

Abstract
The Retinoblastoma protein (Rb) is important in the control of cell proliferation and apoptosis. Its activity is controlled by reversible phosphorylation on several serine and threonine residues. When Rb is hypophosphorylated, it inhibits proliferation by preventing passage through the G1- S phase transition. Hyperphosphorylated Rb promotes cell cycle progression. The role of Rb phosphorylation in the control of apoptosis is largely unknown, although several apoptotic stimuli result in dephosphorylation of Rb. It may be that dephosphorylation of specific amino acids signals apoptosis vs. cell cycle arrest. Using glutamic acid mutagenesis, we have generated 15 single phosphorylation site mutants of Rb to alter serine/threonine to glutamic acid to mimic the phosphorylated state. By calcium phosphate transfection, mutant plasmids were introduced into C33A Rb-null cells, and apoptosis was induced using UV. Apoptosis was measured by ELISA detection of degraded DNA and by immunoblotting to assess proteolytic cleavage of PARP. Our results show that only mutation of threonine-821 to glutamic acid (T821E) blocked apoptosis by 50%, whereas other sites tested had little effect. In Rb-null Saos-2 and SKUT-1 cells, the T821E mutation also blocked apoptosis induced by the cdk inhibitor, Roscovitine, by 50%. In addition, we show that endogenous Rb is dephosphorylated on threonine-821 when cells are undergoing apoptosis. Thus, our data indicates that dephosphorylation of threonine-821 of Rb is required for cells to undergo apoptosis.
Keywords: Rb, phosphorylation, apoptosis, Roscovitine, UV
Introduction
The Retinoblastoma tumor suppressor protein (Rb) is important in the processes of proliferation, apoptosis, differentiation and senescence.1 The role of Rb in proliferation is thought to be regulated by phosphorylation on several amino acid sites found throughout the Rb protein sequence.2 Cyclin-dependent kinase (cdk)-mediated hyperphosphorylation (highly phosphorylated) of Rb stimulates cell cycle progression, whereas hypophosphorylated (less phosphorylated) Rb inhibits progression through the cell cycle. By inhibiting the activity of E2F transcription factors, hypophosphorylated Rb blocks progression from the G1 phase into S phase. Upon growth factor stimulation, the synthesis and accumulation of cyclin proteins leads to cdk activation toward Rb. Rb becomes hyperphosphorylated, which releases E2F, allowing it to stimulate cell cycle progression. Alterations in the Rb pathway that lead to excessive phosphorylation of Rb have been observed in almost all cancer cell types.3,4 In addition to its role in cell proliferation, Rb is important in apoptosis. A role for Rb in apoptosis was initially identified utilizing Rb-null mice which demonstrated that in the absence of Rb, mice exhibited excessive apoptosis of cells in the nervous systems, lens and skeletal muscles.5-7 These results supported the idea that Rb could suppress apoptosis during development.8 In addition, it was found that apoptotic signaling is negatively regulated by Rb. For example, overexpression of Rb inhibits ionizing radiation-induced apoptosis in the Rb-null cell line Saos-2.9 The mechanism of Rb regulation of apoptosis has not been fully elucidated; however, hyperphosphorylation of Rb has been shown to be important for its ability to inhibit apoptosis.10-12 Loss of phosphorylation of the Rb protein has been observed during apoptosis.13-19 Also, the specific activation of Rb-directed phosphatase activity has been shown to be required for apoptosis to occur.13,14,16,20,21 These studies suggest that phosphorylated Rb protects against apoptosis, and dephosphorylation of Rb is involved in triggering cell death. In fact, Rb that lacks nine of the 16 phosphorylation sites, called PSM-RB (phosphorylation site-mutated Rb) does not protect cells from cell death initiated by apoptotic stimuli.12 Thus, it seems likely that phosphorylation at specific sites on Rb is required for Rb to be able to block apoptosis. In the aforementioned study, however, there was no analysis of which specific Rb phosphorylation sites are involved in this process. In fact, there has been no systematic evaluation of specific sites of Rb dephosphorylation with respect to the role of Rb in apoptosis.
The notion that the tumor suppressor and anti-apoptotic functions of Rb are regulated by distinct phosphorylation events has been proposed.22 Mutagenesis of Rb has been utilized to study the function of specific phosphorylation sites of Rb,23,24 which has led to the identification of roles for certain sites in specific processes.25,26 These studies highlight the complexity of Rb regulation by phosphorylation, showing that specific Rb phosphorylation sites (or groups of sites) are responsible for the regulation of the diverse functions of Rb. However, to date the Rb phosphorylation sites that are required to be dephosphorylated in apoptosis have not been elucidated. Therefore, we performed glutamic acid mutagenesis of 15 phosphorylation sites of Rb to identify potential sites that are involved in apoptosis. We describe in this paper our finding that one specific mutation, threonine-821 to glutamic acid (T821E), was sufficient to block apoptosis induced by two stimuli, UV stress and cdk inhibition, in several Rb-null cell types.
Results
Previous studies have shown that Rb is dephosphorylated in response to apoptotic treatments. DNA damaging agents such as etoposide, Ara-C (cytosine arabinoside) and cisplatin, cause dephosphorylation of Rb in asynchronously growing cells, as well as in cells synchronized in S phase.13-16 In addition, UV stress and cdk inhibition that leads to apoptosis results in dephosphorylation of Rb.17,18,21 Here, we show in MCF7 breast cancer cells and HCT116 colon cancer cells that various apoptotic stimuli cause dephosphorylation of Rb (Fig. 1). Asynchronously growing MCF7 cells were exposed to UV (10 J/m2), and after a 4 h incubation, cells were analyzed by immunoblotting. Alternatively MCF7 cells were treated with 25 µM Roscovitine for 24 h prior to the immunoblotting analysis. In addition, HCT116 cells were treated with 10 µM Ara-C or the kinase inhibitor Staurosporine (1 µM) for 24 h, and analyzed by western blotting. These apoptotic treatments led to a decrease in Rb phosphorylation, evidenced by the loss of the slowly migrating band of Rb, as well as an increase in cleavage of poly ADP ribose polymerase (Parp), which is a marker of apoptosis.27 This immunoblotting analysis shows that apoptosis results in an overall loss in phosphorylation of Rb during apoptosis; however this method does not address the specific sites of Rb that are dephosphorylated. Therefore, we utilized glutamic acid mutagenesis and generated 15 Rb mutants each with one phosphorylation site changed to glutamic acid, which mimics the phosphorylated amino acid. Expression plasmids encoding mutant and wild-type Rb were transfected into Rb-null C33A cervical carcinoma cells, which were stimulated to undergo apoptosis by treatment with UV approximately 72 h later. Four hours after UV stress, cellular apoptosis was measured by quantification of DNA degradation by ELISA (Cell Death Detection, Roche), followed by immunoblotting to measure cleavage of Parp and Rb expression. In Figure 2, the results of our screen of 15 mutant phosphorylation sites are shown in three groups: C terminal sites (Fig. 2A), internal sites (Fig. 2B) and N terminal sites (Fig. 2C). Throughout these experiments, UV stress of C33A cells caused a 4- to 8-fold increase in apoptosis. Transfection of all of the Rb plasmids (except the T821E mutant) had little to no effect on induction of apoptosis measured by the quantification of degraded DNA. However, expression of Rb with the T821E mutation blocked apoptosis by 50%, suggesting that dephosphorylation of threonine 821 is required for apoptosis to occur. In addition, by immunoblotting we measured the cleavage of Parp as a marker of apoptosis, and found that only the T821E mutation was able to block the cleavage of Parp in these experiments, indicating that dephosphorylation of T821 of Rb is required for apoptosis to occur.

Figure 1. Apoptosis causes dephosphorylation of Rb. Asynchronously growing MCF7 breast cancer cells were exposed to UV stress (10 J/m2) and 4 h later, analyzed by immunoblotting. MCF7 cells were also treated with 25 µM Roscovitine or DMSO for 24 h prior to immunoblotting analysis. HCT116 colon carcinoma cells (p53−/−) were treated with 10 µM AraC for 24 h or 1 µM Staurosporine for 24 h prior to immunoblotting analysis. Western blots were performed as described in the Materials and Methods. Induction of apoptosis and equal protein loading were confirmed by examining expression of cleaved Parp and β-actin, respectively. Dephosphorylation of Rb was detected as the loss of the slowly migrating, upper Rb band. Data shown is representative of three independent experiments.

Figure 2. Rb expressing the T821E mutation blocks apoptosis induced by UV. Plasmids expressing Rb mutant proteins were generated as described in the Materials and Methods. Rb-null C33A cells were transfected using calcium phosphate precipitation (Invitrogen). In (A), the following plasmids encoding C terminal mutations of Rb were utilized in addition to WT: T826E, T821E, S811E, S807E and S795E. In (B), we utilized plasmids encoding internal Rb mutations in addition to WT: S788E, S780E, S612E, S608E and T373E. In (C), we used plasmids encoding N terminal mutations of Rb in addition to WT: T356E, T252E, S249E, S230E and T5E. After 3 d, cells were treated with UV. Four hours later, cells apoptosis was measured using the Cell Death Detection ELISA (Roche Diagnostics), which detects degraded DNA released from the nucleus into the cytoplasm. The amount of apoptosis (degraded DNA) detected in non-treated control cells was normalized to one. Graph depicts the fold increase in degraded DNA observed due to UV treatment. Error bars represent standard deviation of the mean of triplicate samples, and data shown is representative of three independent experiments. Apoptosis was also measured by immunoblotting analysis of the cleavage of Parp as an indicator of apoptosis. In addition, expression of Rb was verified by immunoblotting in these experiments.
To determine whether dephosphorylation of T821 of Rb is required for other stimuli to cause apoptosis, we utilized the cdk inhibitor, Roscovitine, in additional experiments. In cells treated with Roscovitine, cdk inhibition leads to dephosphorylation of Rb and the C terminal domain of RNA polymerase II.28 Here we employed Saos2 cells which express a C terminal truncated, nonfunctional Rb protein.29 As shown in Figure 3A, Roscovitine treatment causes a 7-fold increase in apoptosis, as measured by the quantification of degraded DNA (Cell Death Detection ELISA). In these experiments, we tested a subset of our plasmids expressing wild-type or mutant Rb and found that the T821E mutation was able to reduce apoptosis by approximately 40%, in contrast to other Rb proteins with mutations at S795 and S788, which did not reduce apoptosis at all. In addition, we further found that Rb expressing the T821E mutation was able to inhibit apoptosis in SKUT-1 cells, an Rb-null uterine cancer cell line. As shown in Figure 3B, when SKUT-1 cells were treated with 25 µM Roscovitine for 24 h, these cells exhibit a 5-fold increase in apoptosis as measured by an ELISA assay that quantifies cleavage of Parp (Cell Signaling Tech). In these experiments, out of the 5 Rb mutants tested, only the T821E mutation inhibited apoptosis by 50%. This finding was corroborated by measuring DNA degradation using the Cell Death Detection ELISA. In Figure 3C, although the overall increase in apoptosis induced due to Roscovitine treatment was modest (1.5-fold increase), expression of the Rb plasmid encoding T821E was able to reverse the induction of apoptosis stimulated by Roscovitine. Wild-type Rb in these experiments does not affect the induction of apoptosis. Thus, we have shown that Rb carrying the T821E mutation expressed in two different cell types is able to inhibit apoptosis induced by the cdk inhibitor, Roscovitine. This data demonstrates that dephosphorylation of T821 is required for apoptosis.

Figure 3. (A) Plasmids encoding WT Rb or Rb with the indicated mutations were transfected into Saos2 cells using Fugene (Roche) according to the manufacturer’s directions. Forty-eight to 72 h later, cells were treated with 25 µM Roscovitine for 24 h. Apoptosis was measured using the Cell Death Detection ELISA (Roche Diagnostics), which detects degraded DNA released from the nucleus into the cytoplasm. The amount of apoptosis (degraded DNA) detected in non-treated control cells was normalized to one. Graph depicts the fold increase in degraded DNA observed due to Roscovitine treatment. Error bars represent standard deviation of the mean of triplicate samples, and data shown is representative of three independent experiments. Rb expression was verified by immunoblotting (data not shown). (B) SKUT-1 cells were transfected using plasmids encoding the indicated Rb mutations using Fugene as described in the Materials and Methods, and 48–72 h later, cells were treated with 25 µM Roscovitine for 24 h. Cleavage of Parp (as an indicator of apoptosis) was measured using an ELISA (Cell Signaling Tech). The amount of apoptosis (cleaved Parp) detected in non-treated control cells was normalized to one. Graph depicts the fold increase in cleavage of Parp observed due to Roscovitine treatment. Error bars represent standard deviation of the mean of triplicate samples, and data shown is representative of two independent experiments. (C) SKUT-1 cells were transfected with plasmids encoding WT Rb or the T821E mutation as described in (B), and treated with Roscovitine and subjected to the Cell Death Detection ELISA as described in (A). The amount of apoptosis (degraded DNA) detected in non-treated control cells was normalized to one. Graph depicts the fold increase in degraded DNA observed due to Roscovitine treatment. Error bars represent standard deviation of the mean of triplicate samples, and data shown is representative of three independent experiments.
We have shown that Rb is dephosphorylated in apoptosis, and that mutation of T821 to glutamic acid inhibits apoptosis induced by UV or cdk inhibition. This suggests that if dephosphorylation of T821 is required for apoptosis, it should occur concomitant with an increase in the cleavage of Parp observed, due to Roscovitine treatment. To determine whether endogenous Rb in cells is dephosphorylated on T821 in response to an apoptosis inducer, we performed dose response experiments using Roscovitine on MCF7 cells. MCF7 cells were treated with a range of concentrations of Roscovitine, and 24 h later cells were analyzed by immunoblotting. In Figure 4, phosphorylation state-specific antibodies toward Rb were utilized to detect phosphorylation of T821, S780 and S608. Only T821, but not S780 or S608, became dephosphorylated at the concentration of Roscovitine (25 µM) that resulted in apoptosis (measured by the detection of cleaved Parp). The overall expression of Rb remains constant, although dephosphorylation of T821 specifically occurs due to the induction of apoptosis. This data shows that T821 is dephosphorylated at the time of Parp cleavage, whereas other Rb phosphorylation sites remain phosphorylated, which indicates that T821 is specifically involved in the induction of apoptosis.
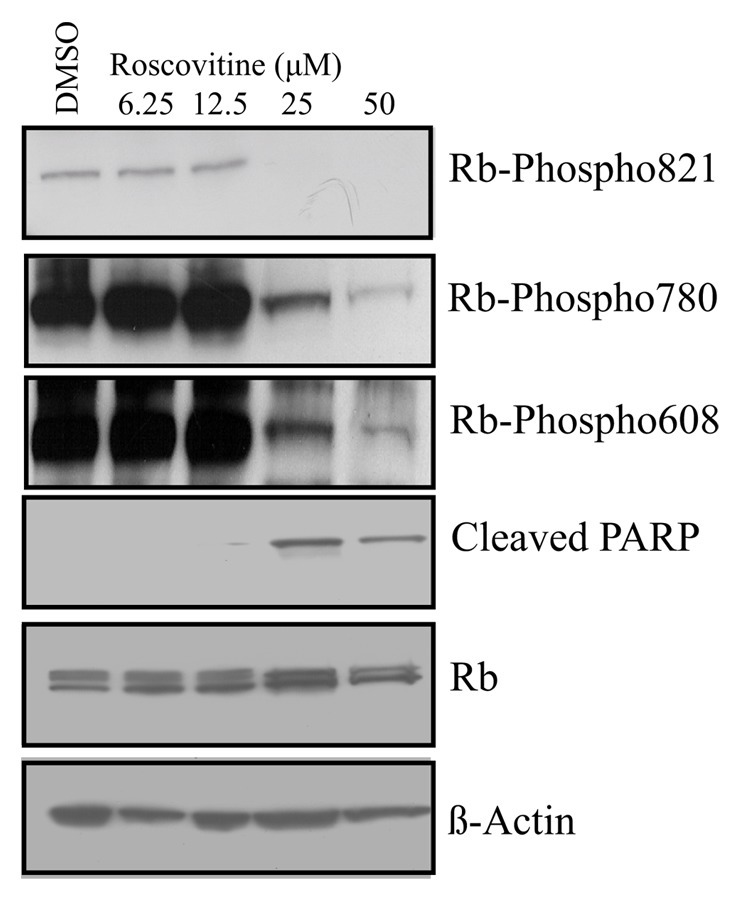
Figure 4. Dephosphorylation of T821 of Rb occurs concomitant with apoptosis. Asynchronously growing MCF7 cells were treated with DMSO as a control, or 6.25 µM, 12.5 µM, 25 µM or 50 µM Roscovitine for 24 h, followed by immunoblotting with the indicated antibodies. Three Rb phosphorylation-specific antibodies were utilized to detect phosphorylation at T821, S780 and S608, and apoptosis was detected using antibodies to cleaved Parp. Equivalent expression of Rb and equal protein loading were confirmed by immunoblotting of Rb and β-actin, respectively. Data shown is representative of three independent experiments and blot shown (for T821, S780 and S608) is of the same (20 sec) exposure of the film.
Discussion
Hyperphosphorylation of Rb is found in most human cancer cells.3,4 This is the result of the overexpressed D and E type cyclins, gene amplification of cdk4 or loss of the cdk inhibitor p16INK4a, resulting in excessive phosphorylation of Rb. Highly phosphorylated Rb in tumors would not only enhance proliferative capacity, but in addition, confer resistance to apoptosis. The data showing that phosphorylated Rb inhibits apoptosis is consistent with this idea.10-12 Further, the observation that Rb is dephosphorylated in apoptosis induced by various stimuli also supports a required role for Rb dephosphorylation in apoptosis.13-19 Because it was unclear which phosphorylation sites of Rb were involved in the apoptotic signal, we utilized glutamic acid mutagenesis to individually change each Rb phosphorylation site to glutamic acid, to mimic the phosphorylated state. A previous study utilizing glutamic acid mutagenesis of the Rb protein employed phosphorylation site mutants that contained different combinations of between 1–12 altered sites.24 However, this study measured the role of phosphorylation sites in the processes of senescence and G1 arrest, and apoptosis was not addressed in this study. Our data presented here indicate that the dephosphorylation of T821 is required for the induction of apoptosis induced by UV stress and cdk inhibition.
To our knowledge this is the first report identifying dephosphorylation of a specific site of Rb as a requirement for apoptosis. In several studies, phosphatase activity has been shown to be required to cause apoptosis by UV stress or chemotherapeutic agents, such as etoposide and Ara-C.13,16,21 In these studies, phosphatase inhibitors blocked both Rb dephosphorylation and apoptosis. Protein phosphatase 1 (PP1) is the primary phosphatase of Rb, responsible for the dephosphorylation of Rb at mitotic exit.30 In proliferating cells, PP1 binds to an interacting subunit called PNUTS (phosphatase nuclear targeting subunit), which regulates its phosphatase activity toward Rb.31,32 Interestingly, T821 of Rb remains phosphorylated throughout M phase and into G1 phase.32,33 However, T821 is rapidly dephosphorylated (before other sites such as S780, S807 and S811) in response to cellular stress, such as hypoxia or Ara-C treatment. This suggests that dephosphorylation of T821 is the signal to Rb for apoptosis. In vitro studies using the PP1 catalytic enzyme (without PNUTS) shows that PP1 readily dephosphorylates T821 before other Rb sites, such as S780, S807 and S811.32 Together, these findings suggest that during normal cell cycle progression from M phase into G1 phase, the PP1 enzyme, in a complex with PNUTS, dephosphorylates Rb on sites other than T821. However, under cellular stresses that lead to apoptosis, PNUTS dissociates from PP1, and phosphatase activity toward T821 of Rb is stimulated. In fact, our studies using siRNA directed toward PNUTS demonstrated that reduced PNUTS expression leads to increased phosphatase activity toward Rb and apoptosis.20 Thus, it may be the case that apoptotic stimuli cause an increase in PP1 activity toward T821 of Rb by regulating the association between PP1 and PNUTS.
What is the mechanism by which phosphorylation of Rb at T821 imparts protection against apoptosis? Phosphorylation of T821 (and T826) of Rb have been shown to inhibit LXCXE motif, containing proteins such as T-Ag, E7 and Elf-1.23 However, phosphorylation of these sites was not shown to regulate the binding of Rb to E2F. E2F can associate with hypophosphorylated or hyperphosphorylated Rb, depending on cellular context.2,34 In addition, Rb regulation of E2F activity can affect cell proliferation and/or apoptosis. In apoptosis, Rb can inhibit the expression of E2F target genes such as the p53 family member, p73, APAF1 and caspases.8 Due to several types of apoptotic stimuli, Rb is cleaved by caspase-8 between amino acids Asp 886-Gly 887, and subsequently degraded.8,35 This C terminal cleavage generates an Rb protein lacking 42 amino acids and is usually preceded by Rb dephosphorylation. Thus, dephosphorylation of T821 of Rb may be a prerequisite for this initial cleavage event. Degradation of Rb would lead to release of Rb binding proteins, some of which have been identified as playing a role in apoptosis.36,37 Two pro-apoptotic proteins that bind to Rb and are inhibited by the interaction are c-ABL and c-Jun N terminal kinase/stress-activated protein kinase (JNK/SAPK). The c-ABL tyrosine kinase is activated by DNA damage or TNF-α and is associated with apoptosis.38 In apoptosis, c-ABL regulates the activity of p53 and p73.39,40 That Rb degradation is required for c-ABL-induced apoptosis was shown using mice thymocytes expressing a cleavage-resistant form of Rb (Rb-MI). In these experiments, degradation-resistant Rb blocked c-ABL activation and apoptosis.41 Another Rb binding protein that promotes apoptosis is JNK/SAPK. The JNK/SAPK pathway mediates intracellular signals for cell death initiated by apoptotic stresses such as UV radiation. For this interaction, it was shown that the C terminal portion of Rb (amino acids 768–928) was required for binding to JNK/SAPK and inhibition of its associated kinase activity.42 Clearly, the C terminal cleaved form of Rb would lack the ability to bind to and inhibit JNK/SAPK. In fact, JNK/SAPK exhibits kinase activity toward T821 of Rb in the presence of full-length Rb,43 suggesting that while Rb inhibits JNK/SAPK pro-apoptotic activity, JNK/SAPK, in turn, promotes the anti-apoptotic activity of Rb by the phosphorylation of T821.
The phosphorylation state of Rb (of specific amino acids such as T821) may play an important role in the stability of Rb, and T821 dephosphorylation may promote the degradation and loss of Rb. Many studies have addressed the fact that Rb loss stimulates apoptosis. For example, in breast cancer cells that lack Rb, E2F1 stimulates apoptosis, rather than proliferation.44 In prostate cancer cells, the tumor suppression activity of miR-449a is dependent upon the presence of functional Rb.45 In addition, loss of Rb in combination with the inactivation of the TSC2 tumor suppressor induces a synergistic cell death.46 Rb has also been shown to be required for the maintenance of differentiated myotubes.47 Thus, if T821 dephosphorylation of Rb induces Rb degradation, apoptosis is the predictable outcome.
Therefore, we conclude that in our analysis of single phosphorylation site mutants of Rb we found that only one, T821E, was able to block apoptosis stimulated by UV stress or cdk inhibition. These data are consistent with the idea that dephosphorylation of T821 occurs only in apoptosis and abrogates the anti-apoptotic function of Rb. Interestingly, Rb family proteins p107 and p130, while highly similar to Rb in the area surrounding T821 and T826, lack T821 and nine adjacent amino acids found in Rb.48 In fact, a role in apoptosis has not been found for p107 or p130,49 further suggesting that dephosphorylation of T821 is the required signal for Rb to allow apoptosis.
Materials and Methods
Cell culture
All the cell lines utilized have been obtained from ATCC unless otherwise specified. Cell culture materials were obtained from Invitrogen unless otherwise indicated. MCF7, C33A and SKUT-1 cells were grown in Dulbecco’s modified Eagle’s media (DMEM), supplemented with 10% fetal bovine serum (FBS), 100 U/ml Penicillin, 100 ug/ml Streptomycin and 2 mM glutamine. HCT116 p53−/− cells (a kind gift from Dr. B. Vogelstein, Johns Hopkins) were maintained in McCoy’s 5A media with 10% FBS and penicillin/streptomycin. Saos2 cells were grown in McCoy’s 5A media, supplemented with 15% FBS and penicillin/streptomycin. Cells were routinely maintained at 37°C in a humidified, 5% CO2-containing atmosphere and were split two–three times weekly to maintain subconfluent cultures.
Transfections
The CMV/Rb plasmid was obtained from Addgene. It was used to generate S/T to E mutations at the following amino acid sites: T5, S230, S249, T252, T356, T373, S608, S612, S780, S788, S795, S807, S811, T821 and T826 (Genewiz, Inc.). Plasmids were introduced into C33A cells using calcium phosphate transfection kits (Invitrogen), performed according to the manufacturer’s directions. Saos2 and SKUT-1 cells were transfected using Fugene (Roche). Cells were harvested 48–72 h post-transfection. All transfections were performed using 10 µg of total DNA/60 mm dish. Expression of wild-type Rb and Rb mutant proteins was routinely verified by immunoblotting.
Immunoblotting
SDS-PAGE and western blotting was performed as described elsewhere.20 In this study, we utilized the following primary antibodies: Rb-phospho-821 (Pierce), Rb-phospho-780, Rb-phospho-608, cleaved PARP, (Cell Signaling Technology); or Rb (IF8, Santa Cruz Biotech); β-Actin (Sigma).
Apoptosis assays
Cells were treated with UV (10 J/m2) on a DNA crosslinker (UVP Inc.). Roscovitine was administered to cells at 25 µM, control cultures received DMSO. The Cell Death Detection ELISA (Roche Diagnostics) was performed as directed by the manufacturer. Briefly, 104 cells from each condition were lysed and subjected to a slow spin centrifugation to pellet nuclei. Extracts from the cytoplasmic fraction were used to detect fragmented DNA, which was measured on a BioRad microplate reader, and experiments were conducted in triplicate. The Parp cleavage ELISA (Cell Signaling Tech) was performed as directed by the manufacturer’s directions.
Acknowledgments
We thank Jack Horne, Andrew Weir and Michael H. Roberts for critical review of the manuscript. This work was supported by NIH 1R15CA143390 awarded to N.A.K.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21693
References
- 1.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–82. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–7. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 3.Mittnacht S. The retinoblastoma protein--from bench to bedside. Eur J Cell Biol. 2005;84:97–107. doi: 10.1016/j.ejcb.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/S1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 5.Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, et al. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–30. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- 6.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 7.Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–94. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 8.Chau BN, Wang JYJ. Coordinated regulation of life and death by RB. Nat Rev Cancer. 2003;3:130–8. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- 9.Haas-Kogan DA, Kogan SC, Levi D, Dazin P, T’Ang A, Fung YK, et al. Inhibition of apoptosis by the retinoblastoma gene product. EMBO J. 1995;14:461–72. doi: 10.1002/j.1460-2075.1995.tb07022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berndt N, Dohadwala M, Liu CW. Constitutively active protein phosphatase 1α causes Rb-dependent G1 arrest in human cancer cells. Curr Biol. 1997;7:375–86. doi: 10.1016/S0960-9822(06)00185-0. [DOI] [PubMed] [Google Scholar]
- 11.Wallace M, Coates PJ, Wright EG, Ball KL. Differential post-translational modification of the tumour suppressor proteins Rb and p53 modulate the rates of radiation-induced apoptosis in vivo. Oncogene. 2001;20:3597–608. doi: 10.1038/sj.onc.1204496. [DOI] [PubMed] [Google Scholar]
- 12.Masselli A, Wang JYJ. Phosphorylation site mutated RB exerts contrasting effects on apoptotic response to different stimuli. Oncogene. 2006;25:1290–8. doi: 10.1038/sj.onc.1209161. [DOI] [PubMed] [Google Scholar]
- 13.Dou QP, An B, Will PL. Induction of a retinoblastoma phosphatase activity by anticancer drugs accompanies p53-independent G1 arrest and apoptosis. Proc Natl Acad Sci USA. 1995;92:9019–23. doi: 10.1073/pnas.92.20.9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morana SJ, Wolf CM, Li J, Reynolds JE, Brown MK, Eastman A. The involvement of protein phosphatases in the activation of ICE/CED-3 protease, intracellular acidification, DNA digestion, and apoptosis. J Biol Chem. 1996;271:18263–71. doi: 10.1074/jbc.271.30.18263. [DOI] [PubMed] [Google Scholar]
- 15.Knudsen KE, Booth D, Naderi S, Sever-Chroneos Z, Fribourg AF, Hunton IC, et al. RB-dependent S-phase response to DNA damage. Mol Cell Biol. 2000;20:7751–63. doi: 10.1128/MCB.20.20.7751-7763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang R-H, Liu CWY, Avramis VI, Berndt N. Protein phosphatase 1alpha-mediated stimulation of apoptosis is associated with dephosphorylation of the retinoblastoma protein. Oncogene. 2001;20:6111–22. doi: 10.1038/sj.onc.1204829. [DOI] [PubMed] [Google Scholar]
- 17.Payton M, Chung G, Yakowec P, Wong A, Powers D, Xiong L, et al. Discovery and evaluation of dual CDK1 and CDK2 inhibitors. Cancer Res. 2006;66:4299–308. doi: 10.1158/0008-5472.CAN-05-2507. [DOI] [PubMed] [Google Scholar]
- 18.Plastaras JP, Kim SH, Liu YY, Dicker DT, Dorsey JF, McDonough J, et al. Cell cycle dependent and schedule-dependent antitumor effects of sorafenib combined with radiation. Cancer Res. 2007;67:9443–54. doi: 10.1158/0008-5472.CAN-07-1473. [DOI] [PubMed] [Google Scholar]
- 19.Jeon HS, Dracheva T, Yang SH, Meerzaman D, Fukuoka J, Shakoori A, et al. SMAD6 contributes to patient survival in non-small cell lung cancer and its knockdown reestablishes TGF-beta homeostasis in lung cancer cells. Cancer Res. 2008;68:9686–92. doi: 10.1158/0008-5472.CAN-08-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Leon G, Sherry TC, Krucher NA. Reduced expression of PNUTS leads to activation of Rb-phosphatase and caspase-mediated apoptosis. Cancer Biol Ther. 2008;7:833–41. doi: 10.4161/cbt.7.6.5839. [DOI] [PubMed] [Google Scholar]
- 21.Popowski M, Ferguson HA, Sion AM, Koller E, Knudsen E, Van Den Berg CL. Stress and IGF-I differentially control cell fate through mammalian target of rapamycin (mTOR) and retinoblastoma protein (pRB) J Biol Chem. 2008;283:28265–73. doi: 10.1074/jbc.M805724200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma D, Zhou P, Harbour JW. Distinct mechanisms for regulating the tumor suppressor and antiapoptotic functions of Rb. J Biol Chem. 2003;278:19358–66. doi: 10.1074/jbc.M301761200. [DOI] [PubMed] [Google Scholar]
- 23.Knudsen ES, Wang JYJ. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271:8313–20. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- 24.Barrientes S, Cooke C, Goodrich DW. Glutamic acid mutagenesis of retinoblastoma protein phosphorylation sites has diverse effects on function. Oncogene. 2000;19:562–70. doi: 10.1038/sj.onc.1203332. [DOI] [PubMed] [Google Scholar]
- 25.Lents NH, Gorges LL, Baldassare JJ. Reverse mutational analysis reveals threonine-373 as a potentially sufficient phosphorylation site for inactivation of the retinoblastoma tumor suppressor protein (pRB) Cell Cycle. 2006;5:1699–707. doi: 10.4161/cc.5.15.3126. [DOI] [PubMed] [Google Scholar]
- 26.Gorges LL, Lents NH, Baldassare JJ. The extreme COOH terminus of the retinoblastoma tumor suppressor protein pRb is required for phosphorylation on Thr-373 and activation of E2F. Am J Physiol Cell Physiol. 2008;295:C1151–60. doi: 10.1152/ajpcell.00300.2008. [DOI] [PubMed] [Google Scholar]
- 27.Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–9. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 28.MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, et al. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005;65:5399–407. doi: 10.1158/0008-5472.CAN-05-0233. [DOI] [PubMed] [Google Scholar]
- 29.Shew J-Y, Lin BTY, Chen P-L, Tseng BY, Yang-Feng TL, Lee W-H. C-terminal truncation of the retinoblastoma gene product leads to functional inactivation. Proc Natl Acad Sci USA. 1990;87:6–10. doi: 10.1073/pnas.87.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson DA, Krucher NA, Ludlow JW. High molecular weight protein phosphatase type 1 dephosphorylates the retinoblastoma protein. J Biol Chem. 1997;272:4528–35. doi: 10.1074/jbc.272.7.4528. [DOI] [PubMed] [Google Scholar]
- 31.Udho E, Tedesco VC, Zygmunt A, Krucher NA. PNUTS (phosphatase nuclear targeting subunit) inhibits retinoblastoma-directed PP1 activity. Biochem Biophys Res Commun. 2002;297:463–7. doi: 10.1016/S0006-291X(02)02236-2. [DOI] [PubMed] [Google Scholar]
- 32.Krucher NA, Rubin E, Tedesco VC, Roberts MH, Sherry TC, De Leon G. Dephosphorylation of Rb (Thr-821) in response to cell stress. Exp Cell Res. 2006;312:2757–63. doi: 10.1016/j.yexcr.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Rubin E, Mittnacht S, Villa-Moruzzi E, Ludlow JW. Site-specific and temporally-regulated retinoblastoma protein dephosphorylation by protein phosphatase type 1. Oncogene. 2001;20:3776–85. doi: 10.1038/sj.onc.1204518. [DOI] [PubMed] [Google Scholar]
- 34.Ianari A, Natale T, Calo E, Ferretti E, Alesse E, Screpanti I, et al. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell. 2009;15:184–94. doi: 10.1016/j.ccr.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang X, Masselli A, Frisch SM, Hunton IC, Jiang Y, Wang JYJ. Blockade of tumor necrosis factor-induced Bid cleavage by caspase-resistant Rb. J Biol Chem. 2007;282:29401–13. doi: 10.1074/jbc.M702261200. [DOI] [PubMed] [Google Scholar]
- 36.Taya Y. RB kinases and RB-binding proteins: new points of view. Trends Biochem Sci. 1997;22:14–7. doi: 10.1016/S0968-0004(96)10070-0. [DOI] [PubMed] [Google Scholar]
- 37.Dick FA. Structure-function analysis of the retinoblastoma tumor suppressor protein - is the whole a sum of its parts? Cell Div. 2007;2:26. doi: 10.1186/1747-1028-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang JY. Regulation of cell death by the Abl tyrosine kinase. Oncogene. 2000;19:5643–50. doi: 10.1038/sj.onc.1203878. [DOI] [PubMed] [Google Scholar]
- 39.Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73α and their collaboration to induce apoptosis. Nature. 1999;399:809–13. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- 40.Goga A, Liu X, Hambuch TM, Senechal K, Major E, Berk AJ, et al. p53 dependent growth suppression by the c-Abl nuclear tyrosine kinase. Oncogene. 1995;11:791–9. [PubMed] [Google Scholar]
- 41.Chau BN, Chen T-T, Wan YY, DeGregori J, Wang JYJ. Tumor necrosis factor alpha-induced apoptosis requires p73 and c-ABL activation downstream of RB degradation. Mol Cell Biol. 2004;24:4438–47. doi: 10.1128/MCB.24.10.4438-4447.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shim J, Park H-S, Kim MJ, Park J, Park E, Cho S-G, et al. Rb protein down-regulates the stress-activated signals through inhibiting c-Jun N-terminal kinase/stress-activated protein kinase. J Biol Chem. 2000;275:14107–11. doi: 10.1074/jbc.275.19.14107. [DOI] [PubMed] [Google Scholar]
- 43.Chauhan D, Hideshima T, Treon S, Teoh G, Raje N, Yoshihimito S, et al. Functional interaction between retinoblastoma protein and stress-activated protein kinase in multiple myeloma cells. Cancer Res. 1999;59:1192–5. [PubMed] [Google Scholar]
- 44.Sun B, Wingate H, Swisher SG, Keyomarsi K, Hunt KK. Absence of pRb facilitates E2F1-induced apoptosis in breast cancer cells. Cell Cycle. 2010;9:1122–30. doi: 10.4161/cc.9.6.10990. [DOI] [PubMed] [Google Scholar]
- 45.Noonan EJ, Place RF, Basak S, Pookot D, Li LC. miR-449a causes Rb-dependent cell cycle arrest and senescence in prostate cancer cells. Oncotarget. 2010;1:349–58. doi: 10.18632/oncotarget.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Searle JS, Li B, Du W. Targeting Rb mutant cancers by inactivating TSC2. Oncotarget. 2010;1:228–32. doi: 10.18632/oncotarget.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ciavarra G, Zacksenhaus E. Multiple pathways counteract cell death induced by RB1 loss: implications for cancer. Cell Cycle. 2011;10:1533–9. doi: 10.4161/cc.10.10.15520. [DOI] [PubMed] [Google Scholar]
- 48.Knudsen ES, Wang JYJ. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem. 1996;271:8313–20. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- 49. DeGregori J. The Rb network. J Cell Sci. 2004;117:3411–3. doi: 10.1242/jcs.01189. [DOI] [PubMed] [Google Scholar]