

Abstract
Osteopetrosis, a disorder of skeletal bone, can cause death during childhood. We previously described a new spontaneous autosomal recessive osteopetrosis mouse mutant, “new toothless” (ntl). In this study, we reported for the first time the identification, cloning and characterization of the coiled-coil domain-containing 154 (CCDC154), a novel gene whose deletion of ~5 kb sequence including exons 1–6 was completely linked to the ntl mutant. The CCDC154 was conserved between mouse and human and is wildly expressed in mouse tissues. The cellular localization of CCDC154 was in the early endosomes. Overexpression of CCDC154 inhibited cell proliferation of HEK293 cells by inducing G2/M arrest. CCDC154 also inhibited tumor cell growth, and the soft agar assay revealed a significant decrease of the colony size of Hela cells upon transfection of CCDC154. Our results indicate that CCDC154 is a novel osteopetrosis-related gene involved in cell cycle regulation and tumor suppression growth.
Keywords: CCDC154, osteopetrosis, cellular localization, cell proliferation, G2/M arrest
Introduction
Osteoclasts are specialized multinucleated cells, which are responsible for degradation of bone mineral and bone matrix.1 Abnormalities of osteoclast differentiation or function are associated with many skeleton disorders, including cancer bone metastasis, osteoporosis and osteopetrosis.2,3 Osteopetrosis, characterized by increased bone density, can cause death during childhood due to ablation of the bone marrow space and skeletal abnormalities in the absence of normal osteoclasts and bone remodeling activity.4 Several mouse genes, such as Gl, Atp6i, Clcn-7, c-Src and Traf-6, when mutated, gave rise to osteopetrosis.5-9 The human homologs for three of these genes (GL, ATP6i and CLCN-7, respectively) are in fact disease-causing genes for human autosomal recessive osteopetrosis.5-7,10 Other genes, like RANKL, PLEKHM1 and NEMO, were also reported to be involved in the genesis of human osteopetrosis.11-13 However, mutations in these genes do not appear to account for all of the cases of this disease,14 suggesting that there are additional disease-causing genes responsible for the human autosomal recessive osteopetrosis.
In our mouse colony, a new spontaneous autosomal recessive osteopetrosis mouse mutant (ntl) was discovered.15 This new mutant is distinct from all known osteopetrosis mutant mice reported to date by a number of features, including their ability to reproduce and their long and twisted nail phenotype. Genetic mapping data placed the mutant allele within a 3.2 cM genetic interval on mouse chromosome 17, a region that contains about 50 genes. A known osteopetrosis gene, Clcn-7, is also present in this region. However, no changes of Clcn-7 were observed, ruling out the possibility that the mutant phenotype is due to an alteration of Clcn-7. Our previous data indicate that this spontaneous mutant represents a unique novel osteopetrosis disease-causing gene.15
Based on our previous results, further experiments were performed to define the candidate gene. In the present study, we provided evidence that a ~5 kb deletion including exons 1–6 of CCDC154, a gene predicted by automated computational analysis (GenBank: XM_139904.7), was co-segregated with the mutant phenotype, suggesting that CCDC154 was a candidate gene leading to severe recessive osteopetrosis. CCDC154 is a novel gene, and little is known about its function. To better understand the roles of CCDC154, the cDNA sequence from mouse and human were cloned and characterized. The CCDC154 represented an extensive expression pattern in mouse tissues and a sublocalization in early endosomes. While exploring the biological functions of the CCDC154 gene, we found that CCDC154 could regulate cell growth. Overexpression of CCDC154 significantly inhibited cell proliferation. The mechanism of cell growth inhibition by CCDC154 was due to induced G2/M arrest, rather than increased apoptosis. CCDC154 also inhibited tumor cell growth. In vitro soft agar assay showed that CCDC154 significantly decreased the colony size of Hela cells. These data suggest that CCDC154 is a novel cell cycle modulator as well as a potential tumor suppressor.
Results
Identification of CCDC154 as a novel osteopetrosis-related gene
An ntl mouse representing a new model of autosomal-recessive osteopetrosis was obtained previously,15 and the mutant allele was mapped to a 3.2 cM genetic interval on mouse chromosome 17. A survey of the annotated genes within this 3.2 cM region revealed a number of potential candidates, including Atp6v0c,16 Cacna1h17 and Rab26.18 However, our preliminary results did not show any changes in these genes (data not shown). Using a RT-PCR approach, we found that CCDC154, a gene predicted by automated computational analysis (GenBank: XM_139904.7), failed to show any signal in the mutant osteoclasts (Fig. 1A). Genomic sequencing revealed that there was a ~5 kb deletion including exons 1–6 of CCDC154 gene in the ntl mice (Fig. 1B). This result was confirmed by the Southern blot analysis (Fig. 1C). Furthermore, we examined 100 offspring from the ntl mice by genomic typing and found that the deletion of CCDC154 was completely linked to the ntl mutant (Fig. 1D). Together, these data strongly suggest that the CCDC154 is the candidate ntl allele.

Figure 1. Identification of CCDC154 as a candidate ntl allele. (A) RT-PCR assay was used to detect the mRNA expression of CCDC154. The templates were extracted from the differentiated mouse osteoclasts. Specific primer pairs were listed in Table S2 and indicated as arrows in (B). Expression of CCDC154 was observed in wild-type (wt, +/+) osteoclasts while the transcript was undetectable in mutant mouse (mut, −/−). (B) Genomic structures of the wt and the mut mouse CCDC154 genes. The restriction sites used for Southern blot analysis are BglII. (C) Southern blot analysis of the wt and the mut CCDC154 genomic DNA. The fragment used as probe (thick horizontal line) is indicated in (B). The wild-type hybridization pattern consists of a ~7.4 kb BglII fragment. The deleted CCDC154 allele shows a shorter fragment of ~2.5 kb. (D) Linkage of CCDC154 to the ntl mutant was detected by genomic typing PCR. Genomic DNA was isolated from mouse tail and PCR was performed with primers mCCDC154 F3 and mCCDC154 R3 (Table S2).
Cloning and characterization of mouse and human CCDC154 genes
To study the function of CCDC154, we cloned and characterized the mouse and human CCDC154 genes. The open reading frames obtained from mouse (GenBank: JN935900) and human (GenBank: JN935901) were 1,995 bp and 2,004 bp in length, encoding 664 and 667 amino acids, respectively (Fig. S1). The coding sequences between mouse and human CCDC154 genes shared 72% identity, and their deduced amino acid sequences had 65% similarity (Fig. 2A). A protein-functional motif search showed that mouse CCDC154 contained six coiled-coil domains, and human CCDC154 contained four domains. In addition, the cyclin-binding motifs and TRAF2-binding motifs were also found (Fig. 2B; Fig. S1). Genomic structure characterization revealed a similar gene organization between mouse and human CCDC154. Both contained 17 exons and 16 introns, and the length of their exons was almost the same (Fig. 2C). Comparison of the three-dimensional structure showed that mouse and human CCDC154 shared a similar tertiary structure, containing an N-terminal domain and a C-terminal domain (Fig. 2D). Together, these data indicate that CCDC154 is conserved between mouse and human. Additionally, sequence alignment displayed a related, high amino acid identity (51–97%) among human, chimpanzee, monkey, orangutan, horse, mouse, rat, pig, hamster and panda (Fig. S2 and Table S1), indicating the conservation of CCDC154 among mammals. Moreover, CCDC154 abided the evolutionary rule as accessed by the phylogenetic analysis, which showed that the human CCDC154 clustered together with the CCDC154 of other primates, while the CCDC154 of mouse and other rodents formed an exclusive group (Fig. S3).

Figure 2. Comparison of mouse and human CCDC154 genes. (A) Alignment of the predicted amino acid sequences of mouse (mCCDC154) and human CCDC154 (hCCDC154). Residues conserved between two proteins are shaded with black. (B) Schematic representation of the predicted domains in mouse and human CCDC154. (C) Genomic structure analysis of mouse and human CCDC154. Coding exons are indicated with black boxes. Non-coding exons are indicated with white boxes. The length of the exons is indicated by the number above. Lines adjacent to exons represent introns. The number below shows the length of introns. (D) The three-dimensional structure comparison of mouse and human CCDC154. The three-dimensional structures of mouse and human CCDC154 were modeled by the PHYRE program with the programmed cell death 6-interacting protein as a template, respectively (Tables S3 and S4). Ribbons represent the α-helixes.
Tissue distribution and subcellular localization of CCDC154
RT-PCR analysis of the tissue distribution of CCDC154 revealed that the mouse CCDC154 mRNA was present in all examined tissues, including brain, heart, lung, liver, spleen, kidney, testis, muscle, intestine and thymus (Fig. 3A), indicating an extensive expression pattern of CCDC154. The subcellular localization of CCDC154 was determined using immunofluorescence staining with different cellular organelle markers. No colocalization of CCDC154 was observed with the lysosome marker Lamp1 and the trans-Golgi protein Gmx33, whereas there was a nearly complete overlap with EEA1, an early endosome marker (Fig. 3B), suggesting a localization of CCDC154 in early endosomes.

Figure 3. Tissue distribution and subcellular localization of CCDC154. (A) Tissue distribution of mouse CCDC154 mRNA. Total RNA was prepared from different tissues of C57BL/6J mouse and subjected to RT-PCR amplification with mouse CCDCD154 or β-actin sequence-specific primers (Table S2). The resulting PCR products were analyzed by 1% agarose gel and visualized by ethidium bromide staining. (B) Subcellular localization of CCDC154. Human (hCCDC154) or mouse CCDC154-RFP fusion construct (mCCDC154) was transfected into HEK293 or NIH3T3 cells. EEA1 and Lamp1 were immunostained with Alexa Fluor 488-labled secondary antibody. Gmx33 was visualized by transfecting the Gmx33-GFP fusion vector. Colocalization was observed by confocal microscope, Vec, pDsRed-N1 vector; Scale bars, 20 μm.
Overexpression of CCDC154 inhibits cell proliferation
To gain insights into the function of CCDC154, we overexpressed CCDC154 in HEK293 and NIH3T3 cells. Transient overexpression of CCDC154 significantly inhibited cell proliferation (Fig. 4). Smaller cell numbers (~50% of control cells) and lower colony formation efficiencies (60–70%) were observed in both mouse and human CCDC154 transient-transfected HEK293 cells (Fig. 4B and C). Transient transfection of mouse CCDC154 in NIH3T3 cells also led to a significant decrease in colony-formation (p < 0.05) (Fig. 4D). Additional support for the inhibitory effect of CCDC154 on cell growth came from the results of HEK293 stable transfectants. The growth rates of human CCDC154 stable clones were 40–70% of the control cells (Fig. 4E and F). And only 50–65% of colonies were formed by the human CCDC154 stable transfectants as compared with the control cells (Fig. 4G). Together, these data suggest that CCDC154 acts as a negative regulator in cell proliferation.

Figure 4. Overexpression of CCDC154 inhibits cell growth. (A) pEGFP-N1 was co-transfected to detect the transfection efficiency. Bar, 50 μm. (B−D) Transient transfection of CCDC154 inhibited human and mouse cell proliferation, as determined by the cell-counting assay and colony-formation assay. Mouse (mC) and human CCDC154 (hC) protein expression was detected by western blot using anti-myc antibody (upper panel). Vec, control vector. (E−G) Stable overexpression of CCDC154 suppressed human cell growth in cell-counting assay, MTT and colony formation assay. Vec is tranfected with control vector. hC-1# and hC-2# are two randomly selected human CCDC154 stable overexpression clones. Data shown are the mean results ± SD of a representative experiment performed in triplicate (n = 3), *p < 0.05, **p < 0.01 and ***p < 0.001.
Interestingly, the suppressive effect of CCDC154 on cell proliferation was also found in tumor cells (Fig. 5). Transient or stable overexpression of human CCDC154 in Hela cells led to a significant reduction in colony formation (Fig. 5A and C). Hela cells stably overexpressing CCDC154 also showed a significantly reduced cell proliferation in MTT assay (Fig. 5B). To further access the effect of CCDC154 on tumorigenesis in vitro, we performed the soft agar assay. Stable transfection of CCDC154 in Hela cells significantly decreased the size of colonies (p < 0.001) (Fig. 5D and E). These data imply that CCDC154 might be a potential tumor suppressor.

Figure 5. CCDC154 suppresses tumor cell growth. (A) Transient transfection of human CCDC154 (hC) inhibited Hela cell colony formation. CCDC154 expression was detected by western blot (upper panel). Vec is control vector. (B) Cell viability was decreased in pooled CCDC154-transfected Hela cells by the MTT assay. (C) Stable overexpression of human CCDC154 declined clonogenic ability of Hela cells. Pooled CCDC154-transfected cells (hC) were used. (D−E) The effect of CCDC154 on tumorigenesis in vitro was evaluated by a soft agar assay. The colony size was quantitated by ImageJ program. Bar, 100 μm. Data shown are the means ± SD of a representative experiment performed in triplicate (n = 3), *p < 0.05, **p < 0.01 and ***p < 0.001.
CCDC154 inhibits cell proliferation by inducing G2/M arrest
To understand the mechanism of CCDC154-induced growth inhibition, the cell apoptosis analysis was performed. However, overexpression of CCDC154 did not induce more apoptosis than the control cells (Fig. S4A and B), suggesting that the growth suppression induced by CCDC154 was not due to the cell apoptosis. Accordingly, overexpression of CCDC154 did not alter the protein expression of p53, Bcl-2 and Bax (Fig. S4C). Next, we tested if the CCDC154-induced growth inhibition on HEK293 cells was caused by a change of the cell cycle. Indeed, flow cytometry (FCM) analysis showed that overexpression of mouse or human CCDC154 significantly induced G2/M arrest (Fig. 6A). The mechanism underlying the G2/M arrest was further accessed by determining the expression of the key regulators of G2/M progress, Cdc2-cyclin B kinase complex. Western blot analysis revealed that overexpression of mouse or human CCDC154 reduced both the protein expression of Cdc2 (40–52%) and cyclin B1 (30–45%, Fig. 6B). In contrast, the expression of Chk1 and p21, the upper negative regulators of Cdc2-cyclin B kinase complex, was increased (Fig. 6B).

Figure 6. CCDC154 regulates G2/M checkpoint. (A) Cell cycle distribution of mouse (left) or human (right) CCDC154 stably transfected HEK293 cells. Data shown are the means ± SD of a representative experiment performed in triplicate (n = 3). Vec, control vector; mC-1# and mC-2#, two randomly selected mouse CCDC154 stable clones; hC-1# and hC-2#, two randomly selected human CCDC154 stable clones. (B) Western blot analysis. Whole-cell lysates isolated from HEK293/mCCDC154, HEK293/hCCDC154 and HEK293/Vec cells were separated on SDS-PAGE gel, transferred into PVDF membranes and incubated either with anti-myc (1:1,000), Cdc2 (1:1,000), cyclin B1 (1:1,000), Chk1 (1:500), p21 (1:200) or β-actin (1:2,000) antibody. Quantitation of protein levels was analyzed by ImageJ program. Control vector was set as 100%.
Functional domain analysis of CCDC154 in cell growth regulation
Derivatives with different deletion were constructed and used to determine the functional domain of CCDC154 (Fig. 7A and B). Colony-formation assay revealed that the derivative No. 3 (amino acids 1–365) containing two cyclin-binding motifs showed almost identical activity to the full-length CCDC154, while the derivative No. 1 with one cyclin-binding motif partially suppressed cell growth (Fig. 7C). However, no suppression was observed by the derivative No. 2 that contains no cyclin-binding motif. These data imply that two cyclin-binding domains in CCDC154 may play key roles in cell-growth suppression. Further structural and mutational studies are needed to inspect this speculation.
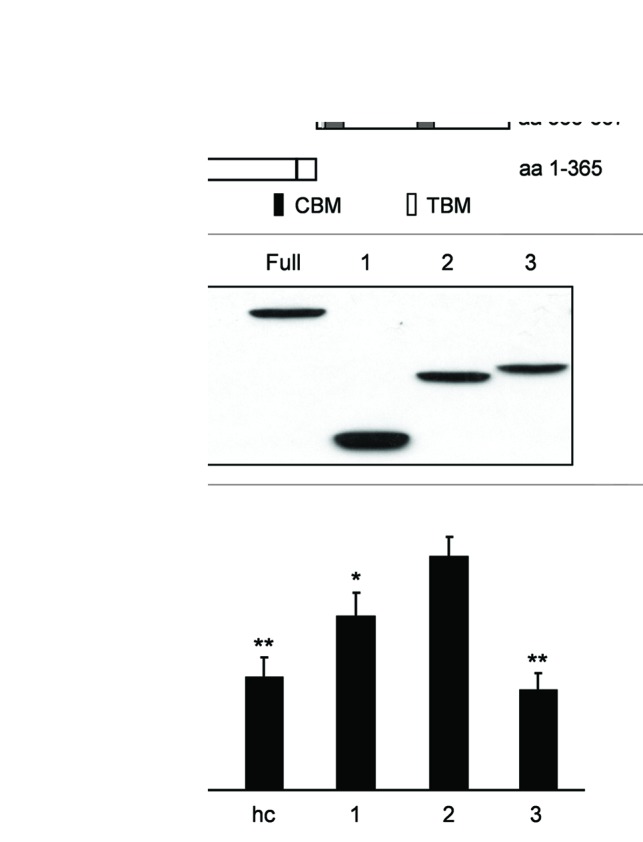
Figure 7. Functional domain analysis of CCDC154. (A) Schematic representation of the CCDC154 deletion derivatives. CCD, coiled-coil domain; CBM, cyclin binding motif; TBM, TRAF2 binding motif. (B) Western blot analysis of the expression of different CCDC154 derivatives indicated in (A). (C) The inhibitory effects of CCDC154 derivatives on HEK293 cell proliferation determined by the colony-formation assay.
Discussion
Previously, we discovered a spontaneous autosomal recessive osteopetrosis ntl mouse mutant, which represented a novel model for type II auto-recessive osteopertrosis with features such as absence of bone resorption, lack of tooth eruption and odontoma-like proliferation near the ends of incisors.15 In the present study, we characterized for the first time the gene and its mutation that was related to this mutant. Evidences that the CCDC154 is the candidate ntl allele were from: (1) RT-PCR showed no detectable mRNA expression of CCDC154 in homozygous mutant mice. (2) Genomic structural comparison of the CCDC154 in wild-type and ntl mice revealed a deletion of ~5 kb sequence, including 1–6 exons of the CCDC154 gene in the mutant mice. (3) The CCDC154 gene was co-segregated with the ntl mutant in 100 offspring of ntl mice as determined by genetic linkage analysis. Definite proof that CCDC154 is the disease-causing gene will come from a phenotype rescue experiment that is progressing in our laboratory.
The CCDC154 is a novel gene and little is known about its function so far. To explore the roles of CCDC154, we cloned the mouse and human CCDC154 genes. Mouse and human CCDC154 shared relatively high amino acid identity, suggesting CCDC154 is conserved in these two species. Further support for this assumption came from comparative studies representing similar genomic and three-dimensional structures between mouse and human CCDC154. Functional domain analysis revealed that the CCDC154 protein has several coiled-coil domains and two cyclin-binding motifs. The coiled-coil is a common motif that consists of two to five α-helices scrolled around each other to form a supercoil.19 The coiled-coil has been reported to be involved in a lot of cell functions like dimerization, protein-protein interaction, molecular targeting, DNA binding and transcriptional regulation.20-23 Recently, the coiled-coil domains have been found in matrilins, which are involved in the development and homeostasis of cartilage and bone. Oligomerization of multisubunit proteins through the coiled-coil domains is considered to be essential for forming functional matrilins.23 In our study, we found the two cyclin-binding motifs were required for cell growth suppression. The cyclin-binding motif, which exists in a number of cyclin-binding proteins, has been reported to have the function of regulating the cyclin-Cdk kinase activity and inducing G2/M arrest.24,25 We showed that overexpression of CCDC154 significantly reduced cyclin B1 and induced G2/M cell cycle arrest. These results, consistent with previous reports, imply that the cyclin-binding motifs play a critical role in cell growth regulation. To verify which domain, the cyclin-binding motifs or the coiled-coil domains of CCDC154, is the key factor for the bone-modeling deficiency, further studies are still needed.
Study of the intercellular localization of CCDC154 suggested its location in early endosomes. Endosomal localization has been reported for other osteopetrosis-related proteins, such as c-Src,26 Clcn-727 and PLEKHM1,12 whose expressions have been also found in the ruffled border. The ruffled border, in many respects, is similar to the late endosome. Many proteins, such as Rab7,28 V-ATPase29 and CI-MPRs,30 that are present in the late endosomes are also found in the ruffled border membranes. Endocytosis and biosynthesis through early endosome or early and late endosomes to the resorption lacuna have been suggested to play a key role in the formation and maintenance of the ruffled border.31 The ntl mice have a normal number of osteoclasts, but they fail to reabsorb bone. A loss of the ruffled border in the ntl osteoclasts has been suggested to account for the impaired bone resorption.15 The localization of CCDC154 in early endosomes implies that CCDC154 might be involved in the formation of ruffled border. Further experiments are needed to address this possibility.
The mouse CCDC154 mRNA was found in the brain, heart, lung, liver, spleen, kidney, testis, muscle, intestine and thymus. This ubiquitous expression of CCDC154 indicates its crucial physiological functions in organisms. To get more details about the roles of this novel gene, we performed functional analysis of CCDC154 in vitro. In this study, we have provided evidences that CCDC154 suppressed cell growth when overexpressed. We reasoned that apoptosis was the cause of this cell growth inhibition. However, our results suggested that the cell suppression is not due to apoptosis, but rather due to a change in the cell cycle. FCM analysis confirmed that overexpression of CCDC154 induced G2/M cell cycle arrest. The alteration of a cell cycle is normally accompanied by the changes of cell cycle-specific signal molecules.32,33 In correspondence with this, the protein level of G2/M regulator, cyclin B/Cdc2 complex, was suppressed with the overexpression of CCDC154. Moreover, we observed an increase in the expression of Chk1 and p21, the upper negative regulators of Cdc2. As CCDC154 represents a wild-border expression pattern in mouse tissues, its expression should be tightly regulated as an elevated protein level results in cell proliferation suppression. More efforts are required to figure out how this balance is controlled.
We have reported the identification and initial characterization of CCDC154, a novel gene whose mutation is related to the osteopetrosis mutant. We have showed that CCDC154 has a broad expression pattern and locates in early endosomes. Overexpression of CCDC154 induces G2/M arrest and suppresses cell growth, indicating that CCDC154 is a novel negative cell cycle regulator and a potential tumor suppressor. The detailed mechanism of CCDC154-induced G2/M arrest is vague, and further studies are required.
Materials and Methods
Mice, cell lines and culture conditions
The ntl mice15 were housed in our specific pathogen-free (SPF) facility. After weaning, the ntl mutant mice were fed with the liquid diet, Peptamin (Nestle Nutrition). HEK293, NIH3T3 and Hela cells (from ATCC) were cultured in DMEM medium (Thermo Scientific) supplemented with, unless otherwise stated, 10% heat-inactivated fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin, at 37°C in a humidified atmosphere containing 5% CO2.
Antibodies
Cyclin B1 (GNS1), Chk1 (G-4) and Lamp1 (H4A3) mouse monoclonals, Cdc2 (C-19) rabbit polyclonal and peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology. Beta-actin (13E5) and anti-myc (71D10) rabbit polyclonals were obtained from Cell Signaling Technology. EEA1 mouse monoclonal was purchased from BD Bioscience. Alexa Fluor 488-labeled secondary antibody for immunofluorescence was from Invitrogen.
Mutation screening, genomic typing and Southern blot analysis
Mutation screening experiments were performed as described before.15 Briefly, splenocytes of 4-wk-old mice were isolated and differentiated into osteoclasts in vitro. Differentiated osteoclasts were collected and the total RNA was extracted using TRIzol reagent (Invitrogen). The total RNA was reverse-transcribed into first-strand cDNA using a SuperScript™ First-Strand kit (Invitrogen). The mRNA expression of mouse CCDC154 was detected by RT-PCR with primers mCCDC154 F1/mCCDC154 R1 and mCCDC154 F2/mCCDC154 R2 (Table S2). Genomic DNA was isolated from mouse tail and the whole region of CCDC154 gene was sequenced by Invitrogen. Genomic DNA was digested with BglII and Southern blot analysis was performed as described previously.34
Gene cloning and construct generation
Total RNA was extracted from mouse testis and reverse transcribed into first-strand cDNA with a gene-specific reverse primer mCCDC154 R4 (Table S2) that was designed according to the predicted sequence of mouse CCDC154 (GenBank: NM_001079929). The open reading frame of mouse CCDC154 was obtained by RT-PCR using primers mCCDC154 F5/mCCDC154 R5 and mCCDC154 F6/mCCDC154 R6 (Table S2), sequenced and subcloned into pcDNA3.1-myc-His-A (Invitrogen). The open-reading frame of human CCDC154 was obtained with a similar strategy from HEK293 cells with primers hCCDC154 F1 and hCCDC154 R1 (Table S2) and inserted into pcDNA3.1-myc-His-A vector (pcDNA3.1-hCCDC154). The reverse sequence of mouse CCDC154 coding region was cloned into pcDNA3.1-myc-His-A as a control vector. To construct the expression vectors carrying RFP at the C-terminal ends of mouse or human CCDC154, the coding regions of mouse and human CCDC154 genes were amplified and subcloned into pDsRed1-N1 vector (Clontech) using XhoI/KpnI restriction sites. The resulting constructs mCCDC154-RFP and hCCDC154-RFP were confirmed by sequencing. The pEGFP-N1 plasmid was from Clontech, and the vector expressing the trans-Golgi protein Gmx33 tagged with GFP was a kind gift from Guangbin Luo (Case Western Reserve University). cDNA sequences encoding residues 1–225, 1–365 and 336–667 of human CCDC154 were amplified by PCR using pcDNA3.1-hCCDC154 as a template, with primers hCCDC154 F1/hCCDC154 R2, hCCDC154 F1/hCCDC154 R3 and hCCDC154 F4/hCCDC154 R1, respectively. The resulting sequences were subcloned into pcDNA3.1-myc-His-A vector.
Sequence analysis
Deduced amino acid sequence was generated by DNAman. Sequence alignment was produced using the ClusterW2 program35 and viewed with GeneDoc. Potential functional motifs and tertiary structures were analyzed with ELM program (www.elm.eu.org/) and Phyre server,36 respectively. Prediction of the coiled-coil domains was done with the following programs: SMART (www.smart.embl-heidelberg.de/), COILS37 and Paircoil2.38 Gene organizations were sorted out by comparing mouse and human CCDC154 cDNA sequences with their genomic sequences, respectively, using the NCBI splign program.
Tissue distribution of mouse CCDC154 mRNA
The expression of mouse CCDC154 mRNA in different tissues was determined by RT-PCR, using β-actin as an external control. Total RNA was isolated from adult mouse brain, heart, lung, liver, spleen, kidney, testis, muscle, intestine and thymus. Two pairs of gene-specific PCR primers were designed for mouse CCDC154 (mCCDC154 F7 and mCCDC154 R7, Table S2) and β-actin (ACT F1 and ACT R1, Table S2) cDNA amplification. Sequence of mouse β-actin was obtained from GenBank database (NM_007393). The PCR products were separated on a 1.0% agarose gel, stained with ethidium bromide and photographed.
Immunofluorescence microscopy
HEK293 or NIH3T3 cells grown on 16-mm coverslips were transfected with 2 μg of DNA plasmids using LipofectamineTM2000 (Invitrogen). Twenty-four hours after transfection, cells were fixed with 4% formaldehyde in phosphate-buffered saline (PBS) for 20 min at room temperature, followed by incubation with 0.1 M glycine for 10 min and permeabilization with 0.1% Triton X-100 in PBS for 20 min. After blocking with 1% bovine serum albumin (BSA) in PBS for 10 min, cells were incubated with primary anti-EEA1 (1:100) or anti-Lamp1 (1:100) diluted in PBS for 1 h, and secondary antibody conjugated to Alexa Fluor 488 for 1 h in the dark. Coverslips were mounted onto glass slides with mounting solution, and slides were viewed under a ZEISS confocal microscope (Oberkochen). Images were obtained using the ZEN 2009 Light Edition software, and data were subsequently processed with the Adobe Photoshop program.
Transient and stable transfection of cells, and establishment of pooled transfectants
HEK293, NIH3T3 or Hela cells cultured in 6-well plates were transfected with 2–5 μg of DNA plasmids using Lipofectamine 2000 reagent (Invitrogen). For stable transfectants, 24 h after transfection, cells were plated into 10-cm plates with medium containing G418 (1,500 μg/ml for HEK293 cells, 800 μg/ml for NIH3T3 cells and 1,000 μg/ml for Hela cells). The medium with G418 was replaced every 3 d. Cells were grown until the clones developed. G418-resistant clones were picked up and further amplified. Stably transfected clones were confirmed by western blot. For pooled transfectants, Hela cells were transfected with CCDC154 as described above, and after 2 wk of selection, G418-resistant cells were pooled together for further experiments.
Western blot analysis
Cells were harvested and lysed in cold lysis buffer (50 mM Tris/HCl, pH 7.6, 150 mM NaCl, 0.1% SDS, 1% Nonidet P-40, 1 mg/ml PMSF) at 4°C for 20 min. Cellular lysates were cleared by centrifugation, and protein concentrations were determined by Bradford method. Equal amounts of total cellular proteins were resolved by 12% SDS-polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Millipore). After blocking, the membranes were incubated with primary and secondary antibodies. Protein bands were visualized by the ECL system (Amersham Biosciences). Anti-myc was used to detect the CCDC154 expression. ImageJ program was used to quantitate digital images from scanned autorads.
MTT and cell-counting assays
The cell viability was determined by the MTT assay as described previously.39 Briefly, equal amounts of cells were plated into 96-well plates and cultured in DMEM medium with 5% FBS. At the indication time points, the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT, Sigma) solution was added into the wells and incubated at 37°C for another 4–6 h. The medium was carefully removed, and the formazan production was dissolved with dimethyl sulfoxide. The absorbance was measured at a weight length of 570 nm with Microplate Reader (BioRad Model 550). For the cell-counting assay, equal amounts of cells were plated into 48-well plates and cultured in DMEM medium with 5% FBS. The cells were counted after 6 d of incubation.
Colony-formation assay
The colony-formation assay was performed to determine the effect of CCDC154 on cell proliferation. Briefly, equal amounts of transfected cells were seeded in 6-well plates. Cells were selected with G418 and allowed to form colonies. After 10–12 d of culture, colonies were stained with 0.1% crystal violet. Colonies containing more than 20 cells were scored as positive.
Soft agar assay
The soft agar assay was performed as described previously to evaluate the effect of CCDC154 on tumorigenesis in vitro.40 Briefly, cells (1 × 104) were resuspended in medium containing 10% fetal bovine serum (FBS) with 0.3% agarose, and layered on top of 0.6% agar in medium supplemented with 20% FBS on 60-mm plates. After 10–20 d of culture at 37°C, plates were stained with 0.005% crystal violet for > 1 h. Colonies were photographed and the relative colony sizes were measured by ImageJ program.
Cell cycle analysis
Cell cycle measurement was done as described before.40 Briefly, 5 × 105 HEK293 cells were seeded in 6-well plates and allowed to adhere. Twenty-four hours later, the cells were harvested by centrifugation at 1,000 rpm for 5 min. The cell pellets were washed twice with PBS followed by fixation with ice-cold 70% ethanol and stored at -20°C overnight. Then, the pellets were washed with cold PBS, suspended in 500 ml PBS containing 50 mg/ml propidium iodide, 0.1 mg/ml RNase A and 0.05% Triton X-100 and incubated at 37°C for 40 min in the dark. The cell cycle distribution was determined on the Becton Dickinson FACSCaliburr. The experiment was repeated thrice under the same conditions.
Statistical analysis
All the data were analyzed via the Student’s t-test. The statistical difference p < 0.05 was considered to be significant.
Obara K, Ohsumi Y. Atg14: a key player in orchestrating autophagy. Int J Cell Biol. 20112011:713435. doi: 10.1155/2011/713435.
Supplementary Material
Acknowledgments
We thank Dr. Guangbin Luo (Case Western Reserve University) for kindly providing us with the ntl mice. This work was supported by the National Natural Science Foundation of China (31071086) and the Initiation Funds for Talent of Wenzhou Medical College (A890001).
Glossary
Abbreviations:
- CCDC154
coiled-coil domain-containing 154
- ntl
“new toothless” osteopetrosis mouse
- FCM
flow cytometry analysis
- SPF
specific pathogen-free
- MTT
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
- FBS
fetal bovine serum
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21642
References
- 1.Väänänen HK, Zhao H, Mulari M, Halleen JM. The cell biology of osteoclast function. J Cell Sci. 2000;113:377–81. doi: 10.1242/jcs.113.3.377. [DOI] [PubMed] [Google Scholar]
- 2.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389–406. doi: 10.1016/S1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 3.Lazner F, Gowen M, Pavasovic D, Kola I. Osteopetrosis and osteoporosis: two sides of the same coin. Hum Mol Genet. 1999;8:1839–46. doi: 10.1093/hmg/8.10.1839. [DOI] [PubMed] [Google Scholar]
- 4.de Vernejoul MC, Bénichou O. Human osteopetrosis and other sclerosing disorders: recent genetic developments. Calcif Tissue Int. 2001;69:1–6. doi: 10.1007/s002230020046. [DOI] [PubMed] [Google Scholar]
- 5.Chalhoub N, Benachenhou N, Rajapurohitam V, Pata M, Ferron M, Frattini A, et al. Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat Med. 2003;9:399–406. doi: 10.1038/nm842. [DOI] [PubMed] [Google Scholar]
- 6.Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, et al. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet. 2000;9:2059–63. doi: 10.1093/hmg/9.13.2059. [DOI] [PubMed] [Google Scholar]
- 7.Li YP, Chen W, Liang Y, Li E, Stashenko P. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet. 1999;23:447–51. doi: 10.1038/70563. [DOI] [PubMed] [Google Scholar]
- 8.Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–24. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-O. [DOI] [PubMed] [Google Scholar]
- 10.Sobacchi C, Frattini A, Orchard P, Porras O, Tezcan I, Andolina M, et al. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum Mol Genet. 2001;10:1767–73. doi: 10.1093/hmg/10.17.1767. [DOI] [PubMed] [Google Scholar]
- 11.Douni E, Rinotas V, Makrinou E, Zwerina J, Penninger JM, Eliopoulos E, et al. A RANKL G278R mutation causing osteopetrosis identifies a functional amino acid essential for trimer assembly in RANKL and TNF. Hum Mol Genet. 2012;21:784–98. doi: 10.1093/hmg/ddr510. [DOI] [PubMed] [Google Scholar]
- 12.Van Wesenbeeck L, Odgren PR, Coxon FP, Frattini A, Moens P, Perdu B, et al. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J Clin Invest. 2007;117:919–30. doi: 10.1172/JCI30328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai S, Abu-Amer W, Karuppaiah K, Abu-Amer Y. Evidence that the kinase-truncated c-Src regulates NF-κB signaling by targeting NEMO. J Cell Biochem. 2011;112:2463–70. doi: 10.1002/jcb.23170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis. 2009;4:5. doi: 10.1186/1750-1172-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu X, Rios HF, Jiang B, Xing L, Kadlcek R, Greenfield EM, et al. A new osteopetrosis mutant mouse strain (ntl) with odontoma-like proliferations and lack of tooth roots. Eur J Oral Sci. 2009;117:625–35. doi: 10.1111/j.1600-0722.2009.00690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perdu B, Odgren PR, Van Wesenbeeck L, Jennes K, Mackay CC, Van Hul W. Refined genomic localization of the genetic lesion in the osteopetrosis (op) rat and exclusion of three positional and functional candidate genes, Clcn7, Atp6v0c, and Slc9a3r2. Calcif Tissue Int. 2009;84:355–60. doi: 10.1007/s00223-009-9229-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heron SE, Khosravani H, Varela D, Bladen C, Williams TC, Newman MR, et al. Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann Neurol. 2007;62:560–8. doi: 10.1002/ana.21169. [DOI] [PubMed] [Google Scholar]
- 18.Tian X, Jin RU, Bredemeyer AJ, Oates EJ, Błazewska KM, McKenna CE, et al. RAB26 and RAB3D are direct transcriptional targets of MIST1 that regulate exocrine granule maturation. Mol Cell Biol. 2010;30:1269–84. doi: 10.1128/MCB.01328-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mason JM, Arndt KM. Coiled coil domains: stability, specificity, and biological implications. ChemBioChem. 2004;5:170–6. doi: 10.1002/cbic.200300781. [DOI] [PubMed] [Google Scholar]
- 20.Poon BP, Mekhail K. Cohesin and related coiled-coil domain-containing complexes physically and functionally connect the dots across the genome. Cell Cycle. 2011;10:2669–82. doi: 10.4161/cc.10.16.17113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obara K, Ohsumi Y. Atg14: a key player in orchestrating autophagy. Int J Cell Biol. 2011;2011:713435. doi: 10.1155/2011/713435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hohl M, Kwon Y, Galván SM, Xue X, Tous C, Aguilera A, et al. The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nat Struct Mol Biol. 2011;18:1124–31. doi: 10.1038/nsmb.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frank S, Schulthess T, Landwehr R, Lustig A, Mini T, Jenö P, et al. Characterization of the matrilin coiled-coil domains reveals seven novel isoforms. J Biol Chem. 2002;277:19071–9. doi: 10.1074/jbc.M202146200. [DOI] [PubMed] [Google Scholar]
- 24.Lee HJ, Chua GH, Krishnan A, Lane DP, Verma CS. Substrate specificity of cyclins determined by electrostatics. Cell Cycle. 2007;6:2219–26. doi: 10.4161/cc.6.18.4706. [DOI] [PubMed] [Google Scholar]
- 25.Chen J, Saha P, Kornbluth S, Dynlacht BD, Dutta A. Cyclin-binding motifs are essential for the function of p21CIP1. Mol Cell Biol. 1996;16:4673–82. doi: 10.1128/mcb.16.9.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Q, Zhang Y, Marden JJ, Banfi B, Engelhardt JF. Endosomal NADPH oxidase regulates c-Src activation following hypoxia/reoxygenation injury. Biochem J. 2008;411:531–41. doi: 10.1042/BJ20071534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kornak U, Kasper D, Bösl MR, Kaiser E, Schweizer M, Schulz A, et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104:205–15. doi: 10.1016/S0092-8674(01)00206-9. [DOI] [PubMed] [Google Scholar]
- 28.Sun Y, Büki KG, Ettala O, Vääräniemi JP, Väänänen HK. Possible role of direct Rac1-Rab7 interaction in ruffled border formation of osteoclasts. J Biol Chem. 2005;280:32356–61. doi: 10.1074/jbc.M414213200. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura I, Sasaki T, Tanaka S, Takahashi N, Jimi E, Kurokawa T, et al. Phosphatidylinositol-3 kinase is involved in ruffled border formation in osteoclasts. J Cell Physiol. 1997;172:230–9. doi: 10.1002/(SICI)1097-4652(199708)172:2<230::AID-JCP10>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 30.Zhao H, Väänänen HK. Pharmacological sequestration of intracellular cholesterol in late endosomes disrupts ruffled border formation in osteoclasts. J Bone Miner Res. 2006;21:456–65. doi: 10.1359/JBMR.051204. [DOI] [PubMed] [Google Scholar]
- 31.Palokangas H, Mulari M, Väänänen HK. Endocytic pathway from the basal plasma membrane to the ruffled border membrane in bone-resorbing osteoclasts. J Cell Sci. 1997;110:1767–80. doi: 10.1242/jcs.110.15.1767. [DOI] [PubMed] [Google Scholar]
- 32.Wang J, Gu Q, Li M, Zhang W, Yang M, Zou B, et al. Identification of XAF1 as a novel cell cycle regulator through modulating G(2)/M checkpoint and interaction with checkpoint kinase 1 in gastrointestinal cancer. Carcinogenesis. 2009;30:1507–16. doi: 10.1093/carcin/bgp155. [DOI] [PubMed] [Google Scholar]
- 33.Dumesic PA, Scholl FA, Barragan DI, Khavari PA. Erk1/2 MAP kinases are required for epidermal G2/M progression. J Cell Biol. 2009;185:409–22. doi: 10.1083/jcb.200804038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu X, Guo H, Molter J, Miao H, Gerber L, Hu Y, et al. Alpha-fetoprotein-thymidine kinase-luciferase knockin mice: a novel model for dual modality longitudinal imaging of tumorigenesis in liver. J Hepatol. 2011;55:96–102. doi: 10.1016/j.jhep.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 36.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–71. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 37.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–4. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 38.McDonnell AV, Jiang T, Keating AE, Berger B. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics. 2006;22:356–8. doi: 10.1093/bioinformatics/bti797. [DOI] [PubMed] [Google Scholar]
- 39.Wang O, Liu S, Zou J, Lu L, Chen L, Qiu S, et al. Anticancer activity of 2α, 3α, 19β, 23β-Tetrahydroxyurs-12-en-28-oic acid (THA), a novel triterpenoid isolated from Sinojackia sarcocarpa. PLoS ONE. 2011;6:e21130. doi: 10.1371/journal.pone.0021130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao WT, Jiang D, Yuan J, Cui YM, Shi XW, Chen CM, et al. HOXB7 as a prognostic factor and mediator of colorectal cancer progression. Clin Cancer Res. 2011;17:3569–78. doi: 10.1158/1078-0432.CCR-10-2533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.