

Abstract
Objective:
The goal of the current investigation was to examine a cohort of symptomatic and asymptomatic LRRK2 mutation carriers, in order to address whether the reported alterations in amyloid β (Aβ) and tau species in the CSF of patients with sporadic Parkinson disease (PD) are a part of PD pathogenesis, the aging process, or a comorbid disease in patients with PD, and to explore the possibility of Aβ and tau as markers of early or presymptomatic PD.
Methods:
CSF Aβ42, total tau, and phosphorylated tau were measured with Luminex assays in 26 LRRK2 mutation carriers, who were either asymptomatic (n = 18) or had a phenotype resembling sporadic PD (n = 8). All patients also underwent PET scans with 18F-6-fluoro-l-dopa (FD), 11C-(±)-α-dihydrotetrabenazine (DTBZ), and 11C-d-threo-methylphenidate (MP) to measure dopaminergic function in the striatum. The levels of CSF markers were then compared to each PET measurement.
Results:
Reduced CSF Aβ42 and tau levels correlated with lower striatal dopaminergic function as determined by all 3 PET tracers, with a significant association between Aβ42 and FD uptake. When cases were restricted to carriers of the G2019S mutation, the most common LRRK2 variant in our cohort, significant correlations were also observed for tau.
Conclusions:
The disposition of Aβ and tau is likely important in both LRRK2-related and sporadic PD, even during early phases of the disease. A better understanding of their production, aggregation, and degradation, including changes in their CSF levels, may provide insights into the pathogenesis of PD and the potential utility of these proteins as biomarkers.
Recent studies, particularly those with large cohorts of subjects, have reported altered CSF levels of amyloid β (Aβ) and tau species in patients with sporadic Parkinson disease (PD).1–6 However, it is unclear whether these alterations are mechanistically important to PD pathogenesis, or represent a comorbidity or aging process. To address this question, Aβ and tau species need to be measured in patients with early (ideally preclinical) PD. Moreover, unique protein alterations in early/preclinical cases, alongside with other early (nonmotor) symptoms,7 could assist in identifying PD at premotor stages.
Studying sporadic PD prior to the onset of clinical symptoms has been impractical, making early intervention exceedingly challenging. Autosomal dominant mutations in the leucine-rich repeat kinase (LRRK2) gene, the most common known genetic cause of parkinsonism, result in a clinical phenotype similar to sporadic PD.8,9 LRRK2 mutation carriers have been extensively characterized by PET studies, demonstrating neurochemical changes similar to sporadic PD cases, as well as detectable dopaminergic dysfunction in asymptomatic carriers.10,11 Thus, subjects with LRRK2 mutations constitute an excellent cohort for studying preclinical and early PD.
In the present study, we examined CSF Aβ1–42 (Aβ42) peptide, total tau (t-tau), and phosphorylated tau (p-tau) in symptomatic and asymptomatic LRRK2 mutation carriers. CSF protein levels were correlated with dopamine denervation (assessed by PET measurements), to determine whether Aβ42 and tau species are altered during early PD processes, and to explore the utility of these proteins as early or presymptomatic PD diagnostic or progression markers.
METHODS
Subjects.
This study focused on preclinical and asymptomatic LRRK2 mutation carriers: 18 of the 26 carriers included in the study were asymptomatic at the time of evaluation. The remaining 8 subjects had clinically confirmed PD; most of them were at early disease stages that permitted long-range travel and limited confounding due to complicated CNS changes associated with advanced PD. Details on subject recruitment can be found in the e-Methods on the Neurology® Web site at www.neurology.org. Demographic and clinical information is listed in table 1 for all subjects.
Table 1.
Summary of demographics and PET values of subjects
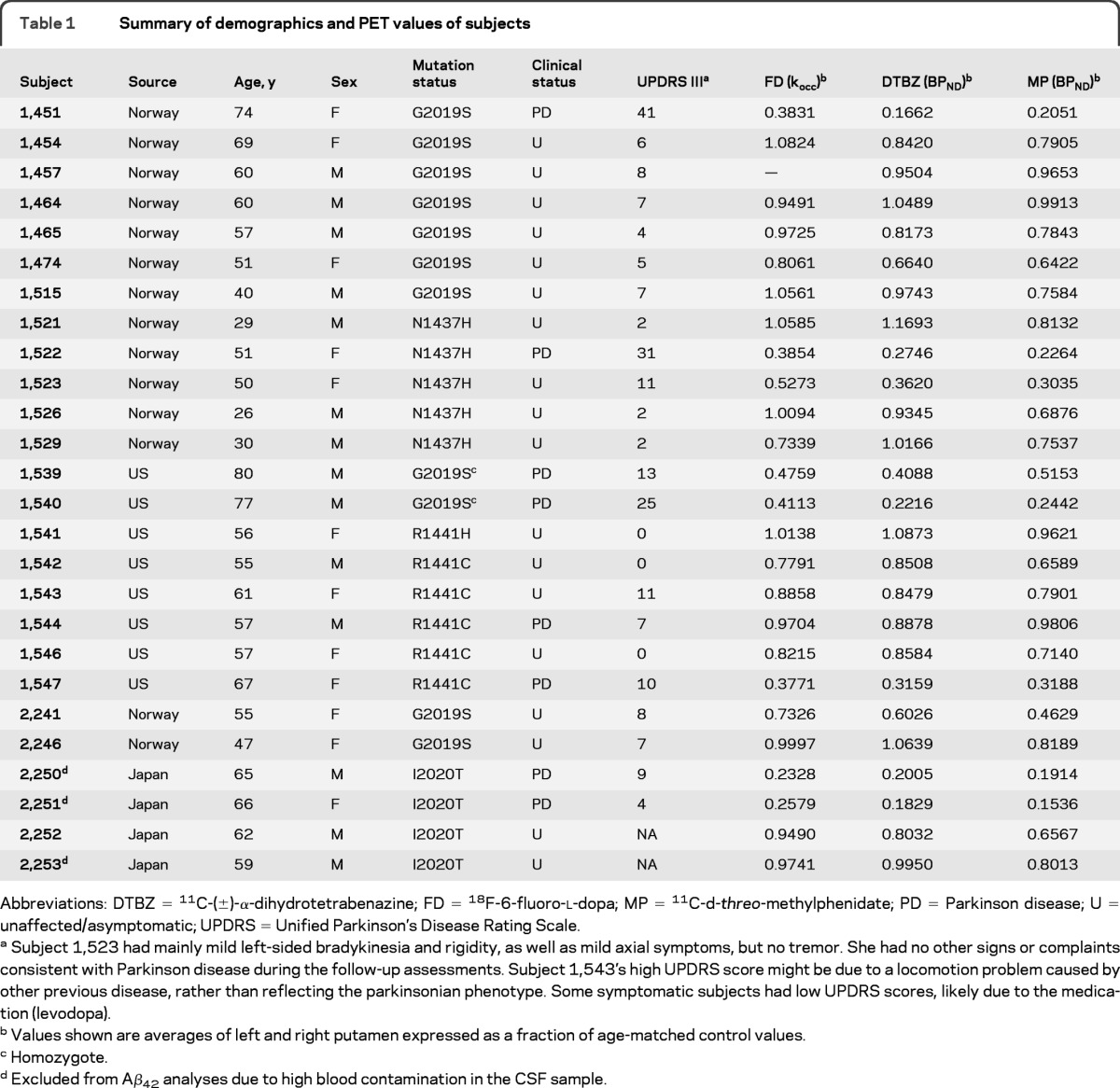
Abbreviations: DTBZ = 11C-(±)-α-dihydrotetrabenazine; FD = 18F-6-fluoro-l-dopa; MP = 11C-d-threo-methylphenidate; PD = Parkinson disease; U = unaffected/asymptomatic; UPDRS = Unified Parkinson's Disease Rating Scale.
Subject 1,523 had mainly mild left-sided bradykinesia and rigidity, as well as mild axial symptoms, but no tremor. She had no other signs or complaints consistent with Parkinson disease during the follow-up assessments. Subject 1,543's high UPDRS score might be due to a locomotion problem caused by other previous disease, rather than reflecting the parkinsonian phenotype. Some symptomatic subjects had low UPDRS scores, likely due to the medication (levodopa).
Values shown are averages of left and right putamen expressed as a fraction of age-matched control values.
Homozygote.
Excluded from Aβ42 analyses due to high blood contamination in the CSF sample.
Standard protocol approvals, registrations, and patient consents.
This study was approved by the institutional review boards of all participating institutions. All individuals provided written informed consent to participate.
PET, CSF samples, and Luminex assays.
Within 1 year of CSF sample collection, each subject was scanned with 3 tracers, 18F-6-fluoro-l-dopa (FD, a marker for the uptake and decarboxylation of levodopa as well as the trapping of dopamine in synaptic vesicles), 11C-(±)-α-dihydrotetrabenazine (DTBZ, a vesicular monoamine transporter 2 [VMAT2] ligand), and 11C-d-threo-methylphenidate (MP, a dopamine transporter [DAT] ligand). CSF Aβ42, t-tau, and p-tau levels of all cases were measured as previously described.2 To minimize any age effects, PET data were normalized to age-matched control values. Details can be found in the e-Methods.
Statistical analysis.
All analyses were performed with PASW Statistics 18.0 (SPSS Inc., Chicago, IL). Nonparametric correlation methods (Kendall rank correlation) were used to assess the relationship between the skewed PET measurements and the CSF biomarkers and to minimize the effects of outliers. See more details in the e-Methods.
RESULTS
PET measurements.
Subjects were scanned for putaminal tracer binding/uptake, and the average values of left and right putamen, expressed as a fraction of age-matched control values, are shown in table 1. Representative PET images from LRRK2 PD patients, asymptomatic mutation carriers, and healthy controls are shown in figure e-1.
Except for subject 1,544, all LRRK2 PD subjects had significantly reduced PET values for all 3 tracers (figure 1 and table 1). Among asymptomatic mutation carriers, 4 subjects had significantly reduced values for all 3 tracers; in subject 1,523, PET abnormalities demonstrated asymmetry, typical of sporadic PD. Subject 1,542 had significantly reduced MP (DAT) binding and FD uptake values only. In subject 1,526, DAT binding was significantly reduced; reduced DTBZ (VMAT2) binding was observed only in the right putamen. Subjects 1,454 and 2,246 showed borderline decrease in right putaminal FD uptake and DAT binding, respectively. All other asymptomatic mutation carriers demonstrated normal uptake/binding of all 3 tracers in the putamen. Subjects 1,465 and 1,543 showed abnormal VMAT2 and DAT binding in the caudate, but not the putamen. Subject 1,457 could not complete the FD scan.
Figure 1. Putaminal tracer binding/uptake in LRRK2 mutation carriers.

The average left and right putaminal tracer binding/uptake values in LRRK2 mutation carriers are given as a fraction of age-matched healthy control values. The means with 95% confidence intervals for asymptomatic (unaffected) subjects and subjects with clinically confirmed LRRK2–Parkinson disease (PD) are also shown. Circles indicate 18F-fluoro-l-dopa (FD) uptake, triangles indicate 11C-dihydrotetrabenazine (DTBZ) binding, and squares indicate 11C-d-threo-methylphenidate (MP) binding.
Correlation of CSF biomarkers with PET data.
Blood contamination of CSF, which occasionally occurs during lumbar puncture collection, can significantly affect CSF levels of Aβ42 (but not t-tau or p-tau).2 To control for this variable, 3 subjects (1 LRRK2 PD and 2 asymptomatic) whose CSF hemoglobin levels were >250 ng/mL were excluded from further analysis of Aβ42.
Similar to our previous report in patients with sporadic PD,2 CSF levels of all analytes were lower in symptomatic LRRK2 mutation carriers compared to asymptomatic carriers (symptomatic [mean ± SD]: Aβ42, 504.1 ± 128.5 pg/mL, t-tau, 24.4 ± 22.6 pg/mL, p-tau, 16.7 ± 3.8 pg/mL; asymptomatic: Aβ42, 601.5 ± 234.1 pg/mL, t-tau, 36.4 ± 32.0 pg/mL, p-tau, 21.1 ± 9.0 pg/mL; individual values can be found in table e-1); but this trend did not reach significance, likely due to the limited number of subjects included. Additionally, because symptomatic subjects were older than asymptomatic carriers (mean 67.1 ± 9.8 vs 51.3 ± 12.3 years), the extent of decrease in CSF tau levels in LRRK2 PD may have also been reduced by the known age-dependent increase in CSF tau levels (Aβ42 is stable as a function of age in healthy controls).2 Nonetheless, the correlations between CSF analyte levels and PET measurements are unlikely to be substantially affected by age because all the PET data were standardized to age-matched control values.
CSF levels of Aβ42 in the entire LRRK2 cohort decreased with decreasing uptake of FD (Kendall tau = 0.316, p = 0.040), indicating that decreasing protein levels correspond with progressive loss of dopamine function (figure 2A). Decreased levels of Aβ42 also corresponded to lower values of the other PET tracers, as did decreased levels of t-tau or p-tau with all PET tracers, but these relationships did not reach significance (figure 2).
Figure 2. Correlation of CSF biomarkers with PET measurements in the entire LRRK2 cohort.

The correlations of protein levels in CSF with putaminal PET values expressed as fractions of age-matched healthy control values are shown. For Aβ42 (A–C), FD: Kendall tau = 0.316, p = 0.040; DTBZ: Kendall tau = 0.223, p = 0.119; MP: Kendall tau = 0.217, p = 0.146. For total tau (t-tau) (D–F), FD: Kendall tau = 0.065, p = 0.656; DTBZ: Kendall tau = 0.182, p = 0.200; MP: Kendall tau = 0.119, p = 0.401. For phosphorylated tau (p-tau) (G–I), FD: Kendall tau = 0.226, p = 0.117; DTBZ: Kendall tau = 0.203, p = 0.151; MP: Kendall tau = 0.190, p = 0.178. Aβ42 values restricted to 23 out of 26 cases due to high blood contamination in CSF, which does not influence t-tau or p-tau values. Note that regression lines were generated from linear regression for visualization only. DTBZ = 11C-dihydrotetrabenazine; FD = 18F-fluoro-l-dopa; MP = 11C-d-threo-methylphenidate.
Because LRRK2 mutations are heterogeneous, this analysis was repeated using the subset of patients with the G2019S mutation (n = 11), the most common variant in our cohort. Remarkably, there was a clear reduction in the levels of all proteins with decreasing PET measurements in the striatum, regardless of the tracer used (figure 3). CSF levels of Aβ42 and p-tau were both reduced with greater degrees of reduced FD uptake (i.e., diminished dopamine function; Aβ42: Kendall tau = 0.556, p = 0.025; p-tau: Kendall tau = 0.597, p = 0.020). In addition, reduced CSF p-tau significantly correlated with reduced DTBZ binding (Kendall tau = 0.597, p = 0.013) and MP binding (Kendall tau = 0.559, p = 0.020), but t-tau only significantly correlated with MP binding (Kendall tau = 0.506, p = 0.037).
Figure 3. Correlation of CSF biomarkers with PET measurements in LRRK2 G2019S mutation carriers.

The correlations of protein levels in CSF with putaminal PET values expressed as fractions of age-matched healthy control values are shown (n = 11). For Aβ42 (A–C), FD: Kendall tau = 0.556, p = 0.025; DTBZ: Kendall tau = 0.455, p = 0.052; MP: Kendall tau = 0.382, p = 0.102. For total tau (t-tau) (D–F), FD: Kendall tau = 0.339, p = 0.192; DTBZ: Kendall tau = 0.331, p = 0.199; MP: Kendall tau = 0.506, p = 0.037. For phosphorylated tau (p-tau) (G–I), FD: Kendall tau = 0.597, p = 0.020; DTBZ: Kendall tau = 0.597, p = 0.013; MP: Kendall tau = 0.559, p = 0.020. Note that the regression lines were generated from linear regression for visualization only. DTBZ = 11C-dihydrotetrabenazine; FD = 18F-fluoro-l-dopa; MP = 11C-d-threo-methylphenidate.
One symptomatic LRRK2 PD subject (1,544) with normal PET measurements demonstrated relatively lowered t-tau and p-tau levels, i.e., behaved more like a patient with PD than a normal control, suggesting that CSF tau might be a more sensitive indicator than the PET measurements.
DISCUSSION
Several independent investigations reported that CSF Aβ levels decreased (but not as substantially as in Alzheimer disease [AD]) in patients with sporadic PD, particularly in those with cognitive impairment.2,5,6,12,13 Additionally, studies with large cohorts (≥50 subjects in either sporadic PD or control group) suggest that, unlike what is typically described for AD, tau levels tend to decrease in PD compared to controls.2–4,6 The obvious trend of decreasing CSF levels of Aβ42 and tau in LRRK2 PD and asymptomatic carriers (figure 2 and 3, table e-1) is in line with these observations. This raises the question of whether decreased concentrations of CSF tau and Aβ might be mechanistically involved in PD pathology. Tau pathology and amyloid plaques, though increasingly reported,14,15 are not typically seen in the brains of patients with PD. Therefore, it is possible that the decrease in CSF tau and Aβ levels occurs as soluble tau/p-tau and Aβ oligomers, often considered more toxic,16 deposit in the brains of patients with PD without formation of amyloid plaques and neurofibrillary (tau) tangles.
Regardless of the cause of decreased CSF Aβ42, t-tau, and p-tau in asymptomatic and symptomatic mutation carriers, our data suggest that metabolism of Aβ and tau in brain is likely dysfunctional in LRRK2-related PD, even before motor symptoms appear. If the same is true of sporadic PD, then tau and amyloid pathology in patients with PD, rather than just indicating comorbidity or general aging processes, might interact with synucleinopathy as a pathologic network that contributes to PD development and progression, even at premotor stages. In addition to experiments showing that both tau and Aβ might directly interact with α-synuclein, facilitating its aggregation,17,18 this argument is further supported by recent independent genome-wide association studies indicating that the SNCA and MAPT genes/regions (encoding α-synuclein and tau, respectively) are consistently associated with an increased risk of developing PD.19,20
Our results, if applicable to sporadic PD, also suggest that Aβ42 and tau could potentially be used as biomarkers for early PD diagnosis. However, since they are not particularly strong predictors of the disease condition, it is likely that a combination of Aβ42 and tau with other markers will be necessary for effective preclinical diagnosis or monitoring disease progression. A major limitation in focusing on LRRK2 mutation carriers is that this approach is logistically difficult in that it requires recruitment from a very limited pool of subjects, lengthy and potentially uncomfortable procedures (PET imaging and lumbar puncture), and in many instances, international travel. Thus, we were only able to study a modest number of participants, and because this was an exploratory study we did not perform statistical correction for multiple comparisons. Therefore, the data need to be interpreted with caution and the results should be replicated, ideally using a longitudinal design with serial PET scans and CSF measurements.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Sydney Thomas for study coordination; Joshua Bradner for data analysis; Dr. Amy Furay for critical review of the manuscript and copyediting; Jennifer Lash for assistance making arrangements for some research subjects involved in this study for their travel to Seattle, WA, and Vancouver, BC; and the patients for their participation and sample donations.
GLOSSARY
- Aβ
amyloid β
- AD
Alzheimer disease
- DTBZ
11C-(±)-α-dihydrotetrabenazine
- FD
18F-6-fluoro-l-dopa
- MP
11C-d-threo-methylphenidate
- p-tau
phosphorylated tau
- PD
Parkinson disease
- t-tau
total tau
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
J.Z. conceived and supervised the project, and drafted the manuscript with M.S., J.O.A., T.S., and K.K.J., J.O.A., K.K.J., Z.K.W., R.J.U., K.H., T.Y., C.P.Z., H.M.K., and J.B.L. were responsible for patient characterization and sample collection. M.S. assisted in experimental design and execution, as well as in data interpretation and statistical analyses. V.S. and A.J.S. were responsible for PET analysis. C.G. worked on CSF sample handling and Luminex assays. J.A. worked on data management and statistical analyses. K.L.E. and K.W.S. were involved in statistical analyses. All authors critically reviewed the manuscript.
STUDY FUNDING
Supported by NIH (AG025327, AG033398, P30ES007033-6364, ES004696-5897, ES012703, ES016873, NS057567, NS060252 to J.Z., P50NS062684 to C.P.Z., J.B.L., and J.Z., NS065070 to C.P.Z., NS057567 and P50NS072187 to Z.K.W. and R.J.U.), Department of Veterans Affairs (to J.B.L. and H.M.K., and 1I01BX000531 to C.P.Z.), Canadian Institutes of Health Research (to A.J.S.), Michael Smith Foundation for Health Research (to A.J.S.), Pacific Alzheimer Research Foundation (to A.J.S.), Canada Research Chairs program (to A.J.S.), TRIUMF (to A.J.S.), Mayo Clinic Florida Research Committee CR program (to Z.K.W. and R.J.U.), the gift from Carl Edward Bolch, Jr. and Susan Bass Bolch (to Z.K.W. and R.J.U.), and Grants-in-Aid from the Research Committee of CNS Degenerative Diseases, the Ministry of Health, Labour and Welfare of Japan (to K.H. and T.Y.).
DISCLOSURE
Dr. Aasly serves on tzhe editorial boards of Parkinsonism and Related Disorders, Parkinson's Disease, and the Journal of the Norwegian Medical Association; and his institution receives annual royalties from Lundbeck Inc. from the licensing of the technology related to PARK8/LRRK2. Dr. Shi reports no disclosures. Dr. Sossi has received research support from Pacific Alzheimer Research Foundation. Dr. Stewart and Dr. Johansen report no disclosures. Dr. Wszolek serves as Co-Editor-in-Chief of Parkinsonism and Related Disorders, Regional Editor of the European Journal of Neurology, and on the editorial boards of Neurologia i Neurochirurgia Polska, Advances in Rehabilitation, the Medical Journal of the Rzeszow University, and Clinical and Experimental Medical Letters; holds and has contractual rights for receipt of future royalty payments from patents re: A novel polynucleotide involved in heritable Parkinson's disease; receives royalties from publishing Parkinsonism and Related Disorders (Elsevier, 2007, 2008, 2009) and the European Journal of Neurology (Wiley-Blackwell, 2007, 2008, 2009); and receives research support from Allergan, Inc., the NIH/NINDS, Mayo Clinic Florida Research Committee CR program, the Pacific Alzheimer Research Foundation (Canada), the CIHR, the Mayo Clinic Florida Research Committee CR program, and a gift from Carl Edward Bolch, Jr., and Susan Bass Bolch. Dr. Uitti serves as an Associate Editor of Neurology®; has received research support from Advanced Neuromodulations Systems and from the NIH; and his institution receives annual royalties from Lundbeck Inc. from the licensing of the technology related to PARK8/LRRK2. Dr. Hasegawa and Dr. Yokoyama report no disclosures. Dr. Zabetian has received research support from NIH (NINDS/NIA), the US Department of Veterans Affairs, the Parkinson's Disease Foundation, and the American Parkinson Disease Association. Dr. Kim reports no disclosures. Dr. Leverenz has served as a consultant for Bayer Schering Pharma, Novartis, and Teva Pharmaceutical Industries Ltd.; has received speaker honoraria from Novartis; and receives research support from the NIH/NIND, the Michael J. Fox Foundation, the Washington Chapter American Parkinson's Disease Association/Northwest Collaborative Care, and the US Department of Veterans Affairs. Dr. Ginghina and J. Armaly report no disclosures. Dr. Edwards has received research support from the NIH and the Centers for Disease Control. K.W. Snapinn has received research support from the NIH, the US Department of Veterans Affairs, and the Centers for Disease Control. Dr. Stoessl serves on a scientific advisory board for Biovail Corporation/Medgenesis; has received funding for travel and speaker honoraria from Novartis, Teva Pharmaceutical Industries Ltd., Allergan, Inc., and Abbott; serves on the editorial boards of Annals of Neurology, Lancet Neurology, and Parkinsonism & Related Disorders; and receives research support from CIHR, the Michael Smith Foundation for Health Research, the Michael J. Fox Foundation, and the Pacific Alzheimer Research Foundation. Dr. Zhang serves on the editorial boards of Neurological Disease, Neurochemical Research, the Journal of Alzheimer's Disease, and Proteomics; and has received research support from the NIH.
REFERENCES
- 1. Shi M, Zhang J. Cerebrospinal fluid α-synuclein, tau and amyloid β in Parkinson's disease. Lancet Neurol 2011; 10: 681 [DOI] [PubMed] [Google Scholar]
- 2. Shi M, Bradner J, Hancock AM, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol 2011; 69: 570– 580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Döring F, Trenkwalder C, Schlossmacher MG. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol 2011; 10: 230– 240 [DOI] [PubMed] [Google Scholar]
- 4. Abdo WF, Bloem BR, Van Geel WJ, Esselink RA, Verbeek MM. CSF neurofilament light chain and tau differentiate multiple system atrophy from Parkinson's disease. Neurobiol Aging 2007; 28: 742– 747 [DOI] [PubMed] [Google Scholar]
- 5. Alves G, Bronnick K, Aarsland D, et al. CSF amyloid-beta and tau proteins, and cognitive performance, in early and untreated Parkinson's disease: the Norwegian ParkWest study. J Neurol Neurosurg Psychiatry 2010; 81: 1080– 1086 [DOI] [PubMed] [Google Scholar]
- 6. Montine TJ, Shi M, Quinn JF, et al. CSF Abeta42 and tau in Parkinson's disease with cognitive impairment. Mov Disord 2010; 25: 2682– 2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu Y, Le W, Jankovic J. Preclinical biomarkers of Parkinson disease. Arch Neurol 2011; 68: 22– 30 [DOI] [PubMed] [Google Scholar]
- 8. Kumari U, Tan EK. LRRK2 in Parkinson's disease: genetic and clinical studies from patients. FEBS J 2009; 276: 6455– 6463 [DOI] [PubMed] [Google Scholar]
- 9. Haugarvoll K, Wszolek ZK. Clinical features of LRRK2 parkinsonism. Parkinsonism Relat Disord 2009; 15 (suppl 3): S205– 208 [DOI] [PubMed] [Google Scholar]
- 10. Adams JR, van Netten H, Schulzer M, et al. PET in LRRK2 mutations: comparison to sporadic Parkinson's disease and evidence for presymptomatic compensation. Brain 2005; 128: 2777– 2785 [DOI] [PubMed] [Google Scholar]
- 11. Nandhagopal R, Mak E, Schulzer M, et al. Progression of dopaminergic dysfunction in a LRRK2 kindred: a multitracer PET study. Neurology 2008; 71: 1790– 1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Compta Y, Marti MJ, Ibarretxe-Bilbao N, et al. Cerebrospinal tau, phospho-tau, and beta-amyloid and neuropsychological functions in Parkinson's disease. Mov Disord 2009; 24: 2203– 2210 [DOI] [PubMed] [Google Scholar]
- 13. Siderowf A, Xie SX, Hurtig H, et al. CSF amyloid β 1–42 predicts cognitive decline in Parkinson disease. Neurology 2010; 75: 1055– 1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Litvan I, Halliday G, Hallett M, et al. The etiopathogenesis of Parkinson disease and suggestions for future research: part I. J Neuropathol Exp Neurol 2007; 66: 251– 257 [DOI] [PubMed] [Google Scholar]
- 15. Wills J, Jones J, Haggerty T, Duka V, Joyce JN, Sidhu A. Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson's disease brains with and without dementia. Exp Neurol 2010; 225: 210– 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol 2007; 8: 101– 112 [DOI] [PubMed] [Google Scholar]
- 17. Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci 2010; 30: 7281– 7289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsigelny IF, Crews L, Desplats P, et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer's and Parkinson's diseases. PLoS ONE 2008; 3: e3135 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19. Pankratz N, Wilk JB, Latourelle JC, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 2009; 124: 593– 605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 2009; 41: 1308– 1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.