 min daily for 14 days) [68]. In addition, one research group demonstrated that in human microvascular endothelial cells, HBO (2.4 ATA for 1 h) activated genes encoding antioxidant and detoxifying enzymes and factors essential for endothelial cell survival [69]. Another laboratory investigation has found that even 15 min of HBO exposure (2.4 ATA) potently stimulated the proliferation of cultured endothelial cells [70], which might also indicate the potential of HBO for regeneration of injured endothelium in the spastic cerebral arteries.
When the layer of endothelial cells is damaged, the underlying vascular smooth muscle cells are exposed to intraluminally acting spasmogens like ET-1. In addition, ET-1 released by invading mononuclear leukocytes may exert a potent vasoconstrictive effect abluminally. Therefore, studies of vasospasm antagonism with HBO treatment should evaluate several possible mechanisms, including protection against endothelial cell loss, anti-inflammation, as well as preservation of nitric oxide-releasing neurons in the adventitia of conductive arteries targeted by oxyhemoglobin after SAH [71].
Inflammation and Cerebral Vasospasm
Brain inflammation plays an important role in the development of cerebral vasospasm after SAH [72, 73]. The extravasated blood induces local inflammatory reaction in the closest vicinity of cerebral arteries. In this setting, cerebral vasospasm develops in response to spasmogens such as ET-1 released from invading leukocytes [74] or thromboxane A2 and serotonin, both released from platelets [75, 76]. It has been demonstrated that SAH increases the tissue expression of several inflammatory mediators, including intercellular adhesion molecule 1 (ICAM-1) and TNFα, in proximity to extravasated blood [77]. From clinical studies and laboratory investigations comes an indication that HBO treatment carries a potential to attenuate cerebral sequelae of vasospasm via its anti-inflammatory properties. HBO has been shown to reduce E-selectin blood levels in patients undergoing cardiopulmonary bypass [78]. This soluble cell adhesion molecule plays a principal role in the development of cerebral vasospasm [79]. HBO may also target other proteins participating in cell adhesion that have been implicated in the mechanisms of CVS. Here belong ICAM-1, CD18, and L-selectin, all found elevated after SAH [80, 81]. In the endothelial cell injury model in vitro, HBO reduced ICAM-1 level along with polymorphonuclear leukocyte adhesion. Increasing oxygen pressure showed an incremental suppression of ICAM-1 expression, with the strongest inhibition seen at 2.5 ATA for 90 min. This effect was mediated through the induction of eNOS [82]. In the setting of cerebral vasospasm, inhibition of adhesion molecules by HBO might reduce recruitment of leukocytes releasing spasmodic mediators into the perivascular space of cerebral arteries. Resident brain inflammatory cells, however, can also be kept in check with this treatment. It has been demonstrated that HBO reduced microgliosis and enhanced astroglial response to experimental ischemia, thus producing a brain protective effect by this mechanism [83].
HBO-Induced Neuroprotection in SAH
It has been postulated that the severity of the acute brain injury will have a profound impact on delayed vasospasm and long-term outcomes after subarachnoid hemorrhage [2]. Several MAPK—including ERK1/2, JNK, and p38, with known roles in the development of vasospasm—were found activated in both brain parenchyma and cerebral vascular tissues acutely after SAH [7]. One study found activated JNK in the cerebral vessels both on days 1 and 7 after SAH [47]. These findings may indicate that JNK activation is required but alone is not sufficient for the development of arterial spasm, which usually does not occur on day 1. Treatment with JNK inhibitor SP600125 reduced angiographic and morphological vasospasm after SAH [47]. Other authors recently demonstrated that HBO produced a lowering effect on JNK activation in the injured brain [84]. Collectively, these findings may suggest that it would be worthwhile to test the effect of HBO on the JNK MAPK pathway in the brain after SAH in the setting of ensuing CVS.
Acute Cerebral Ischemia
Except for minor leaks, SAH has a profound ischemic impact on the brain [85–87]. It has been clinically demonstrated that SAH decreases brain tissue oxygen pressure and pH [88]. The acute cerebral ischemia after SAH has a protective aspect as it can reduce bleeding into the subarachnoid space. However, if the ischemia is prolonged beyond several minutes, it can trigger an injurious cascade of molecular events orchestrated by hypoxia inducible factor-1 (HIF-1) [13, 89]. HIF-1 consists of regulatory α-subunit and constitutively expressed β-subunit (aka aryl hydrocarbon receptor nuclear translocator) [90]. The excessive activation of HIF-1 results in the overexpression of its target genes such as vascular endothelial growth factor (VEGF), responsible for increased BBB permeability, or BCL2/adenovirus E1B 19-kDa protein-interacting protein 3 (BNIP3) and Nip3-like protein X, both mediating apoptosis [91]. Thus, the hypoxic brain injury at the onset of SAH can induce apoptosis of endothelial cells in large arteries by the activation of HIF-1α and BNIP3 [92]. In addition, the pro-apoptotic p53 protein, known to be stabilized by HIF-1α, has been found upregulated after SAH [93]. Since p53 was found elevated in the vasospastic basilar arteries, it has been suggested that p53 may play an important role in the etiology of vasospasm with relation to SAH [85]. The results of other studies may also lend support to the hypothesis that HIF-1α plays an important role in CVS mechanisms. Rats treated with the HIF-1α inhibitor, 2-methoxyestradiol (2ME2), showed reduced expression of VEGF, BNIP3, and proliferating cell nuclear antigen in the basilar arteries. This change was associated with attenuation of vasospasm in the basilar artery after SAH [94]. The same group showed that cerebral vasospasm can also be reduced through pharmacological inhibition of p53 [95].
In the rat endovascular perforation model of SAH, a single HBO treatment (2 h, 2.8 ATA) decreased the level of HIF-1α and its downstream genes, including BNIP3 and VEGF, in cerebral tissues, resulting in amelioration of brain injury and improved functional performance of experimental rats [13]. It is reasonable to hypothesize that p53 and several other HIF-1 downstream targets contributing to delayed vasospasm may be targeted by HBO treatment. Studies have shown that HBO can disrupt protein interactions between HIF-1α and p53 in the neonatal brain challenged by hypoxia [96].
The mechanisms responsible for the acute transient cerebral ischemia after SAH have been reviewed [6]. Given the complexity of these mechanisms, it appears that HBO could combat ischemia not only by increasing oxygen delivery. HBO possesses fibrynolytic properties [97] which could oppose intravascular blood clotting associated with cerebral microcirculation compromise after SAH. The coagulation disturbances develop the moment blood reaches the subarachnoid space [97, 98]. HBO has also been suggested to speed up heme breakdown owing to the induction of heme oxygenase-1 in different experimental systems [99, 100].
Cortical Spreading Depression
The aggravation of ischemic brain injury after SAH can occur due to cortical spreading depression (CSD) characterized by periodically generated waves of cortical depolarization accompanied by decreases in CBF. The presence of both events has been detected after SAH [101]. Their propagation in the cerebral cortex may exacerbate ischemic lesions formed as a result of arterial narrowing. The microarterial spasm and cell necrosis caused by CSD may further aggravate the neurological status of SAH patients [102]. Several factors inducing CSD have been identified in experimental studies including ET-1, oxyhemoglobin, and potassium ions [103]. In addition, one of the major triggers for CSD is a decrease in activity of Na+/K+-ATPase. It has been demonstrated that even its incomplete functional inactivation can cause SD-like anoxic depolarization in the hippocampus [104]. Interestingly, SAH decreases Na+/K+-ATPase activity in the synaptosomal membranes, collected 2 h after the induction of subarachnoid hemorrhage [105]. In an attempt to preserve the enzymatic activity of Na+/K+-ATPase in experimental SAH, Yufu et al. [105] used HBO at 2 ATA for 1 h, started at 30 min after hemorrhage induction. HBO significantly ameliorated a decrease in Na+/K+-ATPase activity, which allowed authors to suggest that HBO may be considered as a beneficial treatment for subarachnoid hemorrhage.
Discussion and Future Perspectives
In this paper, we have reviewed the clinical use of HBO for SAH and outlined the underlying molecular mechanisms investigated for the most part in neuroscience laboratories. The lack of long-term outcomes appears to be the major shortcoming of laboratory investigations. Although randomization was performed in all experimental studies, blinded evaluation was reported only in one instance. The negative studies were not available either for clinical or laboratory investigations, which may suggest that publication bias has occurred.
In addition, the translational significance of laboratory investigations of SAH remains uncertain. First, it is difficult to assess the impact of early brain injury on the development of vasospasm because animal models that accurately replicate both components are lacking [106]. The rodent and canine models producing vasospasm without early neurological deficits do not resemble human SAH where acute brain injury is always present. On the other hand, rat models of early brain injury after SAH can induce only a mild vasospasm inasmuch as the acute brain injury will rather activate molecular factors predisposing vascular tissues to contraction. The new animal model that combines early brain injury and cerebral vasospasm could include endovascular perforation or transient cerebral ischemia followed by a controlled blood injection. The characteristics of vasospasm in such model could be compared to those in blood injection models of SAH in order to dissect the effect of early brain injury on cerebral vasospasm.
There is also a lot of uncertainty about how HBO targets the mechanisms of SAH-induced brain injury and CVS at the molecular level. Although it has been suggested that HBO reduces early brain injury through the downregulation of HIF-1α and its targets [13], a recent paper has shown that elevated brain level of HIF-1α by deferoxamine treatment was associated with reduced arterial narrowing after experimental SAH [89]. However, as mentioned earlier, it has been demonstrated that the HIF-1α inhibitor, 2ME2, was able to reduce cerebral vasospasm [94]. These somewhat conflicting data make it difficult to draw conclusions on the role of HIF-1α in cerebral vasospasm and to establish the optimal regimen of treatment. In addition, while a single HBO session has been shown to reduce HIF-1α levels in the brain [107], repeatedly administered HBO can increase HIF-1α levels in cerebral tissues [108], possibly due to inhibitory action on prolyl hydroxylases (Fig. 2) [109, 110]. Hence, the induction of HIF-1α has been proposed as a major mechanism in HBO preconditioning [108]. Additionally, the role of HIF-1 in brain injury appears to be two-faceted, depending upon the level of its activation [111, 112]. HIF-1 inhibitor study used an endovascular perforation model of SAH that results in highly elevated HIF-1α levels. In such setting, 2ME2 might reduce excessive HIF-1 activation with a subsequent downregulation of pro-apoptotic genes such as BNIP3. On the other hand, deferoxamine study used a blood injection model which by itself causes only a minute increase in the brain stem levels of HIF-1α. In this particular model, deferoxamine could trigger adaptive HIF-1α response, which, through the induction of anti-vasospasm genes like erythropoietin (Epo), attenuated cerebral vascular spasm [89, 113].
Fig. 2.
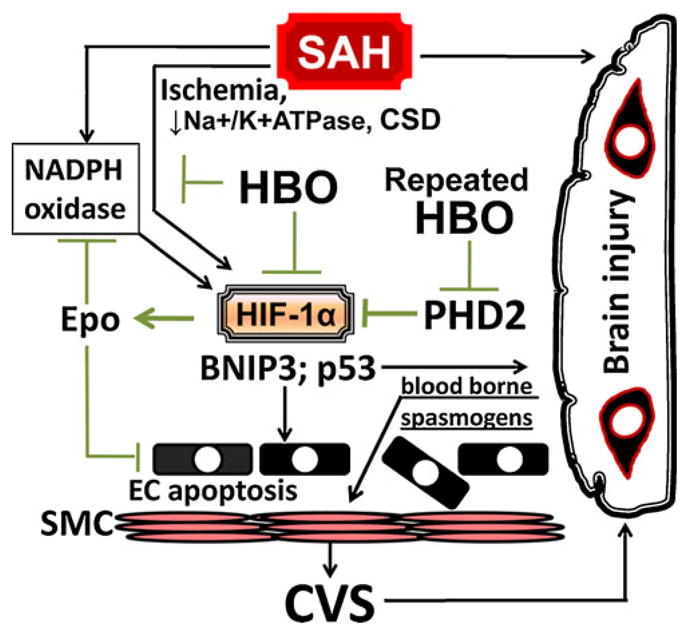
HBO protects against SAH-induced early brain injury. The major effects of HBO include suppression of oxidative stress and downregulation of HIF-1 target genes (BCL2/adenovirus E1B 19-kDa protein-interacting protein 3, vascular endothelial growth factor and p53), which collectively leads to reduced neurovascular injury. In addition, the repeated HBO sessions can induce HIF-1α adaptive genes like erythropoietin, capable of attenuating endothelial apoptosis and reducing vascular contraction
One of the gaps in our current knowledge includes the effects of high partial pressures of oxygen on the ionic channels involved in the prolonged vascular smooth muscle contraction that remain poorly understood. After SAH, both potassium and voltage-dependent calcium channels become dysfunctional [114, 115]. Studies suggest that the increased activity of calcium channels and the suppression of potassium channels may play a role in arterial spasm. Ishiguro et al. [116] have found that oxyhemoglobin suppresses voltage-dependent potassium channels in the rabbit cerebral arteries. Of note is that there is little change in the expression of calcium-activated potassium channels after SAH in laboratory dogs, whereas a significant decrease in voltage-gated potassium channels occurs in the arterial smooth muscle cells due to the impact of the oxyhemoglobin [117].
Hyperbaric oxygen treatment can cause opening of mitochondrial ATP-sensitive potassium channels, thus inducing a neuroprotective effect in the brain targeted by ischemia [118]. The same phenomenon underlies the protective mechanism triggered by oxygen treatment of myocardial injury [119]. However, interactions between oxygen and the ionic channels of cerebral vascular smooth muscle cells after SAH await investigation. To this end, it would be worthwhile to examine the effects of HBO on the capacitative calcium entry, which is one of the mechanisms of prolonged smooth muscle contraction. Capacitative calcium entry follows the depletion of intracellular calcium stores, which activates calcium-permeable store-operated channels (SOC) in the plasma membrane [120]. Zuccarello [121], trying to decipher the mechanism underlying the maintenance of chronic spasm, first suggested that the entry of extracellular calcium into smooth muscle cells may in part be mediated by SOC. Thus, in-depth studies of ionic currents upon HBO treatment are warranted.
Recent reports may suggest yet several other research directions for applications of HBO in the acute brain injury and cerebral vasospasm after SAH. Studies have shown that HBO treatment can diminish the activation of matrix metalloproteinases (MMPs) in the brain. Vetkamp et al. [122] showed reduced serum MMP-9 levels associated with BBB protection following HBO treatment after focal cerebral ischemia. Interestingly, raised serum level of MMP-9 is an important predictor of cerebral vasospasm after aneurysm rupture [123]. Furthermore, Ostrowski et al. [124] showed that preconditioning with HBO reduced ischemic MMP-9 activation. Although the effect of HBO on MMP-9 in SAH has not been specifically studied, the same research group found protection of the BBB with this treatment [13]. Therefore, further studies of HBO on the neurovascular unit after SAH are warranted.
The rescue of vasospasm-induced neurological damage is the major challenge for which the preconditioning approach has been recently advocated [125]. Preconditioning could render cerebral tissues resilient to the delayed brain injury caused by vasospasm. However, hypoxia does not seem to be an optimal preconditioning modality due to a risk of inducing harm to vulnerable patients [126, 127]. In contrast, HBO offers a safer approach and may provide double benefit by combating ischemic brain injury and preconditioning cerebral tissues against the anticipated vasospasm (Fig. 3). Hypothetically, HBO could also precondition cerebral arteries and render them resistant to the detrimental impact of SAH. As demonstrated by Iadecola and colleagues, LPS preconditioning had a beneficial effect on the neurovascular function and ameliorated CBF in the ischemic territories after MCAO [128]. It is, however, unclear to what degree HBO can emulate the protective effect of vascular conditioning induced by other modalities. Recently, however, one research group found the upregulation of molecular chaperones and several genes of the nuclear factor E2-related factor 2 (Nrf2) pathway in human microvascular endothelial cells subjected to HBO under conditions that approximated clinical settings (2.4 atm for 60 min) [129]. Pathways other than HIF-1 or Nrf2 can also condition endothelial cells in response to HBO. For example, HBO induces VEGF in the endothelial cells through ERK, JNK, and c-Jun/AP-1 action, which has been implicated in stimulating angiogenesis [130]. JNK is an established mediator of preconditioning in a variety of experimental settings [131–133]. In summation, these data may suggest that HBO preconditioning is capable of stimulating neurovascular protection and repair of the endothelium in cerebral vasculature [129].
Fig. 3.
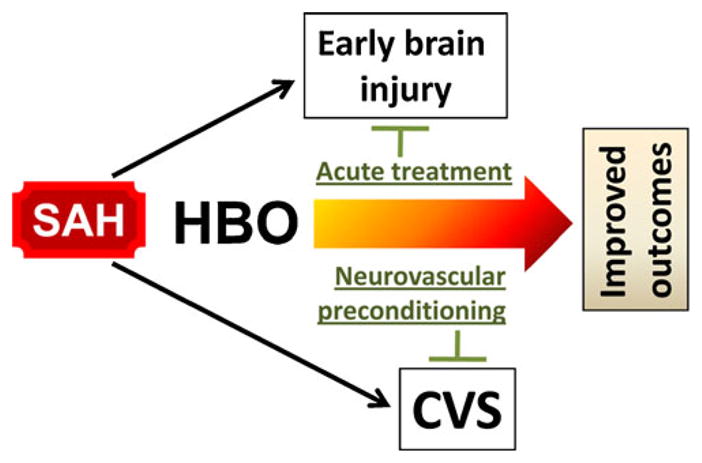
HBO preconditioning hypothetically for subarachnoid hemorrhage (SAH) and cerebral vasospasm. In addition to the treatment of SAH-induced brain injury, hyperbaric oxygen (HBO) is proposed that could be used as a preconditioning modality against anticipated CVS. The induction of preconditioning effect may require repeated HBO exposure, which needs to be taken into consideration when devising treatment regimen
Conclusions
Recently, prominent SAH investigators advocated a “new world of thought” for understanding the mechanism of cerebral vasospasm [11]. The proposed novel therapeutic targets include early brain injury and cortical spreading depression, which contribute to the mortality and morbidity after SAH. In that regard, HBO is a reasonable treatment option with a proven ability to ameliorate early brain injury after SAH. The major hurdle in preclinical SAH research is the lack of animal models that combine acute brain injury and cerebral vasospasm [106]. Consequently, HBO has been evaluated either exclusively in the acute phase of brain injury or after the induction of cerebral vasospasm. Noticeably though, HBO treatment appears to be beneficial in both stages of SAH-induced brain injury.
In summary, the beneficial effects of HBO in SAH may occur through: (1) ameliorating acute brain ischemia and injury to the cerebral vessels, (2) preconditioning cerebral and vascular tissues before cerebral vasospasm develops, and (3) reducing neurological deficits caused by vasospasm. However, due to the paucity of relevant clinical and experimental studies, the role of HBO in the treatment of cerebral vasospasm remains elusive. Further studies are required to determine whether HBO exerts an antagonizing effect on CVS and to better understand the mechanism of HBO treatment in SAH.
Acknowledgments
This study is partially supported by grants from National Institutes of Health: NS53407, NS43338, and HD43120 to J. H. Zhang.
Footnotes
Conflict of Interest The authors declare no conflict of interest pertaining to the submitted work.
Contributor Information
Robert P. Ostrowski, Department of Physiology and Pharmacology, Loma Linda University, 11041 Campus Street, Loma Linda, CA 92350, USA
John H. Zhang, Email: Johnzhang3910@yahoo.com, Department of Physiology and Pharmacology, Loma Linda University, 11041 Campus Street, Loma Linda, CA 92350, USA. Department of Anesthesiology, Loma Linda University Medical Center, Loma Linda, CA 92350, USA. Department of Neurosurgery, Loma Linda University Medical Center, Loma Linda, CA 92350, USA
References
- 1.Keyrouz SG, Diringer MN. Clinical review: prevention and therapy of vasospasm in subarachnoid hemorrhage. Crit Care. 2007;11(4):220. doi: 10.1186/cc5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat Clin Pract Neurol. 2007;3(5):256–63. doi: 10.1038/ncpneuro0490. [DOI] [PubMed] [Google Scholar]
- 3.Macdonald RL, Kassell NF, Mayer S, Ruefenacht D, Schmiedek P, Weidauer S, et al. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke. 2008;39(11):3015–21. doi: 10.1161/STROKEAHA.108.519942. [DOI] [PubMed] [Google Scholar]
- 4.Vajkoczy P, Meyer B, Weidauer S, Raabe A, Thome C, Ringel F, et al. Clazosentan (AXV-034343), a selective endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study. J Neurosurg. 2005;103(1):9–17. doi: 10.3171/jns.2005.103.1.0009. [DOI] [PubMed] [Google Scholar]
- 5.Hansen-Schwartz J, Vajkoczy P, Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm: looking beyond vasoconstriction. Trends Pharmacol Sci. 2007;28(6):252–25. doi: 10.1016/j.tips.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Ostrowski RP, Colohan AR, Zhang JH. Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28(4):399–414. doi: 10.1179/016164106X115008. [DOI] [PubMed] [Google Scholar]
- 7.Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH. Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2004;24(8):916–25. doi: 10.1097/01.WCB.0000125886.48838.7E. [DOI] [PubMed] [Google Scholar]
- 8.Stuart RM, Helbok R, Kurtz P, Schmidt M, Fernandez L, Lee K, et al. High-dose intra-arterial verapamil for the treatment of cerebral vasospasm after subarachnoid hemorrhage: prolonged effects on hemodynamic parameters and brain metabolism. Neurosurgery. 2011 doi: 10.1227/NEU.0b013e318201be47. (in press) [DOI] [PubMed] [Google Scholar]
- 9.Zubkov AY, Rabinstein AA. Medical management of cerebral vasospasm: present and future. Neurol Res. 2009;31(6):626–31. doi: 10.1179/174313209X382331. [DOI] [PubMed] [Google Scholar]
- 10.Dankbaar JW, Slooter AJ, Rinkel GJ, Schaaf IC. Effect of different components of triple-H therapy on cerebral perfusion in patients with aneurysmal subarachnoid haemorrhage: a systematic review. Crit Care. 2010;14(1):R23. doi: 10.1186/cc8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pluta RM, Hansen-Schwartz J, Dreier J, Vajkoczy P, Macdonald RL, Nishizawa S, et al. Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurol Res. 2009;31 (2):151–8. doi: 10.1179/174313209X393564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kasuya H. Clinical trial of nicardipine prolonged-release implants for preventing cerebral vasospasm: multicenter cooperative study in Tokyo. Acta Neurochir Suppl. 2011;110(2):165–7. doi: 10.1007/978-3-7091-0356-2_30. [DOI] [PubMed] [Google Scholar]
- 13.Ostrowski RP, Colohan AR, Zhang JH. Mechanisms of hyperbaric oxygen-induced neuroprotection in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;25 (5):554–71. doi: 10.1038/sj.jcbfm.9600048. [DOI] [PubMed] [Google Scholar]
- 14.Kohshi K, Yokota A, Konda N, Kinoshita Y, Kajiwara H. Intracranial pressure responses during hyperbaric oxygen therapy. Neurol Med Chir (Tokyo) 1991;31(9):575–81. doi: 10.2176/nmc.31.575. [DOI] [PubMed] [Google Scholar]
- 15.Kohshi K, Yokota A, Konda N, Munaka M, Yasukouchi H. Hyperbaric oxygen therapy adjunctive to mild hypertensive hypervolemia for symptomatic vasospasm. Neurol Med Chir (Tokyo) 1993;33(2):92–9. doi: 10.2176/nmc.33.92. [DOI] [PubMed] [Google Scholar]
- 16.Isakov I, Pravdenkova SV, Shchelkovskii VN. Hyperbaric oxygenation in ruptured cerebral aneurysms during the postoperative period. Zh Vopr Neirokhir Im NN Burdenko. 1985;(3):17–21. [PubMed] [Google Scholar]
- 17.Levina OA, Romasenko MV, Krylov V. Therapeutic effects of hyperbaric oxygenation (HBO) on acute cerebral ischemia in patients after intracranial aneurysms clipping. Eur J Underw Hyperb Med. 2002;3:83. [Google Scholar]
- 18.Ohta H, Suzuki E, Hinuma Y, Kawamura S, Nemoto M, Hadeishi H. Effects of hyperoxia, glycerol and ventricular drainage on ICP and CBF in patients with increased ICP due to CSF circulatory-absorbance disturbance. No To Shinkei. 1987;39(3):273–9. [PubMed] [Google Scholar]
- 19.Kawamura S, Ohta H, Yasui N, Nemoto M, Hinuma Y, Suzuki E. Effects of hyperbaric oxygenation in patients with subarachnoid hemorrhage. J Hyperb Med. 1988;3:243–56. [Google Scholar]
- 20.Ugriumov VM, Elinskii MP, Rafikov AM, Kesaev SA. Hyperbaric oxygenation in the complex treatment of patients with aneurysms of the cerebral vessels. Zh Vopr Neirokhir Im NN Burdenko. 1980;(4):49–54. [PubMed] [Google Scholar]
- 21.Kitaoka K, Nakagawa Y, Abe H, Satoh M, Iwakuma T. Hyperbaric oxygenation for the mental conditions following surgery of aneurysm of the anterior communicating artery. Hokkaido Igaku Zasshi. 1983;58(2):154–61. [PubMed] [Google Scholar]
- 22.Takata K, Sheng H, Borel CO, Laskowitz DT, Warner DS, Lombard FW. Simvastatin treatment duration and cognitive preservation in experimental subarachnoid hemorrhage. J Neurosurg Anesthesiol. 2009;21(4):326–33. doi: 10.1097/ANA.0b013e3181acfde7. [DOI] [PubMed] [Google Scholar]
- 23.Sen J, Belli A, Albon H, Morgan L, Petzold A, Kitchen N. Triple-H therapy in the management of aneurysmal subarachnoid haemorrhage. Lancet Neurol. 2003;2(10):614–21. doi: 10.1016/s1474-4422(03)00531-3. [DOI] [PubMed] [Google Scholar]
- 24.Walid SM, Zaytseva NV. Quadruple H therapy for vasospasm. Ann Indian Acad Neurol. 2009;12:22–4. doi: 10.4103/0972-2327.48847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mchedlishvili G, Varazashvili M, Mamaladze A, Momtselidze N. Blood flow structuring and its alterations in capillaries of the cerebral cortex. Microvasc Res. 1997;53(3):201–10. doi: 10.1006/mvre.1997.2012. [DOI] [PubMed] [Google Scholar]
- 26.Dhar R, Zazulia AR, Videen TO, Zipfel GJ, Derdeyn CP, Diringer MN. Red blood cell transfusion increases cerebral oxygen delivery in anemic patients with subarachnoid hemorrhage. Stroke. 2009;40(9):3039–44. doi: 10.1161/STROKEAHA.109.556159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kocaogullar Y, Ustun ME, Avci E, Karabacakoglu A, Fossett D. The role of hyperbaric oxygen in the management of subarachnoid hemorrhage. Intensive Care Med. 2004;30(1):141–6. doi: 10.1007/s00134-003-1916-7. [DOI] [PubMed] [Google Scholar]
- 28.Imperatore F, Cuzzocrea S, Luongo C, Liguori G, Scafuro A, De Angelis A, et al. Hyperbaric oxygen therapy prevents vascular derangement during zymosan-induced multiple-organ-failure syndrome. Intensive Care Med. 2004;30(6):1175–81. doi: 10.1007/s00134-003-2138-8. [DOI] [PubMed] [Google Scholar]
- 29.Huang ZX, Kang ZM, Gu GJ, Peng GN, Yun L, Tao HY, et al. Therapeutic effects of hyperbaric oxygen in a rat model of endothelin-1-induced focal cerebral ischemia. Brain Res. 2007;1153:204–13. doi: 10.1016/j.brainres.2007.03.061. [DOI] [PubMed] [Google Scholar]
- 30.Sharkey J, Butcher SP, Kelly JS. Endothelin-1 induced middle cerebral artery occlusion: pathological consequences and neuroprotective effects of MK801. J Auton Nerv Syst. 1994;49 (Suppl):S177–85. doi: 10.1016/0165-1838(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 31.Gaetani P, Baena R, Grignani G, Spanu G, Pacchiarini L, Paoletti P. Endothelin and aneurysmal subarachnoid haemorrhage: a study of subarachnoid cisternal cerebrospinal fluid. J Neurol Neurosurg Psychiatry. 1994;57(1):66–72. doi: 10.1136/jnnp.57.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansen-Schwartz J, Ansar S, Edvinsson L. Cerebral vasoconstriction after subarachnoid hemorrhage—role of changes in vascular receptor phenotype. Front Biosci. 2008;13:2160–4. doi: 10.2741/2831. [DOI] [PubMed] [Google Scholar]
- 33.Hansen-Schwartz J, Hoel NL, Zhou M, Xu CB, Svendgaard NA, Edvinsson L. Subarachnoid hemorrhage enhances endothelin receptor expression and function in rat cerebral arteries. Neurosurgery. 2003;52(5):1188–94. [PubMed] [Google Scholar]
- 34.Edvinsson L. Cerebrovascular endothelin receptor upregulation in cerebral ischemia. Curr Vasc Pharmacol. 2009;7(1):26–33. doi: 10.2174/157016109787354178. [DOI] [PubMed] [Google Scholar]
- 35.Rothoerl RD, Ringel F. Molecular mechanisms of cerebral vasospasm following aneurysmal SAH. Neurol Res. 2007;29 (7):636–42. doi: 10.1179/016164107X240224. [DOI] [PubMed] [Google Scholar]
- 36.Hirata Y, Yoshimi H, Takaichi S, Yanagisawa M, Masaki T. Binding and receptor down-regulation of a novel vasoconstrictor endothelin in cultured rat vascular smooth muscle cells. FEBS Lett. 1988;239(1):13–7. doi: 10.1016/0014-5793(88)80536-2. [DOI] [PubMed] [Google Scholar]
- 37.Lan C, Das D, Wloskowicz A, Vollrath B. Endothelin-1 modulates hemoglobin-mediated signaling in cerebrovascular smooth muscle via RhoA/Rho kinase and protein kinase C. Am J Physiol Heart Circ Physiol. 2004;286(1):H165–73. doi: 10.1152/ajpheart.00664.2003. [DOI] [PubMed] [Google Scholar]
- 38.Miao L, Dai Y, Zhang J. Mechanism of RhoA/Rho kinase activation in endothelin-1-induced contraction in rabbit basilar artery. Am J Physiol Heart Circ Physiol. 2002;283(3):H983–9. doi: 10.1152/ajpheart.00141.2002. [DOI] [PubMed] [Google Scholar]
- 39.Zhou C, Li Y, Nanda A, Zhang JH. HBO suppresses Nogo-A, Ng-R, or RhoA expression in the cerebral cortex after global ischemia. Biochem Biophys Res Commun. 2003;309(2):368–76. doi: 10.1016/j.bbrc.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 40.Sheth MV, Goodman BE, Friese JL, Eyster KM. Protein kinase and phosphatase activity in the lungs of normoxic versus hyperoxic rats. Exp Lung Res. 1997;23(6):475–94. doi: 10.3109/01902149709039239. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Wang Y, Kim HP, Choi AM, Ryter SW. FLIP inhibits endothelial cell apoptosis during hyperoxia by suppressing Bax. Free Radic Biol Med. 2007;42(10):1599–609. doi: 10.1016/j.freeradbiomed.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 42.Laher I, Zhang JH. Protein kinase C and cerebral vasospasm. J Cereb Blood Flow Metab. 2001;21(8):887–906. doi: 10.1097/00004647-200108000-00001. [DOI] [PubMed] [Google Scholar]
- 43.Koide M, Nishizawa S, Ohta S, Yokoyama T, Namba H. Chronological changes of the contractile mechanism in prolonged vasospasm after subarachnoid hemorrhage: from protein kinase C to protein tyrosine kinase. Neurosurgery. 2002;51(6):1468–74. [PubMed] [Google Scholar]
- 44.Tang H, Hao Q, Rutherford SA, Low B, Zhao ZJ. Inactivation of SRC family tyrosine kinases by reactive oxygen species in vivo. J Biol Chem. 2005;280(25):23918–25. doi: 10.1074/jbc.M503498200. [DOI] [PubMed] [Google Scholar]
- 45.Kusaka G, Kimura H, Kusaka I, Perkins E, Nanda A, Zhang JH. Contribution of Src tyrosine kinase to cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg. 2003;99(2):383–90. doi: 10.3171/jns.2003.99.2.0383. [DOI] [PubMed] [Google Scholar]
- 46.Yamaguchi M, Zhou C, Nanda A, Zhang JH. Ras protein contributes to cerebral vasospasm in a canine double-hemorrhage model. Stroke. 2004;35(7):1750–5. doi: 10.1161/01.STR.0000129898.68350.9f. [DOI] [PubMed] [Google Scholar]
- 47.Yatsushige H, Yamaguchi-Okada M, Zhou C, Calvert JW, Cahill J, Colohan AR, et al. Inhibition of c-Jun N-terminal kinase pathway attenuates cerebral vasospasm after experimental sub-arachnoid hemorrhage through the suppression of apoptosis. Acta Neurochir Suppl. 2008;104:27–31. doi: 10.1007/978-3-211-75718-5_6. [DOI] [PubMed] [Google Scholar]
- 48.Nosko M, Weir BK, Lunt A, Grace M, Allen P, Mielke B. Effect of clot removal at 24 hours on chronic vasospasm after SAH in the primate model. J Neurosurg. 1987;66(3):416–22. doi: 10.3171/jns.1987.66.3.0416. [DOI] [PubMed] [Google Scholar]
- 49.Crowley RW, Medel R, Kassell NF, Dumont AS. New insights into the causes and therapy of cerebral vasospasm following subarachnoid hemorrhage. Drug Discov Today. 2008;13(5–6):254–60. doi: 10.1016/j.drudis.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 50.Guney O, Erdi F, Esen H, Kiyici A, Kocaogullar Y. N-acetylcysteine prevents vasospasm after subarachnoid hemorrhage. Surg Neurol. 2010;73:42–9. doi: 10.1016/j.surneu.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 51.Munakata A, Ohkuma H, Shimamura N. Effect of a free radical scavenger, edaravone, on free radical reactions: related signal transduction and cerebral vasospasm in the rabbit subarachnoid hemorrhage model. Acta Neurochir Suppl. 2011;110(2):17–22. doi: 10.1007/978-3-7091-0356-2_4. [DOI] [PubMed] [Google Scholar]
- 52.Nakamura K, Koga Y, Sakai H, Homma K, Ikebe M. cGMP-dependent relaxation of smooth muscle is coupled with the change in the phosphorylation of myosin phosphatase. Circ Res. 2007;101(7):712–22. doi: 10.1161/CIRCRESAHA.107.153981. [DOI] [PubMed] [Google Scholar]
- 53.Kehl F, Cambj-Sapunar L, Maier KG, Miyata N, Kametani S, Okamoto H, et al. 20-HETE contributes to the acute fall in cerebral blood flow after subarachnoid hemorrhage in the rat. Am J Physiol Heart Circ Physiol. 2002;282(4):H1556–65. doi: 10.1152/ajpheart.00924.2001. [DOI] [PubMed] [Google Scholar]
- 54.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87(1):315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ostrowski RP, Tang J, Zhang JH. Hyperbaric oxygen suppresses NADPH oxidase in a rat subarachnoid hemorrhage model. Stroke. 2006;37(5):1314–8. doi: 10.1161/01.STR.0000217310.88450.c3. [DOI] [PubMed] [Google Scholar]
- 56.Kim DE, Suh YS, Lee MS, Kim KY, Lee JH, Lee HS, et al. Vascular NAD(P)H oxidase triggers delayed cerebral vasospasm after subarachnoid hemorrhage in rats. Stroke. 2002;33 (11):2687–91. doi: 10.1161/01.str.0000033071.99143.9e. [DOI] [PubMed] [Google Scholar]
- 57.Zheng JS, Zhan RY, Zheng SS, Zhou YQ, Tong Y, Wan S. Inhibition of NADPH oxidase attenuates vasospasm after experimental subarachnoid hemorrhage in rats. Stroke. 2005;36 (5):1059–64. doi: 10.1161/01.STR.0000163102.49888.b7. [DOI] [PubMed] [Google Scholar]
- 58.Matsubara M, Hayashi N, Jing T, Titani K. Regulation of endothelial nitric oxide synthase by protein kinase C. J Biochem. 2003;133(6):773–81. doi: 10.1093/jb/mvg099. [DOI] [PubMed] [Google Scholar]
- 59.Hink J, Thom SR, Simonsen U, Rubin I, Jansen E. Vascular reactivity and endothelial NOS activity in rat thoracic aorta during and after hyperbaric oxygen exposure. Am J Physiol Heart Circ Physiol. 2006;291(4):H1988–98. doi: 10.1152/ajpheart.00145.2006. [DOI] [PubMed] [Google Scholar]
- 60.Elayan IM, Axley MJ, Prasad PV, Ahlers ST, Auker CR. Effect of hyperbaric oxygen treatment on nitric oxide and oxygen free radicals in rat brain. J Neurophysiol. 2000;83(4):2022–9. doi: 10.1152/jn.2000.83.4.2022. [DOI] [PubMed] [Google Scholar]
- 61.Chavko M, Braisted JC, Outsa NJ, Harabin AL. Role of cerebral blood flow in seizures from hyperbaric oxygen exposure. Brain Res. 1998;791(1–2):75–82. doi: 10.1016/s0006-8993(98)00083-3. [DOI] [PubMed] [Google Scholar]
- 62.Toda N, Ayajiki K, Okamura T. Cerebral blood flow regulation by nitric oxide: recent advances. Pharmacol Rev. 2009;61(1):62–97. doi: 10.1124/pr.108.000547. [DOI] [PubMed] [Google Scholar]
- 63.Qin Z, Xi G, Keep RF, Silbergleit R, He Y, Hua Y. Hyperbaric oxygen for experimental intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:113–7. doi: 10.1007/978-3-211-09469-3_23. [DOI] [PubMed] [Google Scholar]
- 64.Zhou C, Yamaguchi M, Kusaka G, Schonholz C, Nanda A, Zhang JH. Caspase inhibitors prevent endothelial apoptosis and cerebral vasospasm in dog model of experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2004;24(4):419–31. doi: 10.1097/00004647-200404000-00007. [DOI] [PubMed] [Google Scholar]
- 65.Zubkov AY, Ogihara K, Bernanke DH, Parent AD, Zhang J. Apoptosis of endothelial cells in vessels affected by cerebral vasospasm. Surg Neurol. 2000;53(3):260–6. doi: 10.1016/s0090-3019(99)00187-1. [DOI] [PubMed] [Google Scholar]
- 66.Kimura H, Gules I, Meguro T, Zhang JH. Cytotoxicity of cytokines in cerebral microvascular endothelial cell. Brain Res. 2003;990(1–2):148–56. doi: 10.1016/s0006-8993(03)03450-4. [DOI] [PubMed] [Google Scholar]
- 67.Iseda K, Ono S, Onoda K, Satoh M, Manabe H, Nishiguchi M, et al. Antivasospastic and antiinflammatory effects of caspase inhibitor in experimental subarachnoid hemorrhage. J Neurosurg. 2007;107(1):128–35. doi: 10.3171/JNS-07/07/0128. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Q, Chang Q, Cox RA, Gong X, Gould LJ. Hyperbaric oxygen attenuates apoptosis and decreases inflammation in an ischemic wound model. J Invest Dermatol. 2008;128(8):2102–12. doi: 10.1038/jid.2008.53. [DOI] [PubMed] [Google Scholar]
- 69.Godman CA, Joshi R, Giardina C, Perdrizet G, Hightower LE. Hyperbaric oxygen treatment induces antioxidant gene expression. Ann NY Acad Sci. 2010;1197:178–83. doi: 10.1111/j.1749-6632.2009.05393.x. [DOI] [PubMed] [Google Scholar]
- 70.Tompach PC, Lew D, Stoll JL. Cell response to hyperbaric oxygen treatment. Int J Oral Maxillofac Surg. 1997;26(2):82–6. doi: 10.1016/s0901-5027(05)80632-0. [DOI] [PubMed] [Google Scholar]
- 71.Pluta RM. Dysfunction of nitric oxide synthases as a cause and therapeutic target in delayed cerebral vasospasm after SAH. Acta Neurochir Suppl. 2008;104:139–47. doi: 10.1007/978-3-211-75718-5_28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dumont AS, Dumont RJ, Chow MM, Lin CL, Calisaneller T, Ley KF, et al. Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery. 2003;53 (1):123–33. doi: 10.1227/01.neu.0000068863.37133.9e. [DOI] [PubMed] [Google Scholar]
- 73.Sozen T, Tsuchiyama R, Hasegawa Y, Suzuki H, Jadhav V, Nishizawa S, et al. Role of interleukin-1beta in early brain injury after subarachnoid hemorrhage in mice. Stroke. 2009;40 (7):2519–25. doi: 10.1161/STROKEAHA.109.549592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fassbender K, Hodapp B, Rossol S, Bertsch T, Schmeck J, Schutt S, et al. Endothelin-1 in subarachnoid hemorrhage: an acute-phase reactant produced by cerebrospinal fluid leukocytes. Stroke. 2000;31(12):2971–5. doi: 10.1161/01.str.31.12.2971. [DOI] [PubMed] [Google Scholar]
- 75.Takeuchi H, Tanabe M, Okamoto H, Yamazaki M. Effects of thromboxane synthetase inhibitor (RS-5186) on experimentally-induced cerebral vasospasm. Neurol Res. 1999;21(5):513–6. [PubMed] [Google Scholar]
- 76.Satoh S, Suzuki Y, Harada T, Ikegaki I, Asano T, Shibuya M, et al. The role of platelets in the development of cerebral vasospasm. Brain Res Bull. 1991;27(5):663–8. doi: 10.1016/0361-9230(91)90042-i. [DOI] [PubMed] [Google Scholar]
- 77.Prunell GF, Svendgaard NA, Alkass K, Mathiesen T. Inflammation in the brain after experimental subarachnoid hemorrhage. Neurosurgery. 2005;56(5):1082–92. [PubMed] [Google Scholar]
- 78.Alex J, Laden G, Cale AR, Bennett S, Flowers K, Madden L, et al. Pretreatment with hyperbaric oxygen and its effect on neuropsychometric dysfunction and systemic inflammatory response after cardiopulmonary bypass: a prospective randomized double-blind trial. J Thorac Cardiovasc Surg. 2005;130 (6):1623–30. doi: 10.1016/j.jtcvs.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 79.Lin CL, Dumont AS, Calisaneller T, Kwan AL, Hwong SL, Lee KS. Monoclonal antibody against E selectin attenuates subarachnoid hemorrhage-induced cerebral vasospasm. Surg Neurol. 2005;64(3):201–5. doi: 10.1016/j.surneu.2005.04.038. [DOI] [PubMed] [Google Scholar]
- 80.Bavbek M, Polin R, Kwan AL, Arthur AS, Kassell NF, Lee KS. Monoclonal antibodies against ICAM-1 and CD18 attenuate cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits. Stroke. 1998;29(9):1930–5. doi: 10.1161/01.str.29.9.1930. [DOI] [PubMed] [Google Scholar]
- 81.Polin RS, Bavbek M, Shaffrey ME, Billups K, Bogaev CA, Kassell NF, et al. Detection of soluble E-selectin, ICAM-1, VCAM-1, and L-selectin in the cerebrospinal fluid of patients after subarachnoid hemorrhage. J Neurosurg. 1998;89(4):559–67. doi: 10.3171/jns.1998.89.4.0559. [DOI] [PubMed] [Google Scholar]
- 82.Buras JA, Stahl GL, Svoboda KK, Reenstra WR. Hyperbaric oxygen downregulates ICAM-1 expression induced by hypoxia and hypoglycemia: the role of NOS. Am J Physiol Cell Physiol. 2000;278(2):C292–302. doi: 10.1152/ajpcell.2000.278.2.C292. [DOI] [PubMed] [Google Scholar]
- 83.Gunther A, Kuppers-Tiedt L, Schneider PM, Kunert I, Berrouschot J, Schneider D, et al. Reduced infarct volume and differential effects on glial cell activation after hyperbaric oxygen treatment in rat permanent focal cerebral ischaemia. Eur J Neurosci. 2005;21(11):3189–94. doi: 10.1111/j.1460-9568.2005.04151.x. [DOI] [PubMed] [Google Scholar]
- 84.Liu JR, Zhao Y, Patzer A, Staak N, Boehm R, Deuschl G, et al. The JNK-inhibitor XG-102 enhances the neuroprotection of hyperbaric oxygen after cerebral ischaemia in adult rats. Neuropathol Appl Neurobiol. 2010;36:211–24. doi: 10.1111/j.1365-2990.2009.01047.x. [DOI] [PubMed] [Google Scholar]
- 85.Cahill J, Calvert JW, Solaroglu I, Zhang JH. Vasospasm and p53-induced apoptosis in an experimental model of subarachnoid hemorrhage. Stroke. 2006;37(7):1868–74. doi: 10.1161/01.STR.0000226995.27230.96. [DOI] [PubMed] [Google Scholar]
- 86.Kozniewska E, Michalik R, Rafalowska J, Gadamski R, Walski M, Frontczak-Baniewicz M, et al. Mechanisms of vascular dysfunction after subarachnoid hemorrhage. J Physiol Pharmacol. 2006;57 (Suppl 11):145–60. [PubMed] [Google Scholar]
- 87.Grote E, Hassler W. The critical first minutes after subarachnoid hemorrhage. Neurosurgery. 1988;22(4):654–61. doi: 10.1227/00006123-198804000-00006. [DOI] [PubMed] [Google Scholar]
- 88.Hoffman WE, Wheeler P, Edelman G, Charbel FT, Torres NJ, Ausman JI. Hypoxic brain tissue following subarachnoid hemorrhage. Anesthesiology. 2000;92(2):442–6. doi: 10.1097/00000542-200002000-00026. [DOI] [PubMed] [Google Scholar]
- 89.Hishikawa T, Ono S, Ogawa T, Tokunaga K, Sugiu K, Date I. Effects of deferoxamine-activated hypoxia-inducible factor-1 on the brainstem after subarachnoid hemorrhage in rats. Neurosurgery. 2008;62(1):232–40. doi: 10.1227/01.NEU.0000311082.88766.33. [DOI] [PubMed] [Google Scholar]
- 90.Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiol Bethesda. 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- 91.Schmidt-Kastner R, Aguirre-Chen C, Kietzmann T, Saul I, Busto R, Ginsberg MD. Nuclear localization of the hypoxia-regulated pro-apoptotic protein BNIP3 after global brain ischemia in the rat hippocampus. Brain Res. 2004;1001(1–2):133–42. doi: 10.1016/j.brainres.2003.11.065. [DOI] [PubMed] [Google Scholar]
- 92.Jeon H, Ai J, Sabri M, Tariq A, Shang X, Chen G, et al. Neurological and neurobehavioral assessment of experimental subarachnoid hemorrhage. BMC Neurosci. 2009;10:103. doi: 10.1186/1471-2202-10-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cahill J, Calvert JW, Marcantonio S, Zhang JH. p53 may play an orchestrating role in apoptotic cell death after experimental subarachnoid hemorrhage. Neurosurgery. 2007;60(3):531–45. doi: 10.1227/01.NEU.0000249287.99878.9B. [DOI] [PubMed] [Google Scholar]
- 94.Yan J, Chen C, Lei J, Yang L, Wang K, Liu J, et al. 2-Methoxyestradiol reduces cerebral vasospasm after 48 hours of experimental subarachnoid hemorrhage in rats. Exp Neurol. 2006;202(2):348–56. doi: 10.1016/j.expneurol.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 95.Yan JH, Yang XM, Chen CH, Hu Q, Zhao J, Shi XZ, et al. Pifithrin-alpha reduces cerebral vasospasm by attenuating apoptosis of endothelial cells in a subarachnoid haemorrhage model of rat. Chin Med J Engl. 2008;121(5):414–9. [PubMed] [Google Scholar]
- 96.Calvert JW, Cahill J, Yamaguchi-Okada M, Zhang JH. Oxygen treatment after experimental hypoxia–ischemia in neonatal rats alters the expression of HIF-1alpha and its downstream target genes. J Appl Physiol. 2006;101(3):853–65. doi: 10.1152/japplphysiol.00268.2006. [DOI] [PubMed] [Google Scholar]
- 97.Tjarnstrom J, Holmdahl L, Falk P, Falkenberg M, Arnell P, Risberg B. Effects of hyperbaric oxygen on expression of fibrinolytic factors of human endothelium in a simulated ischaemia/reperfusion situation. Scand J Clin Lab Invest. 2001;61(7):539–45. doi: 10.1080/003655101753218300. [DOI] [PubMed] [Google Scholar]
- 98.Sehba FA, Mostafa G, Friedrich V, Jr, Bederson JB. Acute microvascular platelet aggregation after subarachnoid hemorrhage. J Neurosurg. 2005;102(6):1094–100. doi: 10.3171/jns.2005.102.6.1094. [DOI] [PubMed] [Google Scholar]
- 99.Padgaonkar VA, Giblin FJ, Fowler K, Leverenz VR, Reddan JR, Dziedzic DC. Heme oxygenase synthesis is induced in cultured lens epithelium by hyperbaric oxygen or puromycin. Exp Eye Res. 1997;65(3):435–43. doi: 10.1006/exer.1997.0356. [DOI] [PubMed] [Google Scholar]
- 100.Chang KY, Tsai PS, Huang TY, Wang TY, Yang S, Huang CJ. HO-1 mediates the effects of HBO pretreatment against sepsis. J Surg Res. 2006;136(1):143–53. doi: 10.1016/j.jss.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 101.Pearl JD, Macdonald RL. Vasospasm after aneurysmal subarachnoid hemorrhage: need for further study. Acta Neurochir Suppl. 2008;105:207–10. doi: 10.1007/978-3-211-09469-3_39. [DOI] [PubMed] [Google Scholar]
- 102.Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain. 2009;132(Pt 7):1866–81. doi: 10.1093/brain/awp102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kleeberg J, Petzold GC, Major S, Dirnagl U, Dreier JP. ET-1 induces cortical spreading depression via activation of the ETA receptor/phospholipase C pathway in vivo. Am J Physiol Heart Circ Physiol. 2004;286(4):H1339–46. doi: 10.1152/ajpheart.00227.2003. [DOI] [PubMed] [Google Scholar]
- 104.Balestrino M, Young J, Aitken P. Block of (Na+,K+)ATPase with ouabain induces spreading depression-like depolarization in hippocampal slices. Brain Res. 1999;838(1–2):37–44. doi: 10.1016/s0006-8993(99)01674-1. [DOI] [PubMed] [Google Scholar]
- 105.Yufu K, Itoh T, Edamatsu R, Mori A, Hirakawa M. Effect of hyperbaric oxygenation on the Na+, K(+)-ATPase and membrane fluidity of cerebrocortical membranes after experimental subarachnoid hemorrhage. Neurochem Res. 1993;18(9):1033–9. doi: 10.1007/BF00966765. [DOI] [PubMed] [Google Scholar]
- 106.Titova E, Ostrowski RP, Zhang JH, Tang J. Experimental models of subarachnoid hemorrhage for studies of cerebral vasospasm. Neurol Res. 2009;31(6):568–81. doi: 10.1179/174313209X382412. [DOI] [PubMed] [Google Scholar]
- 107.Li Y, Zhou C, Calvert JW, Colohan AR, Zhang JH. Multiple effects of hyperbaric oxygen on the expression of HIF-1 alpha and apoptotic genes in a global ischemia–hypotension rat model. Exp Neurol. 2005;191(1):198–210. doi: 10.1016/j.expneurol.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 108.Gu GJ, Li YP, Peng ZY, Xu JJ, Kang ZM, Xu WG, et al. Mechanism of ischemic tolerance induced by hyperbaric oxygen preconditioning involves upregulation of hypoxia-inducible factor-1alpha and erythropoietin in rats. J Appl Physiol. 2008;104(4):1185–91. doi: 10.1152/japplphysiol.00323.2007. [DOI] [PubMed] [Google Scholar]
- 109.Cash TP, Pan Y, Simon MC. Reactive oxygen species and cellular oxygen sensing. Free Radic Biol Med. 2007;43(9):1219–25. doi: 10.1016/j.freeradbiomed.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jones NM, Lee EM, Brown TG, Jarrott B, Beart PM. Hypoxic preconditioning produces differential expression of hypoxia-inducible factor-1alpha (HIF-1alpha) and its regulatory enzyme HIF prolyl hydroxylase 2 in neonatal rat brain. Neurosci Lett. 2006;404(1–2):72–7. doi: 10.1016/j.neulet.2006.05.049. [DOI] [PubMed] [Google Scholar]
- 111.Chen W, Ostrowski RP, Obenaus A, Zhang JH. Prodeath or prosurvival: two facets of hypoxia inducible factor-1 in perinatal brain injury. Exp Neurol. 2009;216(1):7–15. doi: 10.1016/j.expneurol.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Halterman MW, Miller CC, Federoff HJ. Hypoxia-inducible factor-1alpha mediates hypoxia-induced delayed neuronal death that involves p53. J Neurosci. 1999;19(16):6818–24. doi: 10.1523/JNEUROSCI.19-16-06818.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grasso G. An overview of new pharmacological treatments for cerebrovascular dysfunction after experimental subarachnoid hemorrhage. Brain Res Brain Res Rev. 2004;44(1):49–63. doi: 10.1016/j.brainresrev.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 114.Jahromi BS, Aihara Y, Ai J, Zhang ZD, Nikitina E, Macdonald RL. Voltage-gated K+ channel dysfunction in myocytes from a dog model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2008;28(4):797–811. doi: 10.1038/sj.jcbfm.9600577. [DOI] [PubMed] [Google Scholar]
- 115.Sobey CG. Cerebrovascular dysfunction after subarachnoid haemorrhage: novel mechanisms and directions for therapy. Clin Exp Pharmacol Physiol. 2001;28(11):926–9. doi: 10.1046/j.1440-1681.2001.03550.x. [DOI] [PubMed] [Google Scholar]
- 116.Ishiguro M, Morielli AD, Zvarova K, Tranmer BI, Penar PL, Wellman GC. Oxyhemoglobin-induced suppression of voltage-dependent K+ channels in cerebral arteries by enhanced tyrosine kinase activity. Circ Res. 2006;99(11):1252–60. doi: 10.1161/01.RES.0000250821.32324.e1. [DOI] [PubMed] [Google Scholar]
- 117.Jahromi BS, Aihara Y, Ai J, Zhang ZD, Weyer G, Nikitina E, et al. Temporal profile of potassium channel dysfunction in cerebrovascular smooth muscle after experimental subarachnoid haemorrhage. Neurosci Lett. 2008;440(1):81–6. doi: 10.1016/j.neulet.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lou M, Chen Y, Ding M, Eschenfelder CC, Deuschl G. Involvement of the mitochondrial ATP-sensitive potassium channel in the neuroprotective effect of hyperbaric oxygenation after cerebral ischemia. Brain Res Bull. 2006;69(2):109–16. doi: 10.1016/j.brainresbull.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 119.Colantuono G, Tiravanti EA, Di Venosa N, Cazzato A, Rastaldo R, Cagiano R, et al. Hyperoxia confers myocardial protection in mechanically ventilated rats through the generation of free radicals and opening of mitochondrial ATP-sensitive potassium channels. Clin Exp Pharmacol Physiol. 2008;35(1):64–71. doi: 10.1111/j.1440-1681.2007.04745.x. [DOI] [PubMed] [Google Scholar]
- 120.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85(2):757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 121.Zuccarello M, Boccaletti R, Tosun M, Rapoport RM. Role of extracellular Ca2+ in subarachnoid hemorrhage-induced spasm of the rabbit basilar artery. Stroke. 1996;27(10):1896–902. doi: 10.1161/01.str.27.10.1896. [DOI] [PubMed] [Google Scholar]
- 122.Veltkamp R, Bieber K, Wagner S, Beynon C, Siebing DA, Veltkamp C, et al. Hyperbaric oxygen reduces basal lamina degradation after transient focal cerebral ischemia in rats. Brain Res. 2006;1076(1):231–7. doi: 10.1016/j.brainres.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 123.McGirt MJ, Lynch JR, Blessing R, Warner DS, Friedman AH, Laskowitz DT. Serum von Willebrand factor, matrix metalloproteinase-9, and vascular endothelial growth factor levels predict the onset of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2002;51(5):1128–34. doi: 10.1097/00006123-200211000-00005. [DOI] [PubMed] [Google Scholar]
- 124.Ostrowski RP, Jadhav V, Chen W, Zhang JH. Reduced matrix metalloproteinase-9 activity and cell death after global ischemia in the brain preconditioned with hyperbaric oxygen. Acta Neurochir Suppl. 2010;106:47–9. doi: 10.1007/978-3-211-98811-4_7. [DOI] [PubMed] [Google Scholar]
- 125.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8(4):398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Keep RF, Wang MM, Xiang J, Hua Y, Xi G. Is there a place for cerebral preconditioning in the clinic? Transl Stroke Res. 2010;1 (1):4–18. doi: 10.1007/s12975-009-0007-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gidday JM. Pharmacologic preconditioning: translating the promise. Transl Stroke Res. 2010;1(1):19–30. doi: 10.1007/s12975-010-0011-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kunz A, Park L, Abe T, Gallo EF, Anrather J, Zhou P, et al. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci. 2007;27(27):7083–93. doi: 10.1523/JNEUROSCI.1645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Godman CA, Chheda KP, Hightower LE, Perdrizet G, Shin DG, Giardina C. Hyperbaric oxygen induces a cytoprotective and angiogenic response in human microvascular endothelial cells. Cell Stress Chaperones. 2010;15:431–42. doi: 10.1007/s12192-009-0159-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee CC, Chen SC, Tsai SC, Wang BW, Liu YC, Lee HM, et al. Hyperbaric oxygen induces VEGF expression through ERK, JNK and c-Jun/AP-1 activation in human umbilical vein endothelial cells. J Biomed Sci. 2006;13(1):143–56. doi: 10.1007/s11373-005-9037-7. [DOI] [PubMed] [Google Scholar]
- 131.Sato H, Bolli R, Rokosh GD, Bi Q, Dai S, Shirk G, et al. The cardioprotection of the late phase of ischemic preconditioning is enhanced by postconditioning via a COX-2-mediated mechanism in conscious rats. Am J Physiol Heart Circ Physiol. 2007;293(4):H2557–64. doi: 10.1152/ajpheart.00858.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang N, Gao G, Bu X, Han S, Fang L, Li J. Neuron-specific phosphorylation of c-Jun N-terminal kinase increased in the brain of hypoxic preconditioned mice. Neurosci Lett. 2007;423 (3):219–24. doi: 10.1016/j.neulet.2007.07.028. [DOI] [PubMed] [Google Scholar]
- 133.Granziera C, Thevenet J, Price M, Wiegler K, Magistretti PJ, Badaut J, et al. Thrombin-induced ischemic tolerance is prevented by inhibiting c-Jun N-terminal kinase. Brain Res. 2007;1148:217–25. doi: 10.1016/j.brainres.2007.02.025. [DOI] [PubMed] [Google Scholar]