

Abstract
Objective:
Memory decline commonly occurs among elderly individuals. This observation is often attributed to early neurodegenerative changes in the hippocampus and related brain regions. However, the contribution of vascular lesions, such as brain infarcts, to hippocampal integrity and age-associated memory decline remains unclear.
Methods:
We studied 658 elderly participants without dementia from a prospective, community-based study on aging and dementia who received high-resolution structural MRI. Cortical and subcortical infarcts were identified, and hippocampal and relative brain volumes were calculated following standard protocols. Summary scores reflecting performance on tasks of memory, language, processing speed, and visuospatial function were derived from a comprehensive neuropsychological battery. We used multiple regression analyses to relate cortical and subcortical infarcts, hippocampal and relative brain volume, to measures of cognitive performance in domains of memory, language, processing speed, and visuospatial ability.
Results:
Presence of brain infarcts was associated with a smaller hippocampus. Smaller hippocampus volume was associated with poorer memory specifically. Brain infarcts were associated with poorer memory and cognitive performance in all other domains, which was independent of hippocampus volume.
Conclusions:
Both hippocampal volume and brain infarcts independently contribute to memory performance in elderly individuals without dementia. Given that age-associated neurodegenerative conditions, such as Alzheimer disease, are defined primarily by impairment in memory, these findings have clinical implications for prevention and for identification of pathogenic factors associated with disease symptomatology.
Memory decline is frequent among the elderly and it is most often attributed to dysfunction and atrophy of the hippocampus and related mesial temporal lobe structures.1–3 Subclinical brain infarcts are present in about one-third of older adults, and are associated with a 2-fold risk of dementia and a steeper decline in age-associated cognitive function.4–6 However, the effects of subclinical brain infarcts on hippocampal integrity and age-related memory decline are not well understood.
Memory deficits have been described following brain infarcts. Cortical infarcts can result in diverse deficits depending on size and location of lesion and can produce cognitive decline in multiple domains, including perceptual speed and memory.7,8 Subcortical infarcts can result in executive dysfunction,9 although strategic thalamic or limbic system infarcts can result in memory loss.10,11 Clinically, it can be difficult to differentiate between cognitive decline due to ischemic brain injury vs incipient Alzheimer disease (AD). By the time significant memory decline and dementia are present, pathologic evaluations reveal mixed pathology in over 50% of cases.11 Despite the heterogeneous profile of deficits seen after clinical strokes, silent infarcts are thought to affect primarily the cognitive domains of processing speed and executive function.5 Although memory deficits may be a downstream consequence of poor learning efficiency secondary to the speed/executive dysfunction, the memory deficits seen in elderly people with silent brain infarcts are typically not attributed to infarcts alone but rather to hippocampal atrophy,12 which is considered a marker of AD.13 Memory function is dependent on the hippocampus, but also on the complex network of its input and output pathways required for appropriate information processing.14 The presence of infarcts likely has a negative effect on information flow due to structural disturbance in cortex and white matter, and may also negatively impact hippocampal function or structure due to the fact that the hippocampus is very sensitive to ischemia.15,16 There are few studies examining the impact of brain infarcts and hippocampal volume simultaneously in relation to memory performance. Thus, it remains unclear whether infarcts and hippocampus atrophy are related, and contribute to memory decline independently.
We hypothesized that brain infarcts and diminished hippocampus volume are each independently associated with poorer memory, reflecting the important contribution of both the integrity of the hippocampus proper as well as the more diffuse brain networks that are a part of hippocampal projection system. In addition, we hypothesized that brain infarcts and hippocampus volume are associated with a unique profile of cognitive deficits.
METHODS
Participants.
Participants came from a community-based prospective cohort study of aging and dementia in adults age 65 and older residing in northern Manhattan (Washington Heights-Inwood Columbia Aging Project).17 Beginning in 2004, participants who did not have dementia at their previous follow-up visit were invited to participate in a neuroimaging study.18 Of these 1,841 participants, 769 (41.8%) underwent MRI. Frequencies and reasons why participants did not undergo imaging were reported elsewhere.18
Standard protocol approvals and patient consents.
All procedures were approved by a local Institutional Review Board. Written informed consent was obtained from all participants.
Neuropsychological assessment.
Participants were administered a neuropsychological battery comprising 15 standardized tests.19 Composite scores were developed using factor analysis, which identified factors of memory, language, processing speed, and visuospatial ability.20,21 Based on factor loadings, summary variables reflecting performances in specific domains were derived as composite z scores: 1) memory: Selective Reminding Test (SRT)22: total recall, delayed recall, and delayed recognition; 2) language: Boston Naming Test25, Letter Fluency Test; Category Fluency test; Wechsler Adult Intelligence Scale–R Similarities subtest26; and Boston Diagnostic Aphasia Examination Repetition and Comprehension subtests25; 3) executive functioning/processing speed: Color Trails 1 and Color Trails 227; 4) visuospatial ability: Benton Visual Retention Test recognition and matching variables,28 Rosen Drawing Test29, and Identities and Oddities subtest.26 In secondary analyses, we considered specific aspects of the SRT, including learning, long-term recall (i.e., number of words recalled on consecutive trials without selective reminding), delayed free recall, and delayed recognition.
MRI.
Image acquisition was performed on a 1.5 Tesla scanner (Philips Intera) at Columbia University. The following images were acquired axially: T2-weighted fluid-attenuated inversion recovery (FLAIR) images (repetition time [TR]: 11,000 msec; echo time [TE]: 144.0 msec; inversion time [TI]: 2,800 msec; field of view [FOV]: 25 cm; 256 × 192 pixel matrix, 3 mm section thickness); proton density/T2-weighted double-echo (TR: 2,675 msec; TE: 12/92 msec; FOV: 220 cm; 256 × 192 pixel matrix, 4 mm section thickness); T1-weighted (TR: 20 msec; TE: 2.1 msec; FOV: 240 cm; 256 × 160 pixel matrix, 1.3 mm section thickness).
Relative brain volume.
Images were transferred to the Imaging of Dementia and Aging Laboratory at the University of California, Davis, for morphometric analysis. Total brain and cranial volumes were derived manually on FLAIR images as previously described.18,23 Relative brain volume was the ratio of total brain volume to intracranial volume.
Hippocampal volume.
The T1-weighted images were reoriented in the coronal plane perpendicular to the long axis of the left hippocampus.18 Borders of the hippocampus were traced manually with simultaneous monitoring in the sagittal and axial views.18 Intrarater reliability in the right and left hippocampi was good (intraclass correlation coefficient 0.98 and 0.96). We examined total relative hippocampal volume (i.e., total hippocampal/intracranial volume).
Brain infarct assessment on MRI.
Lesions 3 mm or larger qualified for consideration as brain infarcts. Signal void seen on the T2-weighted images was interpreted to indicate a vessel. Other necessary characteristics included CSF density on the T1-weighted image and separation from the circle of Willis vessels and perivascular spaces. Previously published reliability estimates among raters have been good.24 Cortical infarcts were defined as those in the frontal, parietal, temporal, and occipital cortices, and those in the extreme capsule (as defined in our sample, this region falls adjacent to or overlapping with insular cortex). Subcortical infarcts were defined as those in putamen, globus pallidus, caudate, thalamus, internal and external capsule, brainstem, and frontal, parietal, occipital, and temporal white matter.
Statistical method.
The characteristics of the cortical, subcortical, and no infarct groups were compared with respect to demographic data (age, sex, ethnicity), relative hippocampal volume, relative brain volume, and vascular disease history/risk factor variables (i.e., APOE ϵ4, hypertension, diabetes mellitus, heart disease, clinical stroke, smoking, and alcohol abuse) using χ2 analysis and general linear models.
A series of multiple regression analyses examined associations between infarct and hippocampal volume. A single infarct variable was created reflecting presence of at least 1 cortical or 1 subcortical infarct (0 = no infarct, 1 = infarct) and relative hippocampal volumes were compared between the 2 groups, with demographic factors held as covariates. The analyses were rerun comparing the 3 infarct groups (i.e., no infarcts, subcortical infarcts only, and cortical infarcts only).
A series of multiple regression analyses examined the association of subcortical infarcts, cortical infarcts, and hippocampal volume with cognitive composite scores. The MRI measures, including infarcts and hippocampal volume, were entered simultaneously as the primary independent variables. Subcortical and cortical infarct variables were created (where 1 = present and 0 = absent) and entered simultaneously into the model. The variables were dummy coded with individuals with neither subcortical nor cortical infarcts as the reference. Separate models evaluated each cognitive domain score as dependent variables. The models included demographic factors and total relative brain volume as covariates and were repeated with the additional vascular risk factor covariates. The vascular risk factors were coded as absent (0) or present (1). For ethnicity, Hispanic and African Americans were dummy coded, with non-Hispanic/non–African American coded as a reference group.
Finally, we used multiple regression to examine whether infarcts and hippocampus volume relate to specific aspects of memory performance. Hippocampal volumes, subcortical and cortical infarcts, and demographic variables were entered simultaneously as predictor variables. Separate simple and fully adjusted models were run for the learning, long-term recall, delayed recall, and delayed recognition parts of the SRT.
RESULTS
Sample.
Of 769 participants with available structural MRI data, 52 met criteria for dementia at the clinical evaluation closest to the neuroimaging study and were excluded from the analyses, leaving 717 subjects. Seventeen participants with evidence of both cortical and subcortical strokes and 5 with infarct ratings that could not be completed reliably were excluded. Of the remaining 695 participants, hippocampus volume was calculated in 658. Participants who did not have hippocampal volumes calculated were similar in age (F1,694 = 0.398, p = 0.598), education (F1,694 = 2.079, p = 0.150), sex (χ2[1] = 1.196, p = 0.274), and race/ethnicity (χ2[3] = 1.862, p = 0.602) as those with hippocampal volumes calculated. There were 484 individuals in the “no infarct” group, 132 in the subcortical infarct group, and 42 individuals in the cortical infarct group (table 1). There were no differences in the demographic and vascular risk profiles or relative brain volumes among the 3 infarct groups. Individuals with subcortical and cortical infarcts were more likely to have a clinical history of stroke and those without infarct had larger relative hippocampal volumes than those with subcortical infarcts. Determination of mild cognitive impairment (MCI)25 was made in 643 participants; there was a greater proportion of individuals meeting MCI criteria among those with cortical infarcts than among those without infarct. Some participants (n = 26) had infarcts in regions that were not considered in our cortical/subcortical classification (most were cerebellar infarcts, n = 21). We ran separate analyses on this subgroup, and decided to retain these individuals in the “no infarct” group for 3 reasons. First, individuals with cerebellar infarcts did not have reduced hippocampal volumes relative to individuals without any infarcts. Similarly, there was no association of cerebellar infarcts with cognitive functioning in any of the 4 cognitive domains. Finally, we ran sensitivity analyses by excluding them from the primary analyses and the findings remained unchanged.
Table 1.
Clinical and demographic characteristics of study subjects
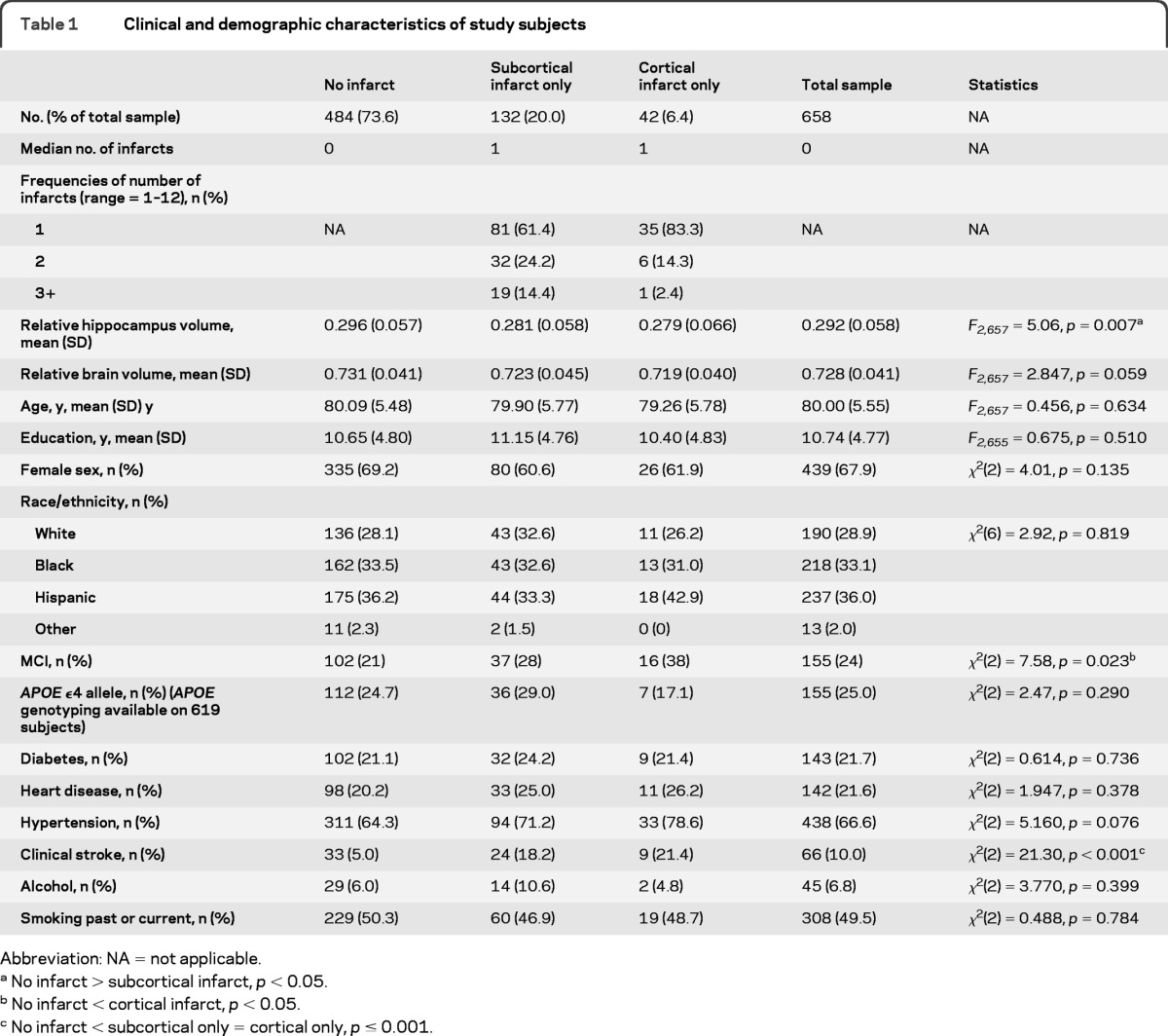
Abbreviation: NA = not applicable.
No infarct > subcortical infarct, p < 0.05.
No infarct < cortical infarct, p < 0.05.
No infarct < subcortical only = cortical only, p ≤ 0.001.
Hippocampus is smaller in elderly people without dementia with brain infarcts.
Individuals with any brain infarct had smaller hippocampi than those without infarct (mean [SD] = 0.2838 [0.059] vs 0.296 [0.057], F1,642 = 5.256, p = 0.022); none of the demographic or vascular risk factors were associated with hippocampal volume. When comparing the 3 infarct groups, a significant main effect (F2,642 = 4.149, p = 0.016) indicated that those with cortical infarcts (mean [SD] = 0.2798 [0.0655]) had similar relative hippocampal volumes as those with subcortical infarcts (mean [SD] = 0.2811 [0.0581]; p = 0.893), who, in turn, had smaller relative hippocampal volumes than individuals without infarct (mean [SD] = 0.2962 [0.05704]; p = 0.012). We repeated this analysis with total relative brain volume as an additional covariate to ensure that the observation is not a reflection of total brain atrophy and the pattern remained the same: individuals with cortical infarcts had similar hippocampal volume as those with subcortical infarcts (p = 0.244), who, in turn, had smaller hippocampal volumes than those without any infarct (p = 0.031). In a separate analysis, we examined the relationship between infarcts in the vascular territory supplying the hippocampus (posterior cerebral artery [PCA]) and hippocampus volume. Although none of the subjects had infarcts in the hippocampal formation, infarcts elsewhere in the same vascular territory have been reported to be associated with dysfunction of the hippocampus.16 However, we found no association between infarcts within PCA territory and hippocampal volume (overall model: F = 0.800, p = 0.165, presence of PCA infarct: β = 0.011, p = 0.165).
Hippocampus volume and brain infarcts are independently associated with memory.
Smaller hippocampus, cortical, and subcortical infarcts were all associated with poorer memory (table 2). As these variables were entered simultaneously into the model, their associations with memory were independent of each other. In the model adjusted for demographic factors, lower hippocampus volume was associated with poorer performance only in the memory domain, whereas infarcts were associated with worse performance in multiple cognitive domains. Sex, race/ethnicity, and education were related to cognitive performance, as previously reported26–28 (table 2). In the fully adjusted model, the association of cortical infarcts with memory reduced to trend level (p = 0.060), but the effect size did not change notably (−0.211 vs −0.218). Otherwise the findings remained as described above. History of diabetes was associated with poorer performance in language, executive/speed, and visuospatial domains. History of smoking was associated with better performance on memory testing.
Table 2.
The association of subcortical infarct, cortical infarct, and relative hippocampal volume with cognitive functioning in 4 domains, controlling for total brain atrophy (i.e., relative brain volume) and demographic variables (separate models were run for each cognitive outcome; variables in the left column are the model predictors; values reported are unstandardized β weights [SE]) (top) and the same models rerun to include additional vascular disease covariates (bottom)
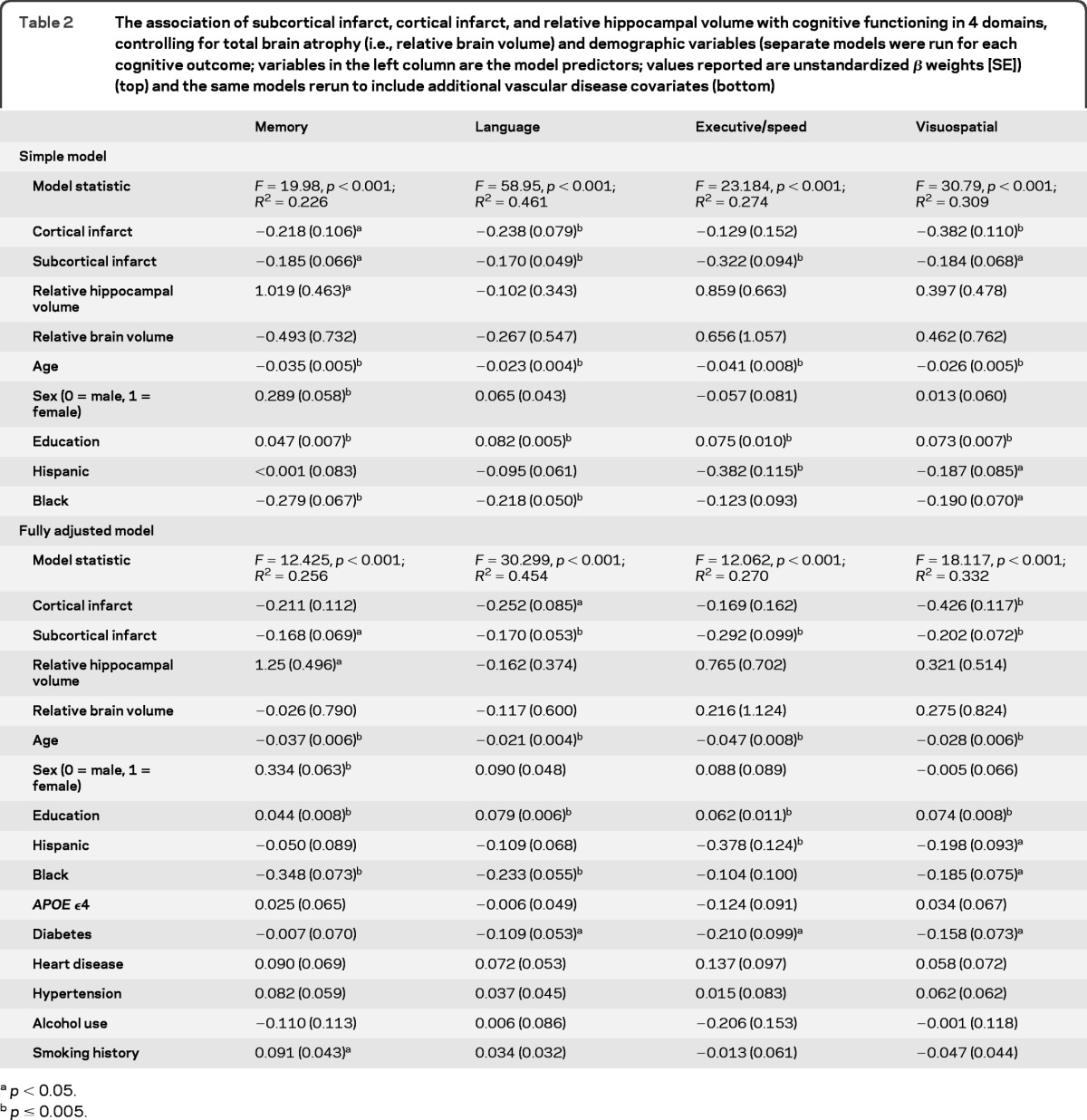
p < 0.05.
p ≤ 0.005.
Infarcts and hippocampal volume are associated with different aspects of memory performance.
Cortical and subcortical infarcts and smaller hippocampal volume were associated with specific aspects of memory performance (table 3). Hippocampal volume was associated with the long-term recall and delayed recognition aspects of the SRT and with delayed free recall at trend level. Subcortical infarcts correlated with performance on learning, long-term recall, and delayed free recall. Cortical infarcts were associated with performance on SRT delayed recognition. In the fully adjusted model, although the association between subcortical infarcts and delayed free recall was reduced to nonsignificance, the pattern of results and effect sizes remained similar.
Table 3.
The profile of memory subtest performance in association with infarcts and hippocampus volume (values reported are unstandardized β weights [SE]) (top) and the same models rerun to include additional vascular disease covariates (bottom)
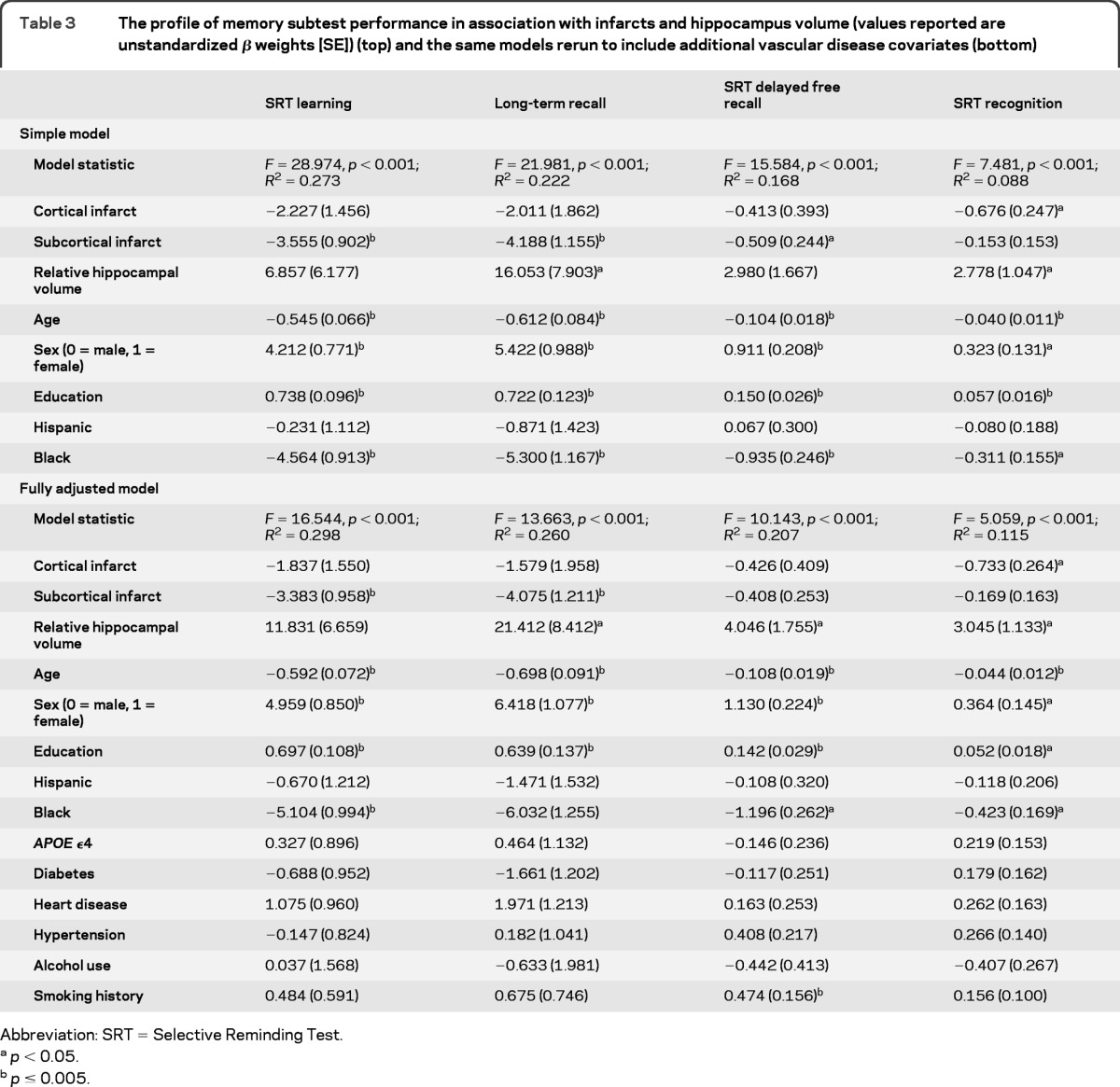
Abbreviation: SRT = Selective Reminding Test.
p < 0.05.
p ≤ 0.005.
DISCUSSION
We found that brain infarcts are associated with a smaller hippocampus, and that both infarcts and hippocampal volume are independently associated with global memory functioning. Furthermore, infarcts and hippocampal volume are associated with specific aspects of memory functioning, which are overlapping but unique.
The traditional view is that vascular disease and AD are frequent causes of dementia.29 The clinical profile of vascular dementia is thought of as primarily a decline in nonmemory domains of cognition, in particular executive function and perceptual speed, and any significant memory decline is attributed to coexisting AD and AD-associated hippocampal atrophy. Although the current study did not examine individuals with dementia per se, the implication of our data is significant in that they suggest that history of brain infarcts can lead to a phenotype that is typically thought of as prodromal AD—that is, infarcts are associated with poorer memory, and smaller hippocampus, not just poorer performance in nonmemory domains. Brain infarcts, particularly subcortical ones, are associated primarily with executive dysfunction, whereas hippocampal volume is correlated with memory performance.9,30,31 Cortical infarcts, in addition to being associated with deficits in processing speed, are also associated with worse episodic memory, but these findings have been interpreted with caution.7,8 A significant gap has remained in the literature: to our knowledge, ours is the first study to examine whether memory is associated with the presence of prior infarcts independently of hippocampal atrophy.
Memory processing is dependent on communication of the hippocampus with diffuse brain networks, including association cortices, and subcortical structures such as the thalamus, the basal forebrain, and the amygdala. Infarcts causing injury to parts of this network likely cause dysfunction in some aspect of memory processing and performance. Memory deficits could also be secondary to destruction of brain parenchyma, granular cortical atrophy, or other cortical changes due to infarcts themselves or hypoperfusion,32 or a consequence of deficits in other cognitive domains. Another possibility is that the ischemia that causes infarcts can also affect the hippocampus. The CA1 subfield of the hippocampus is most susceptible to ischemia, and infarcts are related to lower blood volume in this region.16 Thus, it is also possible that infarcts and neurodegenerative disease affect the hippocampal formation in distinct and complementary ways that result in similar clinical phenotypes.33
Interestingly, the phenotype may be only grossly identical—with prominent memory deficit—but under the surface there appears to be a unique effect of infarcts and hippocampus volume on memory. Hippocampal volume was associated specifically with memory performance, whereas infarcts were associated with poorer performance in all cognitive domains. When we examined specific aspects of memory testing, we found that although overlapping to some degree, the performance profiles associated with smaller hippocampus, cortical, and subcortical infarcts are unique. Hippocampal volume is associated with performance on long-term recall, delayed recognition, and delayed free recall. This finding is consistent with classically described hippocampus-dependent functions.14 Subcortical infarcts, on the other hand, showed a significant association with performance on learning, long-term recall, and delayed recall. Although we cannot rule out that executive dysfunction9 is contributing to the learning and memory deficit observed, participants with subcortical infarcts show evidence of deficits in learning and long-term memory. Cortical infarcts were associated with performance on delayed recognition of the selective reminding test. It should be noted that infarct data were coded categorically as present or absent; quantitative volumetric analysis of cerebral infarcts may yield more precise information about the relationship between the severity of cerebrovascular disease and cognition.
In addition to the observed hippocampus independent effect on memory performance, we found that infarcts are associated with a smaller hippocampus above and beyond whole-brain atrophy, and in turn that a smaller hippocampus is associated with poorer memory performance. These findings suggest that memory deficits may occur with brain infarcts secondary to both extrahippocampal and hippocampal processes—two independent sources of memory dysfunction. The etiology of hippocampal atrophy in aging is generally thought to be neurodegeneration related to AD or sclerosis.34,35 The observed associations of infarcts and hippocampal volume with cognitive function in our study could be explained by either or a combination of these processes. Numerous studies have suggested an association or interaction between cerebrovascular disease and AD,36,37 possibly due to accelerated AD pathology, such as increased deposition of amyloid. Hippocampal sclerosis in those with brain infarcts could be secondary to transient or chronic hypoperfusion related to vascular disease, or wallerian degeneration secondary to cortical injury in brain regions communicating with the hippocampus.32,38 Our observation that hippocampal atrophy is not related to infarcts within the vascular territory supplying the hippocampus may support the latter hypothesis.
A recent study among 425 elderly subjects with neuropathologic examination found an association of cortical microinfarcts with worse semantic and episodic memory8,9 and there was no interaction of microinfarcts with AD pathology. Taken together with our results, these observations further support the notion that brain infarcts exert a negative effect on memory function independently of markers of incipient AD, including hippocampal atrophy. The observation that brain infarcts have an effect on memory and other cognitive domains independently of changes in hippocampus has significant implications with regard to a critical need for stroke prevention. In a prospectively followed community sample of elderly subjects, more than 50% of those with dementia had multiple pathologies on autopsy, whereas among those without dementia over 80% have single or no pathology.11 This observation suggests that dementia is a cumulative effect of “multiple hits” that most often include AD, Parkinson/Lewy body pathology, and brain infarcts; focusing on prevention of one of the “hits” may decrease the incidence of dementia. The majority of individuals in our study who were found to have brain infarcts on MRI had no clinical history of stroke. In this community cohort, the sensitivity of stroke self-report for a diagnosis of stroke on MRI is only 32.4%.39 Therefore, brain infarction is largely a silent injury, and aggressive clinical screening and neuroradiologic examination would be needed to identify individuals with and at risk for development of brain infarcts. Brain infarcts are a largely preventable brain injury, with clearly identified risk factors, and prevention programs. A public health push toward emphasizing stroke prevention may significantly decrease incidence of dementia.
GLOSSARY
- AD
Alzheimer disease
- FLAIR
fluid-attenuated inversion recovery
- FOV
field of view
- MCI
mild cognitive impairment
- PCA
posterior cerebral artery
- SRT
Selective Reminding Test
- TE
echo time
- TI
inversion time
- TR
repetition time
AUTHOR CONTRIBUTIONS
Drafting/revising the manuscript for content: Drs. Blum and Brickman. Study concept or design: Drs. Blum, Luchsinger, Mayeux, and Brickman. Analysis or interpretation of data: Drs. Blum, Luchsinger, Schupf, Stern, DeCarli, Small, Mayeux, and Brickman. Acquisition of data: Drs. Manly, Schupf, and Brown. Statistical analysis: Drs. Blum and Brickman. Study supervision and coordination. Drs. Manly, Schupf, Brown, and Mayeux. Obtaining funding: Drs. Mayeux and Brickman.
DISCLOSURE
Dr. Blum reports no disclosures. Dr. Luchsinger serves on scientific advisory boards for Nutricia and Pennington Research Institute, LSU; serves on the editorial board of the Journal of Alzheimer's Disease; receives publishing royalties for Diabetes and the Brain (Humana Press, 2009); and receives research support from the NIH (NIMHD, NIA, NIDDK, NICHD), the Institute for the Study of Aging, the American Diabetes Association, and the Alzheimer's Association. Dr. Manly serves as an Associate Editor for the Journal of the International Neuropsychological Society and receives research support from the NIH/NIA and the Alzheimer's Association. Dr. Schupf has served as a consultant for Elan Corporation and receives research support from the NIH and the Alzheimer's Association. Dr. Stern has served as a consultant for Allergan Inc., Cephalon, Inc., Elan Corporation, Eisai Inc., Pfizer Inc, Ortho-McNeil Neurologics®, Merck Serono, GlaxoSmithKline, Eli Lily and Company, Bayer Schering Pharma, and Janssen; and receives research support from the NIH (NIA, NINDS). Dr. Brown receives research support from the NIH. Dr. DeCarli serves as Editor-in-Chief for Alzheimer Disease and Associated Disorders; serves as a consultant for Takeda Pharmaceutical Company Limited, Avid Radiopharmaceuticals, Inc., Bayer Schering Pharma, and Avanir Pharmaceuticals; and receives research support from the NIH (NIA, NHLBI). Dr. Small serves on a scientific advisory board for BCI; receives research support from the NIH; and receives Board of Directors compensation and license fee payments from BCI. Dr. Mayeux receives research support from the NIH. Dr. Brickman serves on the editorial boards of the Journal of the International Neuropsychological Society and Neuropsychology Review; serves as a consultant for ProPhase LLC.; and receives research support from the NIH and the Alzheimer's Association.
REFERENCES
- 1. Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol 2001; 58: 1395– 1402 [DOI] [PubMed] [Google Scholar]
- 2. Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science 1997; 278: 412– 419 [DOI] [PubMed] [Google Scholar]
- 3. Golomb J, de Leon MJ, Kluger A, George AE, Tarshish C, Ferris SH. Hippocampal atrophy in normal aging: an association with recent memory impairment. Arch Neurol 1993; 50: 967– 973 [DOI] [PubMed] [Google Scholar]
- 4. Schneider JA, Wilson RS, Cochran EJ, et al. Relation of cerebral infarctions to dementia and cognitive function in older persons. Neurology 2003; 60: 1082– 1088 [DOI] [PubMed] [Google Scholar]
- 5. Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med 2003; 348: 1215– 1222 [DOI] [PubMed] [Google Scholar]
- 6. Reitz C, Luchsinger JA, Tang MX, Manly J, Mayeux R. Stroke and memory performance in elderly persons without dementia. Arch Neurol 2006; 63: 571– 576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saczynski JS, Sigurdsson S, Jonsdottir MK, et al. Cerebral infarcts and cognitive performance: importance of location and number of infarcts. Stroke 2009; 40: 677– 682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA. Microinfarct pathology, dementia, and cognitive systems. Stroke 2011; 42: 722– 727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carey CL, Kramer JH, Josephson SA, et al. Subcortical lacunes are associated with executive dysfunction in cognitively normal elderly. Stroke 2008; 39: 397– 402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tatemichi TK, Desmond DW, Prohovnik I. Strategic infarcts in vascular dementia. A clinical and brain imaging experience. Arzneimittelforschung 1995; 45: 371– 385 [PubMed] [Google Scholar]
- 11. Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007; 69: 2197– 2204 [DOI] [PubMed] [Google Scholar]
- 12. van de Pol LA, Korf ES, van der Flier WM, et al. Magnetic resonance imaging predictors of cognition in mild cognitive impairment. Arch Neurol 2007; 64: 1023– 1028 [DOI] [PubMed] [Google Scholar]
- 13. Jack CR, Jr., Petersen RC, Xu YC, et al. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology 1999; 52: 1397– 1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manns JR, Eichenbaum H. Evolution of declarative memory. Hippocampus 2006; 16: 795– 808 [DOI] [PubMed] [Google Scholar]
- 15. Schmidt-Kastner R, Freund TF. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 1991; 40: 599– 636 [DOI] [PubMed] [Google Scholar]
- 16. Wu W, Brickman AM, Luchsinger J, et al. The brain in the age of old: the hippocampal formation is targeted differentially by diseases of late life. Ann Neurol 2008; 64: 698– 706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tang MX, Cross P, Andrews H, et al. Incidence of AD in African-Americans, Caribbean Hispanics, and Caucasians in northern Manhattan. Neurology 2001; 56: 49– 56 [DOI] [PubMed] [Google Scholar]
- 18. Brickman AM, Schupf N, Manly JJ, et al. Brain morphology in older African Americans, Caribbean Hispanics, and whites from northern Manhattan. Arch Neurol 2008; 65: 1053– 1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stern Y, Andrews H, Pittman J, et al. Diagnosis of dementia in a heterogeneous population: development of a neuropsychological paradigm-based diagnosis of dementia and quantified correction for the effects of education. Arch Neurol 1992; 49: 453– 460 [DOI] [PubMed] [Google Scholar]
- 20. Siedlecki KL, Honig LS, Stern Y. Exploring the structure of a neuropsychological battery across healthy elders and those with questionable dementia and Alzheimer's disease. Neuropsychology 2008; 22: 400– 411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Siedlecki KL, Manly JJ, Brickman AM, Schupf N, Tang MX, Stern Y. Do neuropsychological tests have the same meaning in Spanish speakers as they do in English speakers? Neuropsychology 2010; 24: 402– 411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buschke H, Fuld PA. Evaluating storage, retention, and retrieval in disordered memory and learning. Neurology 1974; 24: 1019– 1025 [DOI] [PubMed] [Google Scholar]
- 23. DeCarli C, Maisog J, Murphy DG, Teichberg D, Rapoport SI, Horwitz B. Method for quantification of brain, ventricular, and subarachnoid CSF volumes from MR images. J Comput Assist Tomogr 1992; 16: 274– 284 [DOI] [PubMed] [Google Scholar]
- 24. DeCarli C, Massaro J, Harvey D, et al. Measures of brain morphology and infarction in the Framingham Heart Study: establishing what is normal. Neurobiol Aging 2005; 26: 491– 510 [DOI] [PubMed] [Google Scholar]
- 25. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004; 256: 183– 194 [DOI] [PubMed] [Google Scholar]
- 26. Heaton RK, Grant I, Matthews CG, Adams KM. Differences in neuropsychological test performance associated with age, education, and sex. In: Neuropsychological Assessment of Neuropsychiatric Disorders. New York: Oxford University Press; 1986: 100– 120 [Google Scholar]
- 27. Manly JJ, Jacobs DM, Sano M, et al. Cognitive test performance among nondemented elderly African Americans and whites. Neurology 1998; 50: 1238– 1245 [DOI] [PubMed] [Google Scholar]
- 28. Brickman AM, Siedlecki KL, Muraskin J, et al. White matter hyperintensities and cognition: testing the reserve hypothesis. Neurobiol Aging 2011; 32: 1588– 1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grabowski TJ, Damasio AR. Definition, Clinical Features and Neuroanatomical Basis of Dementia. Cambridge, UK: Cambridge University Press; 2004: 1– 33 [Google Scholar]
- 30. Gainotti G, Acciarri A, Bizzarro A, et al. The role of brain infarcts and hippocampal atrophy in subcortical ischaemic vascular dementia. Neurol Sci 2004; 25: 192– 197 [DOI] [PubMed] [Google Scholar]
- 31. Fein G, Di Sclafani V, Tanabe J, et al. Hippocampal and cortical atrophy predict dementia in subcortical ischemic vascular disease. Neurology 2000; 55: 1626– 1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jellinger KA. The pathology of ischemic-vascular dementia: an update. J Neurol Sci 2002; 203–204: 153–157 [DOI] [PubMed] [Google Scholar]
- 33. Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease: The Nun Study. JAMA 1997; 277: 813– 817 [PubMed] [Google Scholar]
- 34. Jagust WJ, Zheng L, Harvey DJ, et al. Neuropathological basis of magnetic resonance images in aging and dementia. Ann Neurol 2008; 63: 72– 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chui HC, Zarow C, Mack WJ, et al. Cognitive impact of subcortical vascular and Alzheimer's disease pathology. Ann Neurol 2006; 60: 677– 687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 2005; 65: 545– 551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Honig LS, Tang MX, Albert S, et al. Stroke and the risk of Alzheimer disease. Arch Neurol 2003; 60: 1707– 1712 [DOI] [PubMed] [Google Scholar]
- 38. Desikan RS, Sabuncu MR, Schmansky NJ, et al. Selective disruption of the cerebral neocortex in Alzheimer's disease. PLoS One 2010; 5: e12853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reitz C, Schupf N, Luchsinger JA, et al. Validity of self-reported stroke in elderly African Americans, Caribbean Hispanics, and whites. Arch Neurol 2009; 66: 834– 840 [DOI] [PMC free article] [PubMed] [Google Scholar]