

Abstract
Olfactory sensory neurons, located in the nasal epithelium, detect and transmit odorant information to the central nervous system. This requires that these neurons form specific neuronal connections within the olfactory bulb and express receptors and signaling molecules specific for these functions. This protocol describes a primary olfactory sensory neuron culture technique that allows in vitro investigation of olfactory sensory neuron differentiation, axon outgrowth, odorant receptor expression and function. Olfactory epithelium is obtained from the nasal cavity and enzymatically treated to reduce stroma tissue. Dissociated olfactory sensory neurons are cultured directly on a layer of cortical astrocytes to support their survival. Using this method, cultured olfactory sensory neurons maintain their bipolar morphology and express odorant signal transduction molecules which are specific for olfactory sensory neurons.
Keywords: Olfactory epithelium, bipolar, astrocytes, lentivirus, odorant receptor
UNIT INTRODUCTION
Olfactory sensory neurons (OSNs) are receptor neurons for odorant detection. There are many characteristics unique to OSNs including the genes they express, odorant detection specificity, regulation of neuronal differentiation and physiological properties. Although transgenic animal models provide an effective approach, an in vitro system is often needed for efficient genetic and pharmacological manipulations and monitoring gene functions (Mombaerts, 2006). It has been difficult to establish successful OSN cultures, particularly for mice, for which transgenic animals are available. The technical difficulty in culturing primary sensory neuron is inconsistent survival rate and aberrant differentiation of the cultured neurons. Thus to have successful OSN cultures these two parameters must be overcome. Obtaining enough viable cells is augmented when working with mice due to the smaller size of the structure, although this problem is often unavoidable, since the majority of transgenic models are in this species.
Studies of OSN axon outgrowth require cultured primary neurons to survive at low density and have a similar bipolar morphology as they would in vivo. However, when OSNs are cultured on substrate coated coverslips at low density, they typically do not survive beyond 48 hours in serum-free media. Using OSN cultures to study gene function imposes additional challenges, as surviving neurons cannot be easily manipulated by commercial transfection reagents. The lack of a primary OSN culture technique has meant that odorant receptor function studies have primarily been studied in heterologous systems, in which co-expression of chaperones and signaling molecules to facilitate membrane targeting of odorant receptors and reconstitute signaling pathways is required (Zhuang and Matsunami, 2007).
This protocol describes a method to improve survival of OSNs by culturing directly on a layer of cortical astrocytes. In addition, the use of Waymouth’s medium with N2 supplement supports OSNs at low density that maintain their bipolar morphology. Under these conditions, olfactory sensory cultures can be maintained for as long as 10 days. Odorant receptors and major signaling molecules are expressed by these cultured OSNs. Olfactory marker protein expression can also be detected when cultures are maintained longer than 8 days.
BASIC PROTOCOL
Culture of Primary Mouse Olfactory Sensory Neurons
In this protocol, we describe techniques to isolate olfactory neuroepithelium, dissociate and culture OSNs. Olfactory epithelia with the underlining stroma are harvested from the nasal cavity by dissecting away the surrounding cartilage tissue. After enzymatic treatment, stroma is removed from the olfactory epithelia by microdissection. Olfactory epithelial pieces now consist of primarily OSNs, precursors and sustentacular cells. We refer to this tissue as olfactory neuroepithelium. OSNs are dissociated from the olfactory neuroepithelium and cultured on a feeder layer of astrocytes. The identities and morphologies of OSNs are validated by immunostaining.
Material List
C57BL/6 embryos at the desired age range (E15-E20)
Dissection tools:
1 51/2″ scissors
1 forceps
1 41/2″ fine scissors
2 No. 5 forceps
1 Micro scissors
70% ethanol
Culture prepared 12 mm glass coverslips (see support protocol 1 for preparation)
24 well plate
2.5% Avertin (see recipes)
10 cm sterile Petri dish
CMF-HBSS/HEPES (See recipes)
OSN Medium (see recipes)
Mild trypsinization solution (see recipes)
DNase I
Fetal Bovine Serum
Sterile glass Pasteur pipettes (flame polished and not polished)
15 ml Falcon tube
35mm culture dishes
Tabletop centrifuge
Stereo dissecting microscope
Microscope and Hemocytometer
Cortical astrocytes (see support protocol 2)
Horizontal Laminar Flow Culture Hood
Preparing for the Culture
Clean glass coverslips for astrocytes, see support protocol 1.
Cortical astrocytes were prepared and stored in frozen aliquots, see support protocol 2
At 7-10 days before the culture day, thaw an aliquot of astrocytes and plate 2 × 104 cells onto each coverslip in 24 well plates.
-
Astrocytes are maintained in Glia Medium, 500 μl per well, at 37°C with 5% CO2 until confluent.
Confluent astrocytes serve as a feeder layer and substrata to OSNs. They can be used as long as two weeks after becoming confluent without causing significant changes in the survival of OSNs
Make CMF-HBSS/HEPES and OSN Medium according to the recipes and store at 4°C.
Dissecting Olfactory Epithelium
6. Sterilize dissection tools (e.g. by soaking in 70% Ethanol for 20 minutes, take the tools out and put them in the culture hood to dry).
7. Anesthetize timed pregnant mouse with 2.5% Avertin via intraperitoneal injection (20 μl/g body weight).
-
8. Make sure that institutional and IACUC guidelines are followed for the selected method of anesthesia.
See Ayadi et al. (2011) for an overview on mouse breeding and additional background information.
-
9. When the pregnant female mouse is fully anesthetized, open the abdominal cavity by making a vertical cut along the midline of the abdominal wall. Take out the entire uterus with embryos; put in a 10 cm sterile Petri dish, cover and place the dish on ice. This procedure can be done on a clean bench.
From this step forward, all dissection steps must be performed in a horizontal laminar flow culture hood. Aseptic techniques must be strictly followed.
10. One embryo is taken out at a time. Decapitate the embryo and transfer the head to a clean 35mm dish filled with cold CMF-HBSS/HEPES.
11. Make a midline cut through the head, open and with the midline side facing up. Identify nasal cavity, which is located rostral and ventral to the olfactory bulb. The nasal septum will fall to one side. Use microscissors to dissect out the nasal epithelium and the surrounding cartilage (Figure 1A-B).
12. Under the dissecting microscope, dissect away the cartilage surrounding and underneath the olfactory epithelium and take out the nasal septum. This step can be omitted if longer Dispase treatment (see step 14 below) is applied (Figure 1C-E).
13. Pieces of olfactory epithelium (between 0.1 to 2mm in size) with the underlining stroma are collected and transferred to a clean dish with 2ml of CMF-HBSS/HEPES and kept on ice.
Figure 1.

Dissection and isolation of olfactory neuroepithelium. A. Dorsal view of a neonatal mouse head. A line through the midsagittal plan indicate where the cut is. B. Sagittal view of the opened nasal cavity. Nasal epithelium (OE) is circled. Olfactory bulb (OB) is located superior to the nasal cavity. C-D. Different views of the dissected nasal epithelia with cartilage tissue attached. Arrowhead in C points to nasal septum. E. Olfactory epithelium with underlining stroma. Arrowhead in E points to separated stroma, which appear opaque under the stereomicroscope. F. Isolated olfactory neuroepithelial appear transparent under the steromicroscope. Bar = 1mm in A and B; 1mm in C and D; 2mm in E and F.
Separating Olfactory Neuroepithelium
14. After olfactory epithelia are collected from the entire litter, add Dispase to a final concentration of 2 mg/ml to the dish where olfactory epithelial pieces are collected.
-
15. Incubate at room temperature for 40 min or longer.
Dispase is a gentle protease, which is used to loosen connective tissue. The duration of Dispase digestion needs to be defined empirically, as its enzymatic activities vary between lots. Extend the duration of Dispase digestion if needed.
-
16. Using forceps, transfer olfactory epithelial pieces to a different 35 mm dish filled with OSN medium and DNase I (500 U/ml). ).
OSN medium is used to support better survival of the olfactory epithelium through further dissection. DNase I is used to digest genomic DNA released from damaged cells. If olfactory epithelial pieces appear to be sticky and stringy, more DNase I may be added.
17. Using No. 5 fine tip forceps, carefully separate the nasal stroma underlining the olfactory neuroepithelium. The olfactory neuroepithelium is a layer of transparent tissue while the stroma appears more opaque and irregular (Figure 1E-F).
-
18. Transfer dissected olfactory neuroepithelial pieces to a new dish with OSN culture medium. Incubate at 37°C with 5% CO2 for 2-3 hours.
This step allows OSNs to migrate towards the surface of the pseudo-stratified olfactory epithelium. Movement of OSNs facilitates the following dissociation step. Two hours is the minimum time required for optimal dissociation of the olfactory epithelium. The incubation can be kept as long as 8 hours.
Dissociating Olfactory Sensory Neurons
19. Transfer olfactory neuroepithelial pieces to a 15ml Falcon tube using a glass Pasteur pipette. Take caution to prevent the tissue sticking to the inner wall of the pipette.
20. Spin down the olfactory neuroepithelium in a tabletop centrifuge at 200x g for 2 minutes.
21. Aspirate OSN medium; add 1ml of Mild trypsinization solution with 0.5 mM EDTA, flick to resuspend olfactory epithelial pieces, and incubate for 10 minutes in 37°C water bath. Gently swirl a few times.
22. To terminate trypsin activity, add 110 μl Fetal Bovine Serum.
-
23. To dissociate olfactory neuroepithelium, use a flame polished Pasteur pipette with pore size approximately 0.3 mm in diameter to triturate the tissue for 10-15 times.
Note: trituration should be done by picking up the solution with tissue pieces and “shooting” the solution out along the wall of the Falcon tube. This must be done gently to avoid damaging and/or killing cells. Avoid generating bubbles. Refer to the Troubleshooting section of the Commentary for further details.
24. To pellet dissociated cells, spin at 200x g for 5 minutes; remove supernatant, and add 0.5 ml of OSN culture medium.
25. Determine cell density using a hemocytometer.
Plating and Culturing Olfactory Sensory Neurons
26. Before plating, take out the 24 well plate with confluent astrocytes on the coverslips in the wells. Change Glia Medium to OSN Medium.
27. Plate 1 × 105 cells on the confluent astrocytes for each well.
28. OSN cultures are maintained at 37°C with 5% CO2.
29. 24 hours after plating, change 50% of the OSN medium with fresh warm medium. From then on, half of the medium is replaced every other day.
SUPPORT PROTOCOL 1
Preparation of glass coverslips
This protocol describes steps to prepare glass coverslips for astrocyte culture. Purchased glass coverslips need to be treated with strong basic solution to unify surface charges and clean the surface for better cell attachment. After cleaning, coverslips are coated with Laminin and Poly-D-lysine.
Material List
No. 1 12mm circular glass coverslip
10 M NaOH
70% Ethanol
Coverslip staining racks
Laminin stock, 1 mg/ml
Poly D-lysine stock, 10 mg/ml
- 24 well plate
- Put glass coverslips on the rack and place the rack in a glass container. Wash coverslips with tap water.
- Soak glass coverslips in 10 M NaOH for 15 minutes.
- Discard NaOH and run tap water for 1 hour.
- Wash with ddH2O extensively for 3 hours with several changes.
- Soak in 70% Ethanol for 15 minutes.
- Place the coverslip rack in the laminar flow culture hood to dry
- Cleaned and dried coverslips can be stored in a sterile petri dish.
- To coat coverslips, mix 50 μl of Laminin stock and 50 μl of Poly-D-Lysine stock into 10 ml of ddH20. Mixing should be done in the laminar flow culture hood. Aseptic techniques should be followed for all following steps.
- Place one coverslip per well into the 24 well plate.
- Add 500 μl of the mixed Laminin-Lysine solution in each well and let sitting for 2 hours in room temperature.
- Rinse the coverslip with sterile ddH2O for 5 minutes, repeating this step twice.
- Add 500 μl of culture media (DMEM, for example). Coated coverslips can be kept in DMEM at 37°C with 5% CO2 for up to 2 weeks.
SUPPORT PROTOCOL 2
Purification and culturing mouse cortical astrocytes
This protocol describes techniques to purify and culture cortical astrocytes. The method is modified from the Weintein protocol (Weinstein, 2001). Cortical tissues from neonatal mice are dissected and dissociated. Culture medium and conditions are selected to favor the survival of astrocytes. Steps are taken to eliminate the presence of neurons and oligodendrocytes. Purified astrocytes are plated on glass coverslips for olfactory sensory neuron culture. The purity of each batch is validated by immunocytochemistry.
Material List
5-10 neonatal C57 BL/6 mouse pups (P0 to P3)
Glia Medium (see recipes)
Trypsinization solution (see recipes)
DNase I
- Dissection tools:
- 1 51/2″ scissors
- 1 forceps
- 1 41/2″ fine scissors
- 2 No. 5 forceps
- 1 Micro scissors
- 1 Micro spatula (Ted Pella, #13510)
1mM AraC
75 cm2 flask
Cryovials
DMSO
Fetal Bovine Serum
Microscope and Hemocytometer
Falcon cell strainer, 70 μm
Dissecting and Dissociating Cerebral Cortices
Sterilze dissection tools by soaking in 70% ethanol for 20 minutes. Air dry in the culture hood.
Neonatal pups are anesthestized by placing them in cut glove fingers and embedding in crushed ice with heads above ice for 5 minutes or according to institutes ACUC guidelines.
After the pups are fully anesthetized, wipe the skin with 70% ethanol, and decapitate.
Quickly take out the brain by cutting along the midline and peel off the skin and the skull covering the brain, scoop out the brain with a micro spatula.
Isolate cerebral cortices in CMF-HBSS/HEPES.
Under the stereomicroscope in the horizontal laminar flow hood, strip away the meninges and transfer tissue to another 35mm dish.
Mince the cortical tissue with micro scissors.
Transfer minced tissue to a 50 ml Falcon tube using a sterilized Pasteur pipette.
Pellet tissue in a table top centrifuge at 200x g for 2 minutes.
Replace with trypsinization solution (1ml per brain), flick the tube to resuspend the tissue, incubate in 37°C water bath for 15 minutes. Swirl several times during trypsin digestion.
Add 1/10 volume of Fetal Bovine Serum to stop trypsin activity.
Triturate with a 5 ml pipette to dissociate tissue.
Filter cell suspension through a Falcon cell strainer (70μm) into a 50 ml Falcon tube.
Pellet cells at 200x g for 5 minutes, discard supernatant and replace with 1ml of Glia Medium.
Determine cell density with a hemocytometer.
Purifying Astrocytes
16. Plate 1×107 cells into each 75 cm2 cell culture flask in 10 ml of Glia Medium
17. Cultures are maintained at 37°C with 5% CO2.
18. Replace media 24 hours after plating, and subsequently every third day.
19. Check confluency of the culture at 9 days after plating.
-
20. If cultures are confluent, seal the lid of the flask by tightly closing and wrapping with Parafilm, place flasks on a rotary platform and shake at 37°C overnight at a speed just below foam forming (about 275 rpm).
This step is typically done under non-sterile condition. To prevent contamination, make sure flasks are completely sealed and placed in a secondary containment. Spray with 70% ethanol before putting them back to the culture hood. Neurons are typically not attached very tight compared to glial cells. Shaking the culture will detach any neurons present in the flask.
21. After shaking, replace with fresh media. Add 200 μl of 1mM AraC into 10 ml of medium.
22. After 48 hours of incubation, replace with Glia Medium and allow cells to recover.
-
23. Purified astrocytes can be stored as frozen aliquots. To freeze astrocytes, treat the flask with trypsinization solution for 10 minutes until they detach, stop trypsin activity with 1/10 volume of Fetal Bovine Serum, transfer cells into a 50 ml Falcon tube and pellet at 200x g for 5 minutes, resuspend the pellet with1ml Glia Medium plus 50 μl DMSO, aliquot and store in liquid nitrogen.
Purity of astrocytes can be evaluated by plating on coverslips as described below and immunostaining for astrocyte, oligodendrocyte and neuronal markers (see supporting protocol 3).
Culturing Astrocytes on glass coverslip
24. Take out frozen aliquots of astrocytes, submerge the cryovials in 37°C water bath and shake constantly until thaw.
25. Transfer astrocytes into a 15 ml Falcon tube and centrifuge at 200x g for 5 minutes.
26. Replace DMSO containing medium with 1ml of Glia Medium.
27. Place coverslips (see supporting protocol 1 for preparation) into 24 well plate.
28. Plate 2 × 104 astrocytes into each well.
29. Incubate astrocytes in 500 μl of Glia Medium at 37°C with 5% CO2 until confluent on the coverslip
30. Confluent astrocytes can be kept for as long as 2 weeks for the use of OSN culture.
SUPPORT PROTOCOL 3
Immunocytochemistry of OSN culture
This protocol is used to study the morphology and gene expression of cultured OSNs. A standard immunocytochemistry procedure is described. Antibodies used to identify OSNs and astrocytes are listed.
Material List
Coverslip rack
Petri dishes
Parafilm
4% paraformaldehyde
Horse serum
Antibodies: see protocol for detail
Secondary antibodies: see protocol for detail
Phosphate Buffered Saline (PBS)
Phosphate Buffered Saline with 0.1% Triton X-100 (PBS+TX)
Glass slides
- Fluoromount G (Southern Biotech, Cat#0100-01)
-
Aspirate media from the OSN culture. Add 1ml of warm 4% paraformaldehyde to each well and fix for 15 minutes at room temperature.4% paraformaldehyde should be kept at 4°C. Before use, warm an aliquot in 37°C water bath. Adding warmed fixative to the culture allows best preservation of the neuronal morphology.
- Rinse coverslips in the well with 1ml PBS+TX for 3 times for 5 minutes each.
- Block with 5% normal horse serum in PBS+TX for 30 minutes at room temperature, using 1ml per well.
-
Remove coverslips from the culture plate, rinse with PBS and incubate with primary antibodies in PBS for 1 hour.Coverslips can be picked up by a pair of curved forceps. Rinsing can be done by dipping into PBS a couple of times. For antibody incubation, coverslips are placed on the Parafilm lined Petri dish. Parafilm lining prevents the solution to spill over and holds the antibody solution on the coverslips. As little as 25 μl of antibody solution is enough to cover the coverslip. The Petri dish should be covered to further prevent drying.Primary antibodies used to identify OSNs are: type III β-tubulin (Sigma), 1:250; NCAM (BD Biosciences), 1:25; Golf (Santa Cruz Biotechnologies), 1:100; AC3 (Santa Cruz Biotechnologies), 1:100; GAP43 (Calbiochem), 1:200.
- Place coverslips on the coverslip rack. Make sure to mark the side where cells are. Rinse 3 times for 5 minutes each in PBS.
- Transfer coverslips back to the Parafilm lined Petri dish. Incubate with secondary antibodies for 45 minutes at room temperature. Fluorophore conjugated secondary antibodies are used for this step.
- Rinse with PBS, and dip in H2O
- Mount coverslips with the cell side upside down on a drop of Fluoromount G on a glass slide.
-
REAGENTS AND SOLUTIONS
- 2.5% Avertin
- To make 100% Avertin, mix 10 g of 2,2,2-tribromoethyl alcohol (Aldrich T4,840-2) with 10 ml of tert-amyl alcohol (Aldrich 24,048-6). Cover with foil to prevent light exposure. Store at 4°C for up to 6 month.
- To make 2.5% Avertin, Dilute 1 ml of 100% Avertin with 39 ml of H2O. Cover with foil to prevent light exposure. Store at 4°C for up to 2 weeks.
- OSN Medium
- 100 ml Waymouth’s MB 752/1 medium (1x, Life Technologeis)
- 1ml N2 supplement (100x, Life Technologies)
- 100 μl Gentamicin (50 mg/ml, Life Technologies)
- Store for 1 month at 4°C
- CMF-HBSS/HEPES
- 50 ml Calcium and Magnesium free Hank’s Balanced Salt Solution (10x)
- 5 ml 1M HEPES pH7.2
- 5 ml Penicillin-Streptomycin solution (10,000U penicillin and 10,000 μg streptomycin/ml, Life Technologies)
- H2O to 500 ml
- Filter sterilize and store at 4°C up to 3 month
- Trypsinization solution (0.2% trypsin)
- 1.6 ml 2.5% Trypsin (Life Technologies)
- 10,000 U DNase I (Sigma)
- CMF-HBSS/HEPES to 20 ml
- Adjust pH to 7.2, if necessary
- Aliquot and store at -20°C for up to 6 month
- Mild Trysinization Solution (0.05% trypsin)
- 0.4 ml 2.5% Trypsin (Life Technologies)
- 10,000 U DNase I (Sigma)
- CMF-HBSS/HEPES to 20 ml
- Adjust pH to 7.2, if necessary
- Aliquote and store at -20°C for up to 6 month
- Glia Medium
- 45 ml Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies)
- 5 ml Fetal Bovine Serum
- 1 ml Penicillin-Streptomycin solution
- Store at 4°C up to 1 month
COMMENTARY
Background Information
Olfactory sensory neurons are populated in the pseudostratified olfactory epithelium lining the nasal cavity. The ciliary processes of OSNs are exposed and are vulnerable to environmental insults. In adult animals, OSNs are constantly replaced (Farbman, 1992). Olfactory stem cells, namely horizontal and globose basal cells, are located in the basal layer of the olfactory epithelium (Packard et al., 2011). Basal cells produce neuronal progenitor cells that will differentiate into immature and subsequently mature OSNs. Cells representing different stages of OSN differentiation can be found in the olfactory epithelium. While globose and horizontal basal cells can be identified by Sox2 and Keratin 14 respectively, neuronal progenitors are marked with Mash1 and NeuroD (Chen et al., 2008). Olfactory marker protein is expressed in the mature OSN and immature neurons express GAP43 and NCAM.
Olfactory marker protein expression in culture has been reported in a few studies (Grill and Pixley, 1997). When OSNs are dissociated, mature OSNs do not survive beyond 48 hours in reported studies including the protocol described here. Cortical astrocytes have been used to support survival of dissociated cortical neurons (Goslin et al., 1998). When OSNs are plated directly on cortical astrocytes, survival rate is greatly improved and maturation of OSNs may be observed by the expression of OSN-specific markers like Golf and AC3 (Figure 1) (Chen et al., 2008). Other successful culture methods are reported which supplement the culture media with growth factors (Grill and Pixley, 1997; Moon et al., 2002). Different types of feeder layers have also been used to support OSN survival (Ekberg et al., 2011). Detailed comparison of different culture conditions for survival and differentiation of OSNs have not been done yet.
Critical Parameters and Troubleshooting
Isolating olfactory neuroepithelium
Isolation of olfactory neuroepithelium is a critical step and also is the most time consuming. This step limits how many embryos one can do in each round of culture. It is recommended to only work with 7-15 embryos at a time until the technique is well established. The entire duration of the dissection for all embryos, from taking out the olfactory epithelium to completely separating and discarding the stroma, should not be longer than 4 hours.
Dispase digestion is the key for efficient separation of stroma from the neuroepithelium. The duration of the digestion can be determined by taking out a piece of the olfactory epithelium and trying to separate. Dispase is a gentle protease, at the define concentration, the tissue can be digested up to 90 minutes without obvious effect for survival.
Identification of the neuroepithelium from the stroma takes a lot of practice. Olfactory neuroepithelium appears as a uniform layer of transparent tissue. It tends to curl up towards the stroma side. The nasal stroma is less transparent and softer in touch. It pulls easily way from the olfactory neuroepithelium when digestion is properly done. The olfactory neuroepithelial pieces can be picked up gently by forceps.
Dissociation
Olfactory epithelial cells are tightly associated with each other by gap junctions and cell-cell adhesion, particularly sustentacular cells. Dissociation of the olfactory epithelium is done first by incubating at 37°C for 2-3 hours. Neurons in the olfactory epithelium migrate and some appears as a cluster of cells on the surface of the tissue pieces. This step reduces the degree of mechanical dissociation that follows and therefore allows better survival of OSNs. This incubation step also selects for sensory neurons, as sustentacular cells do not migrate and remain tightly connected.
Trypsinization of olfactory epithelium is important for culture survival as well. Trypsin treatment needs to be closely timed. The mechanical trituration should not be done excessively. Excessive dissociation will result in death of a large number of cells and stringy tissue can be found indicating cell death. Add more DNase when stringy tissue is detected to rescue. Discard any non-dissociated tissue pieces at the end.
Anticipated Results
Survival rate and OSN morphology
During the first 24 hours, there will be a large amount of cell death. All mature OSNs, expressing olfactory marker protein, will not survive under this culture condition. Olfactory marker protein expression is absent at 2 days in vitro (DIV). The survival rate is at 20-30% of the plated cells between 3 to 5 DIV. The rate of survival is not correlated with the age of the embryos. However, higher density of the culture supports better survival.
Neurons with multipolar morphology are observed at 1 DIV. At 5 DIV, the majority of neurons in culture are bipolar. Though single axons are often observe for each neuron, axons tend to branch at the end. More branching are observed in areas where neurons are close to each other. The short dendritic process sometimes form branch as well (Chen et al., 2008). If bipolar morphology is desired, carefully controllig the density of the culture is the key.
Marker expression
OSNs are identified by expression of specific markers using immunocytochemistry. Neuronal identity is validated by the expression of type III β-tubulin and NCAM. Olfactory specific markers are Golf, AC3 and cyclic nucleotide gated channel A2 subunit. Expression of these genes should be detected by either immunocytochemistry or RT-PCR (Figure 2). At 3 DIV, NeuroD expression is also detected in some cells but Mash1 expression is not observed. Olfactory marker protein expression is not detected between 3 to 7 DIV. At 8 DIV, a few OMP positive cells appear in the culture.
Figure 2.
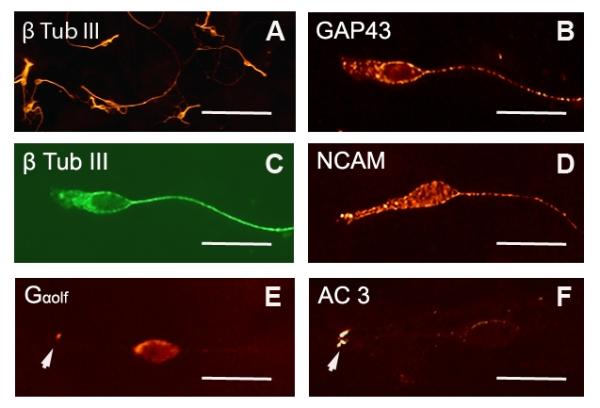
Morphology and gene expression of cultured OSNs. A. OSNs are identified by expression of type III β-tubulin. Sensory neurons are cultured at low density with each individual neuron extending processes without contacting other neurons. Majority of cultured neurons are bipolar in morphology. B-D. At 3 DIV, Cultured OSNs exhibit bipoloar morphology with a short thick dendritic process and a long thin axonal process. These neurons express neuronal markers, GAP43 (B), type III b-tubulin (C) and NCAM (D). E-F. At 6 DIV, cultured OSNs express odorant signaling molecules, Golf (E) and AC3 (F). Bar, 60 μm in A; 20 μm in B-F.
Manipulating gene expression
To manipulate gene expression in cultured OSNs, gene gun transfection approach has been demonstrated (Moon et al., 2002). We reported a lentiviral mediated gene transfer method that gave 100% infection of cultured OSNs. Lentiviral vectors expressing either shRNA or transcripts of choice can be infected into cultured olfactory sensory neurons by adding viral particles at the time of plating. Adding 106 pfu of lentiviruses in each well of 24 well plate results in a 100% infection of cultured neurons. Confluent astrocytes appear not to be infected by lentiviruses when examined at 5 DIV.
Time considerations
Several procedures need to be coordinated for OSN cultures. Cortical astrocytes should be prepared before ordering timed pregnant animals for OSN culture. After astrocytes are obtained, it is important to validate the quality of astrocytes to make sure there is no neuronal contamination. Confirm the availability of embryos before seeding astrocytes onto coverslips. It is important to seed enough astrocyte to ensure they become confluent in the desired amount of time. It is recommended that one practice seeding astrocytes before OSN techniques are attempted.
OSN cultures take a whole day. Plan for 2 hours for the initial dissection. After Dispase treatment, it will take 2-3 hours to dissect neuroepithelium for 10 embryos. Dissociation and plating could take as long as 6-8 hours.
Literature Cited
- Ayadi A, Ferrand G, Cruz I.G.d., Warot X. Mouse breeding and colony management. Curr. Protoc. Mouse Biol. 2011;1:239–264. doi: 10.1002/9780470942390.mo100214. [DOI] [PubMed] [Google Scholar]
- Chen H, Dadsetan S, Fomina AF, Gong Q. Expressing exogenous functional odorant receptors in cultured olfactory sensory neurons. Neural Develop. 2008;3:22. doi: 10.1186/1749-8104-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekberg JA, Amaya D, Chehrehasa F, Lineburg K, Claxton C, Windus LC, Key B, Mackay-Sim A, St John JA. OMP-ZsGreen fluorescent protein transgenic mice for visualisation of olfactory sensory neurons in vivo and in vitro. J. Neurosci. Methods. 2011;196:88–98. doi: 10.1016/j.jneumeth.2011.01.008. [DOI] [PubMed] [Google Scholar]
- Farbman AI. Cell Biology of Olfaction. Cambridge University Press; New York: 1992. [Google Scholar]
- Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing Nerve Cells. The MIT Press; London: 1998. pp. 339–369. [Google Scholar]
- Grill RJ, Pixley SK. In vitro generation of adult rat olfactory sensory neurons and regulation of maturation by coculture with CNS tissues. J. Neurosci. 1997;17:3120–3127. doi: 10.1523/JNEUROSCI.17-09-03120.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P. Axonal wiring in the mouse olfactory system. Annu. Rev. Cell. Dev. Biol. 2006;22:713–737. doi: 10.1146/annurev.cellbio.21.012804.093915. [DOI] [PubMed] [Google Scholar]
- Moon C, Yoo JY, Matarazzo V, Sung YK, Kim EJ, Ronnett GV. Leukemia inhibitory factor inhibits neuronal terminal differentiation through STAT3 activation. Proc. Natl. Acad. Sci. U. S. A. 2002;99:9015–9020. doi: 10.1073/pnas.132131699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard A, Schnittke N, Romano RA, Sinha S, Schwob JE. {Delta}Np63 regulates stem cell dynamics in the mammalian olfactory epithelium. J. Neurosci. 2011;31:8748–8759. doi: 10.1523/JNEUROSCI.0681-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein DE. Isolation and purification of primary rodent astrocytes. Curr. Protoc. Neurosci. 2001;1:3.5.1–3.5.9. doi: 10.1002/0471142301.ns0305s00. [DOI] [PubMed] [Google Scholar]
- Zhuang H, Matsunami H. Synergism of accessory factors in functional expression of mammalian odorant receptors. J. Biol. Chem. 2007;282:15284–15293. doi: 10.1074/jbc.M700386200. [DOI] [PubMed] [Google Scholar]