

Abstract
Migration stimulating factor (MSF) is a genetically truncated N-terminal isoform of fibronectin that is highly expressed during mammalian development in fetal fibroblasts, and during tumor formation in human cancer-associated myofibroblasts. However, its potential functional role in regulating tumor metabolism remains unexplored. Here, we generated an immortalized fibroblast cell line that recombinantly overexpresses MSF and studied their properties relative to vector-alone control fibroblasts. Our results indicate that overexpression of MSF is sufficient to confer myofibroblastic differentiation, likely via increased TGF-b signaling. In addition, MSF activates the inflammation-associated transcription factor NFκB, resulting in the onset of autophagy/mitophagy, thereby driving glycolytic metabolism (L-lactate production) in the tumor microenvironment. Consistent with the idea that glycolytic fibroblasts fuel tumor growth (via L-lactate, a high-energy mitochondrial fuel), MSF fibroblasts significantly increased tumor growth, by up to 4-fold. Mechanistic dissection of the MSF signaling pathway indicated that Cdc42 lies downstream of MSF and fibroblast activation. In accordance with this notion, Cdc42 overexpression in immortalized fibroblasts was sufficient to drive myofibroblast differentiation, to provoke a shift towards glycolytic metabolism and to promote tumor growth by up to 2-fold. In conclusion, the MSF/Cdc42/NFκB signaling cascade may be a critical druggable target in preventing “Warburg-like” cancer metabolism in tumor-associated fibroblasts. Thus, MSF functions in the metabolic remodeling of the tumor microenvironment by metabolically reprogramming cancer-associated fibroblasts toward glycolytic metabolism.
Keywords: TGF-beta signaling, aerobic glycolysis, cancer-associated fibroblasts, metabolic coupling, migration stimulating factor (MSF), myofibroblast, tumor stroma
Introduction
An important relationship exists between tumor cells and their local extracellular microenvironment.1-4 Indeed, tumor-associated stromal cells critically influence cancer progression and metastasis.1-4 Thus, tumor progression is the product of interactions between cancer cells and adjacent stromal cells, such as immune cells, endothelia and fibroblasts, although the exact mechanism(s) still remain poorly understood.5-7
More specifically, stromal myo fibroblasts are now considered active metabolic drivers of tumor growth.8 In our recent studies, we proposed that stromal fibroblasts fuel epithelial tumor cells via a unilateral transfer of energy-rich nutrients from the tumor stroma to cancer cells.9 In accordance with this assertion, the recycled nutrients produced by stromal fibroblasts, via autophagy/mitophagy, provide a steady-stream of energy-rich metabolites to cancer cells, inducing mitochondrial biogenesis.10-15
Normal stromal fibroblasts are converted into carcinoma-associated fibroblasts (CAFs) by complex interactions with adjacent cancer cells.5,16-19 These CAFs show a fetal-like phenotype, characterized by the expression of molecules typically expressed during embryonic development. In addition, CAFs develop a myofibroblast phenotype, with the expression of smooth muscle cell markers and the local production of transforming growth factor β (TGF-β), which can actively spread the CAF phenotype.20-26
Fetal-like fibroblasts and myo fibroblasts are also both viewed as “activated fibroblasts,” due to their increased expression of both ECM components and inflammatory cytokines.27-34 Fetal-like fibroblasts also secrete a soluble, genetically truncated form of fibronectin, termed migration stimulating factor (MSF).27-34 Interestingly, MSF is highly expressed in both fetal epithelial and stromal cells and in cancer patients, but its expression is somehow suppressed in normal adults.27-34
Detailed molecular characterization of MSF indicates that it is a 70-kDa protein that is essentially identical to the N-terminal domain of full-length fibronectin, with the addition of an MSF-specific 10 amino-acid C-terminal sequence.35,36 MSF changes the behavior of many target cell populations (fibroblasts, vascular and epithelial cells) by stimulating migration/invasion, matrix remodelling and neo-angiogenesis.37-46
Here, we generated a new hTERT-immortalized fibroblast cell line overexpressing MSF in order to clarify the functional role of MSF in driving the cancer-associated fibroblast phenotype. Now, we demonstrate that MSF-expressing fibroblasts create an autophagic/catabolic tumor stroma, which then provides high-energy nutrients to epithelial cancer cells via a paracrine mechanism.
Results
To directly assess the role of MSF in tumor growth, we stably overexpressed MSF in an immortalized human fibroblast cell line (hTERT-BJ1 cells) (Fig. 1A). Empty vector (Lv-105) control fibroblasts were produced in parallel. Figure 1A shows that transduction with MSF lentiviral particles successfully increased the stable expression of the MSF protein.
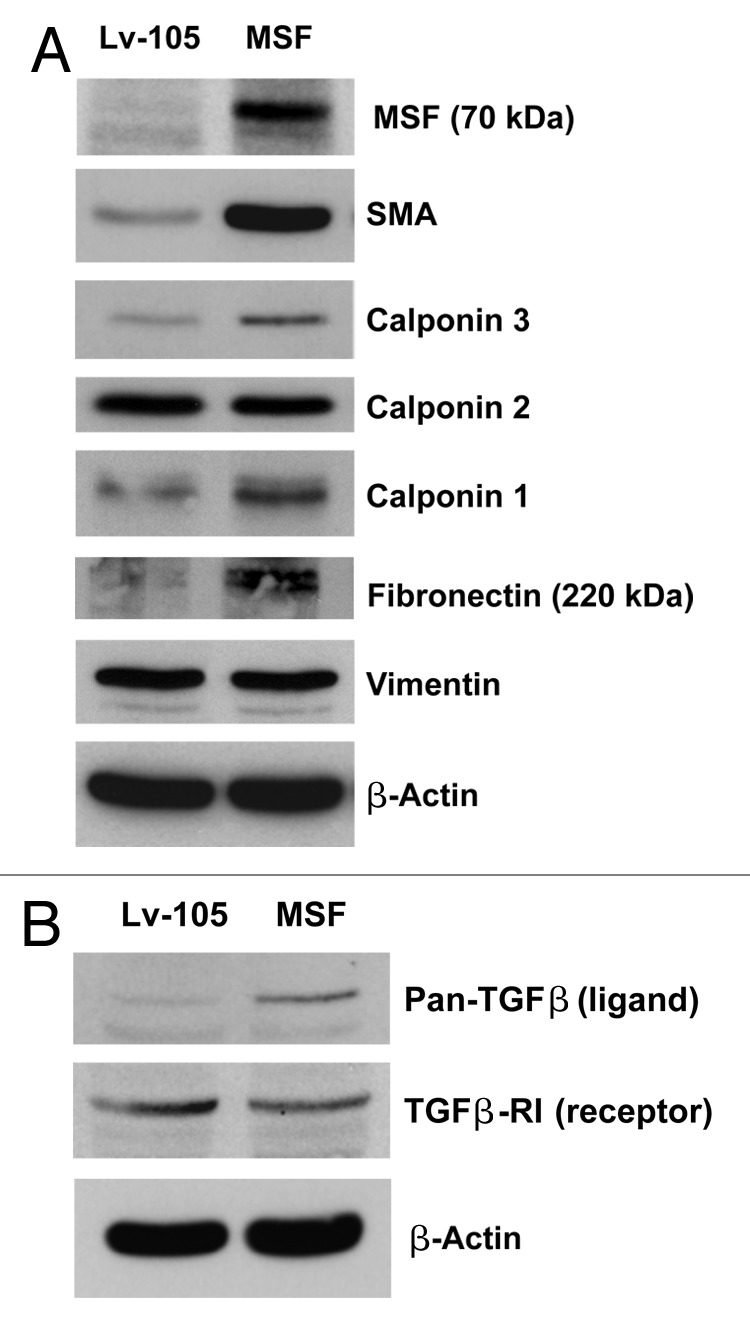
Figure 1. MSF over expression confers the cancer-associated fibroblast phenotype, characterized by the expression of myo fibroblast markers and activated TGF-β signaling. (A) To assess the functional role of MSF stromal expression in tumor growth, we stably overexpressed the MSF protein in immortalized human fibroblast cell line (hTERT-BJ1 cells). Successful overexpression of the MSF protein was verified by immunoblot analysis. Empty vector control (Lv-105) or MSF fibroblasts were also subjected to immunoblot analysis with different myofibroblast marker proteins. Equal protein loading was assessed by immunoblotting with β-actin. Note that MSF overexpression strongly induces the expression of myofibroblast markers such as α-SMA, calponin 1 and 3 and fibronectin, while no increases in vimentin protein expression were observed in the fibroblasts. (B) MSF overexpression induces the activation of TGF-β signaling. Note that immunoblot analysis of control and MSF fibroblasts reveals that TGF-β ligand protein expression is significantly increased in MSF fibroblasts. Conversely, MSF fibroblasts are characterized by a reduction in the protein expression of TGFβ-RI (type I receptor), as compared with control fibroblasts.
Fibroblasts overexpressing MSF develop a cancer-associated fibroblast phenotype, characterized by the expression of myofibroblast marker proteins and activated TGF-β signaling
Cancer-associated fibroblasts exhibit a myo fibroblastic phenotype, characterized by the synthesis of intracellular smooth muscle markers, in particular α-smooth muscle actin (α-SMA). To evaluate if MSF expression promotes myo fibroblastic differentiation, MSF-expressing fibroblasts were subjected to immunoblot analysis, using a panel of myo fibroblastic markers (Fig. 1A). The results show that MSF is indeed sufficient to induce the increased protein expression of SMA, Calponin (particularly, isoforms 1 and 3) and Fibronectin (full-length).
Several lines of evidence indicate that activated fibroblasts increase their expression and secretion of TGF-β, thereby promoting tumor growth. Thus, we next tested if MSF overexpression upregulates the expression of TGF-β. Consistent with this hypothesis, Figure 1B shows that MSF-overexpressing fibroblasts are characterized by an increase in TGF-β expression and a downregulation of its receptor, TGFβ-RI, both indicative of activated TGF-β signaling.
Fibroblasts overexpressing MSF migrate to a significantly greater extent than do control cells, and they also function as chemo-attractants, stimulating cancer cell migration
MSF is a potent motogenic factor, which is able to stimulate the migration of fibroblasts, epithelial as well as endothelial cells.35,46 Here, we demonstrate that MSF overexpression stimulates the migration of fibroblasts, validating the motogenic activity of the MSF protein (Fig. 2A). MSF could also influence the migration of cancer cells, by acting on these cells as a chemo-attractant. In support of this notion, Figure 2B shows that cancer cells, in the presence of MSF-overexpressing fibroblasts, migrate to a greater extent (1.4-fold) than do cancer cells in presence of normal control fibroblasts.
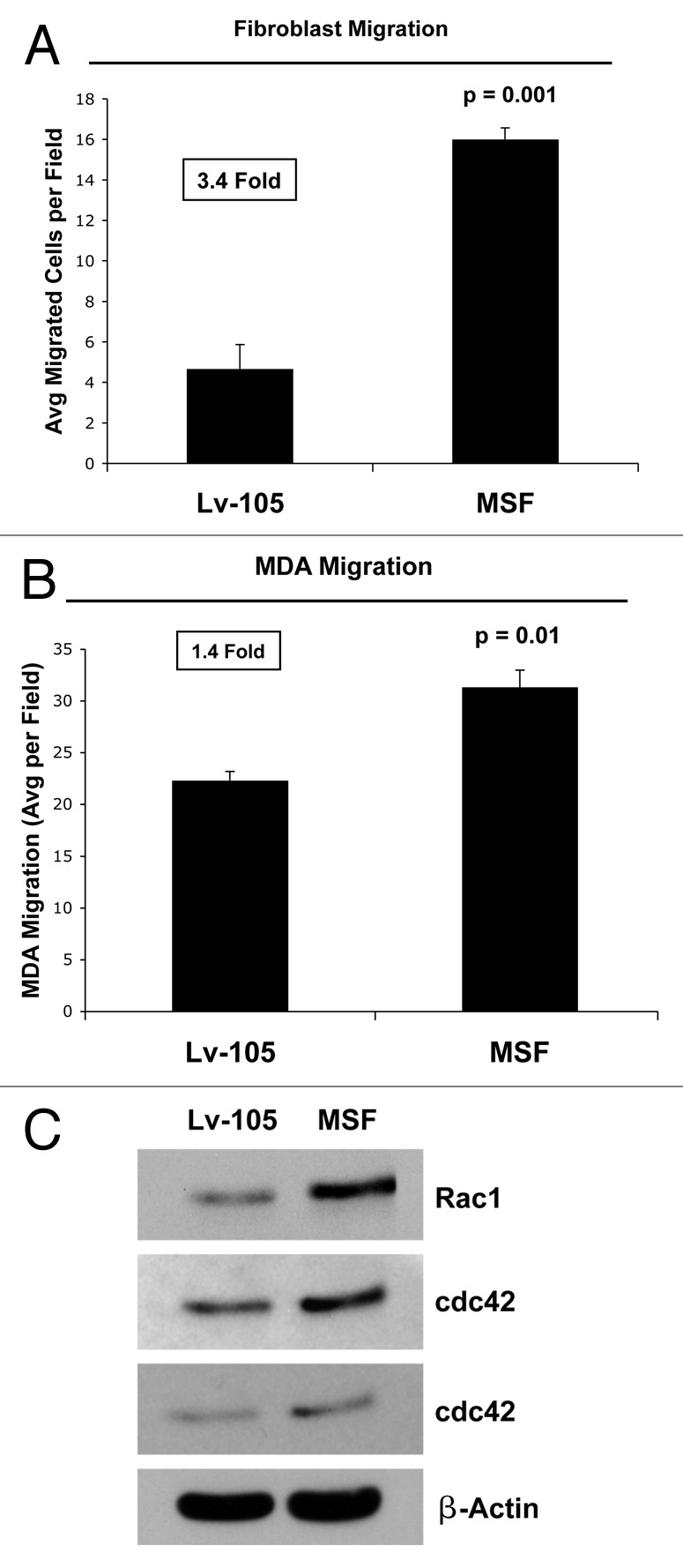
Figure 2. MSF stimulates the migration of fibroblasts and acts as chemo-attractant, stimulating cancer cell migration. (A) The migration of fibroblasts across an 8 µm pore uncoated membrane was assessed after a 6-h period, using a modified “Boyden Chamber” assay, employing Transwell cell culture inserts. Interestingly, the motility of fibroblasts overexpressing MSF was increased by ~3.4-fold, as compared with fibroblasts transfected with the empty-vector (Lv-105); p = 001 relative to control migration (Student’s t-test). (B) MSF overexpression stimulates cancer cell migration. To assess whether MSF fibroblasts can function as chemo-attractants, MDA-MB-231 cells were placed in the upper chambers and fibroblasts [empty vector (Lv-105) fibroblasts or MSF fibroblasts] were seeded into the lower chambers. Note that MSF fibroblasts promote cancer cell migration by ~1.4-fold. p = 0.01, control vs. MSF fibroblasts (Student’s t-test). (C) MSF is sufficient to increase the expression of the two small GTPases, namely Rac1 and Cdc42. Cells [empty vector (Lv-105) fibroblasts or MSF fibroblasts] were subjected to immunoblot analysis using antibodies directed against the two small GTPases. Note that MSF increases the expression of proteins, Rac1 and Cdc42.
This migratory activity could be induced by the increased expression and/or activation of proteins that play a fundamental role in cytoskeletal organization. Small GTPases, such as Rac1 and Cdc42, play a central role in regulating cell movement and migration by interacting with other proteins that more directly confer cytoskeletal rearrangements. As predicted, Figure 2C shows that MSF overexpression upregulates the expression levels of both these small-GTPases, which are associated with remodeling the actin cytoskeleton.
Fibroblasts overexpressing MSF activate NFκB, exhibit the induction of autophagy and cell cycle arrest
Small GTPases are strong activators of the transcription factor NFκB,47,48 so we next validated that MSF is able to induce not only the upregulation of Cdc42 and Rac1, but also the activation of NFκB. As shown in Figure 3A, MSF overexpression resulted in increased levels of p-NFκB, suggesting that MSF could influence the stromal fibroblasts through the activation of a number of different signaling pathways, including the NFκB signaling pathway.
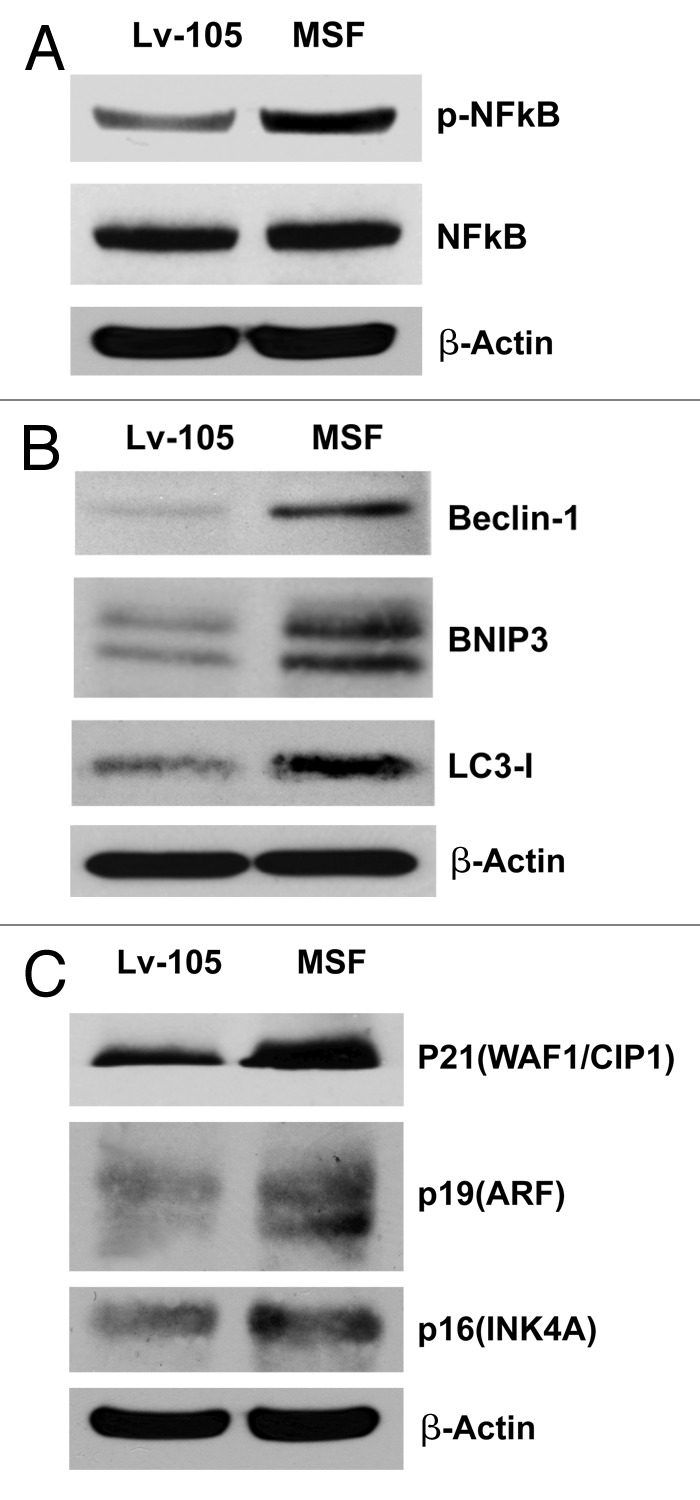
Figure 3. MSF overexpression in fibroblasts promotes the activation of NFκB, increases autophagy, and induces CDK inhibitors. (A) Since the human Rac-1 and Cdc42 proteins efficiently induce the transcriptional activity of nuclear factor kappaB (NF-kappaB), we next evaluated if MSF is able to induce not only the upregulation of the two small GTPases, but also the activation of NFκB. Note that immunoblot analysis of control or MSF fibroblasts revealed that the levels of p-NFκB are significantly increased in MSF fibroblasts, as compared with control fibroblasts. (B) NFκB can induce autophagy, so we verified if activation of NFκB by MSF overexpressed fibroblasts is sufficient to induce autophagy. Cells were lysed and subjected to immunoblot analysis using antibodies directed against a panel of autophagy markers. β-actin was used as equal loading control. Note that MSF increases the expression of several key autophagy markers, namely Beclin-1, BNIP3, and LC3-I. (C) To determine if MSF expression is also associated with senescence, we investigated whether known CDK inhibitors are upregulated. Immunoblot analysis shows that p21 (WAF1/CIP1), p19(ARF) and p16(INK4A) are all upregulated, consistent with cell cycle arrest and/or the onset of senescence.
NFκB plays a pivotal role as a signal integrator, which controls the autophagic process. For this purpose, we evaluated if the activation of NFκB in stromal MSF fibroblasts is sufficient to promote the autophagic process. Therefore, fibroblasts overexpressing MSF were analyzed by immunoblot analysis, using a panel of autophagy markers. Figure 3B shows that MSF increases the expression of several classical autophagy markers, such as Beclin1, BNIP3 and LC3-I. These results suggest that MSF augments or activates the autophagic process in stromal fibroblasts, probably via increased activation of the NFκB pathway. This pro-autophagic phenotype is associated with cell cycle arrest, as evidenced by the upregulation of CDK inhibitors, such as p21(CIP1/WAF1), p19(ARF) and p16(INK4A) (Fig. 3C).
Under hypoxic conditions, MSF fibroblasts generate elevated levels of L-lactate and show decreased mitochondrial activity, consistent with a shift toward glycolytic metabolism
We have previously shown that stromal fibroblasts promote and fuel tumor growth via activation of an autophagic program in the tumor stroma.8,9,12-15 Autophagy leads to the generation of recycled catabolic nutrients that can be used to power the anabolic growth of cancer cells. Because L-lactate is a critical fuel that provides continued energetic support for cancer cells, we next examined if MSF fibroblasts are able to induce L-lactate accumulation. As shown in Figure 4A, fibroblasts overexpressing MSF display increased L-lactate production (~2-fold; p = 0.004; 1.5-fold p = 0.03, values expressed as nmoles/µg or pmoles/cells, respectively). However, the ability of MSF fibroblasts to secrete L-lactate was observed only under hypoxic conditions.

Figure 4. MSF overexpression in fibroblasts induces a shift toward glycolytic metabolism. (A) Under hypoxic conditions, MSF fibroblasts secrete elevated levels of L-lactate, consistent with increased aerobic glycolysis. As shown in the figure, MSF fibroblasts secrete increased levels of L-lactate (~2-fold, p = 0.004; normalized for protein content; ~1.5-fold, p = 0.03; normalized for cell number), relative to control fibroblasts processed in parallel. This suggests that MSF-overexpressing fibroblasts could provide essential energetic support for adjacent tumor epithelial cells, via the secretion of L-lactate, which is a critical mitochondrial fuel for the tumor growth. (B) MSF overexpression induces a decrease in mitochondrial activity, as visualized using MitoTracker staining. Note that MSF decreases mitochondrial activity, both under normoxic condition (upper panels) and under hypoxic condition (lower panels). MitoTracker (red); nuclei/DAPI (blue). Original magnification, 60×. (C) MSF activates the Akt pathway. Control or MSF fibroblasts were subjected to immunoblot analysis with antibodies to phospho-Akt, phospho-mTOR and phospho-p70 S6 kinase. The phospho-Akt, phospho-mTOR and phospho-p70 S6 kinase immunoblots were then reprobed with Akt, mTOR and p70 S6 kinase antibodies, respectively. Note that MSF overexpression is associated with Akt pathway activation. This reaction is probably an anti-apoptotic response, to protect the fibroblasts from autophagy-induced cell death.
That L-lactate accumulation is indicative of a shift toward predominantly glycolytic metabolism. This observation was validated by assessing the status of mitochondrial activity in MSF fibroblasts. Figure 4B shows decreased mitochondrial activity, as predicted, as visualized using MitoTracker staining. Note that MSF induces a dramatic reduction in MitoTracker staining, indicative of a loss of healty functional mitochondria, both under normoxic, as well as hypoxic conditions.
As shown in Figure 4C, MSF overexpression leads to Akt activation, which likely protects these cells against apoptosis. MSF fibroblasts were subjected to immunoblot analysis, using phospho-specific antibodies directed against different protein components of the Akt pathway.
Note that MSF induces the activation of Akt-downstream effectors, such as phospho-mTOR and phospho-p70S6 kinase, both involved in protein biosynthesis. Akt normally activates mTOR, leading to p70S6K activation. Activation of Akt pathway by MSF in stromal fibroblasts may lead to activation of protein synthesis, as a compensatory mechanism to prevent apoptotic cell death in cells undergoing constitutive autophagy/mitophagy.
Fibroblast overexpressing MSF promote tumor growth, without any increases in tumor angiogenesis
Because MSF fibroblasts are able to increase L-lactate production and have a strong autophagic phenotype, we evaluated whether MSF is able to promote tumor growth. For this purpose, we developed a human tumor xenograft model. MSF-overexpressing fibroblasts were co-injected with MDA-MB-231 breast cancer cells into the flanks of immunodeficient nude mice. Figure 5A demonstrates that MSF overexpression in stromal fibroblasts is sufficient to promote tumor growth, as evidenced by significant increases in both tumor weight and volume.

Figure 5. MSF overexpression in fibroblasts dramatically promotes breast cancer tumor growth, without affecting tumor angiogenesis. (A) Control or MSF fibroblasts were co-injected with MDA-MB-231 breast cancer cells into the flanks of nude mice. After 4 weeks post-injection, the resulting tumors were harvested. Interestingly, relative to control fibroblasts (Lv-105), MSF fibroblasts increased tumor weight by ~2.5-fold (p = 0.03) and tumor volume by ~4-fold (p = 0.01). n = 10 tumors per experimental group. (B) MSF overexpressing fibroblasts do not increase tumor angiogenesis. Frozen tumor sections were immunostained with anti-CD31 antibodies to quantify vessel density. As shown in the figure, note that the tumor promoting effect of MSF fibroblasts are independent of angiogenesis, as no significant increases in vessel density were observed.
Stromal expression of MSF may contribute to tumor pathogenesis by a number of mechanism(s), including the stimulation of angiogenesis. To address this issue, frozen tissue sections derived from tumor xenografts were subjected to immunostaining with a well-established vascular marker, namely CD31. As shown in Figure 5B, MSF overexpression in stromal fibroblasts does not have a significant effect on tumor neo-vascularization, indicating that the tumor-promoting effects of MSF in cancer-associated fibroblasts are independent of tumor angiogenesis.
SMA, Rac1 and Cdc42 overexpression in fibroblasts induces myo fibroblast differentiation
We demonstrated above that MSF fibroblasts show increased expression of SMA and two small GTPase proteins, namely Rac1 and Cdc42. To determine if there is a cause-effect relationship here, we employed a genetic approach by overexpressing SMA, Rac1, and Cdc42 in an immortalized human fibroblast cell line (Fig. 6AC) Then, these fibroblast cell lines were subjected to immunoblot analysis, employing a panel of myo fibroblast markers, in order to characterize their phenotype.
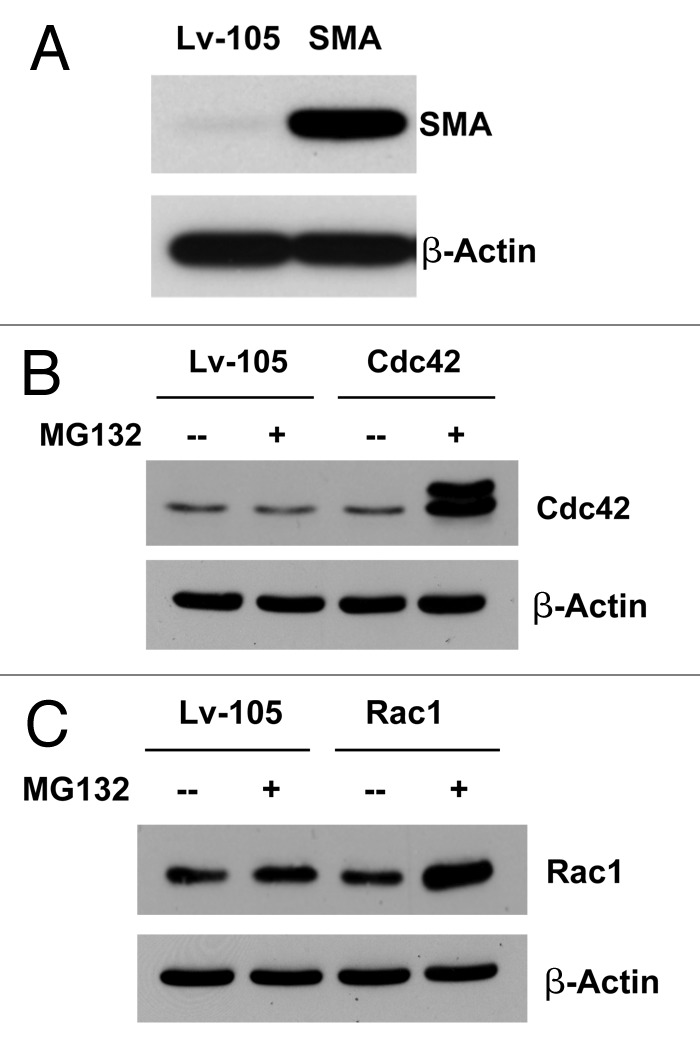
Figure 6. Recombinant expression of SMA, Rac1, and Cdc42 in immortalized fibroblasts. To assess the role of SMA, Rac1, and Cdc42 in the tumor microenvironment, we stably overexpressed SMA, Rac1, and Cdc42 in the immortalized human fibroblast cell line (hTERT-BJ1 cells). Successful protein overexpression of SMA, Rac1, and Cdc42 was validated by immunoblot analysis (A–C). In order to better visualize Cdc42 and Rac1 overexpression, we treated fibroblasts with a protease inhibitor (MG132; 10 µM for 16 h).
Note that Rac1- and Cdc42-overexpressing fibroblasts display the upregulation in SMA protein expression (Fig. 7A), and all three overexpressing cell lines show increases in the calponin and vimentin (Fig. 7B), consistent with a myo fibroblast phenotype. Similarly, both GTPases, Rac1 and Cdc42, were able to induce the reorganization of the F-actin cytoskeleton, as evidenced by an increase in the density of actin stress fibers, as visualized by Phalloidin staining (Fig. 7C); note the bundles of parallel fibers aligned along the cell axis.
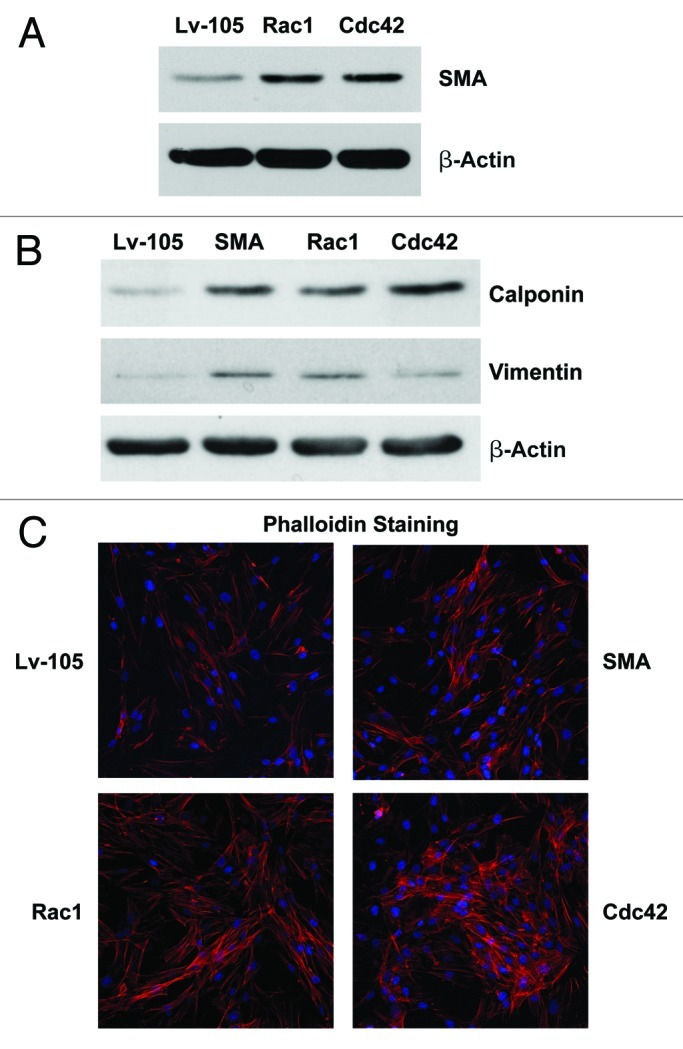
Figure 7. Overexpression of SMA, Rac1, and Cdc42 confers the myo fibroblast phenotype. (A) Rac1- and Cdc42- overexpressing fibroblasts display the upregulation of SMA protein expression. (B) Rac1 and Cdc42 induce the expression of other myofibroblast markers, namely calponin and vimentin. (C) Rac1 and Cdc42 regulate the organization of the actin cytoskeleton, inducing a reorganization of F-actin, as suggested by the increase of actin stress fibers. Equal cell numbers were plated on glass cover sides, and after 24 h, the cells were fixed and actin filaments and nuclei were stained with Phalloidin (red) and DAPI (blue), respectively. Note that SMA-, Rac1- and Cdc42- overexpressing fibroblasts display an increased number of stress fibers, as compared with vector-alone control cells.
Cdc42-overexpression induces NFκB-activation, with increased autophagy and a shift toward glycolytic metabolism
Small GTPases are strong activators of the transcription factor NFκB.47,48 Thus, we evaluated the effects of expressing SMA, Rac1 and cdc42 in fibroblasts, on the status of NFκB and p-NFκB. Our results demonstrate that the p-NFκB protein levels are significantly increased only in Cdc42-overexpressing fibroblasts (Fig. 8A).

Figure 8. Cdc42-overexpression promotes the activation of NFκB, induces increased autophagy and glycolytic metabolism. (A) GTPases are strong activators of the transcription factor NFκB. As shown in figure, note that immunoblot analysis of control or SMA-, Rac1- and cdc42- overexpressing fibroblasts reveals that p-NFκB protein levels are significantly increased, exclusively in Cdc42 fibroblasts, as compared with control fibroblasts. (B) To validate that Cdc42 induces an autophagic program, cells were subjected to immunoblot analysis using several autophagy markers. β-actin was used as an equal loading control. Note that Cdc42 increases the expression of the autophagy markers, such as Beclin-1, BNIP3, LAMP-1 and Cathepsin B (37kDA). (C) Cdc42-overexpressing fibroblasts display a predominantly glycolytic metabolism, as demonstrated by increased L-Lactate production, under hypoxic conditions. Cells cultured for 48 h under hypoxic conditions (0.5% O2) were treated with or without metformin (1 mM), and the results are expressed as ratio between treated vs. untreated cells. (D) The shift toward a predominantly glycolytic metabolism is also demonstrated by decreased mitochondrial activity, as visualized using MitoTracker. Note that Cdc42 significantly decreases mitochondrial activity, as compared with vector alone control or SMA overexpressing fibroblasts. MitoTracker (red); nuclei/DAPI (blue). Original magnification, 60x.
For this and all subsequent experiments, we chose to examine only the fibroblasts overexpressing SMA and Cdc42; SMA was used as a negative control and Cdc42 was used, as it is the GTPase that activates NFκB. To evaluate if this Cdc42-driven NFκB-activation promotes autophagy, fibroblasts overexpressing SMA and Cdc42 were subjected to immunoblot analysis, using a panel of autophagy markers.
Figure 8B demonstrates that Cdc42 overexpression in fibroblasts drives the increased expression of mitophagy (BNIP3) and autophagy markers (Beclin-1, LAMP1 and Cathepsin B).
Also, we evaluated if Cdc42-overexpressing fibroblasts are able to induce L-lactate accumulation and a shift toward glycolytic metabolism. Figure 8C demonstrates that Cdc42 expression is sufficient to induce an ~80% increase in L-lactate production, under hypoxic condition and after treatment with Metformin, a specific inhibitor of mitochondrial complex I. This shift toward glycolytic metabolism was further validated by MitoTracker staining, showing that Cdc42 expression strongly decreases mitochondrial activity under hypoxic conditions (Fig. 8D).
Stromal expression of Cdc42 promotes increased tumor growth in vivo
To evaluate if Cdc42 expression in stromal cells is able to promote tumor growth in vivo, we used a human tumor xenograft model. Control, SMA or Cdc42 fibroblasts were co-injected with MDA-MB-231 breast cancer cells in the flanks of immunodeficient nude mice. Figure 9A shows that overexpression of Cdc42 in stromal fibroblasts consistently promotes tumor growth, over a 25-d time course. Figure 9B shows that, at 4 weeks post-injection, Cdc42 fibroblasts increased tumor volume by ~1.75-fold, as compared with vector-alone control fibroblasts cells, directly demonstrating that stromal Cdc42 is able to support tumor growth in vivo.
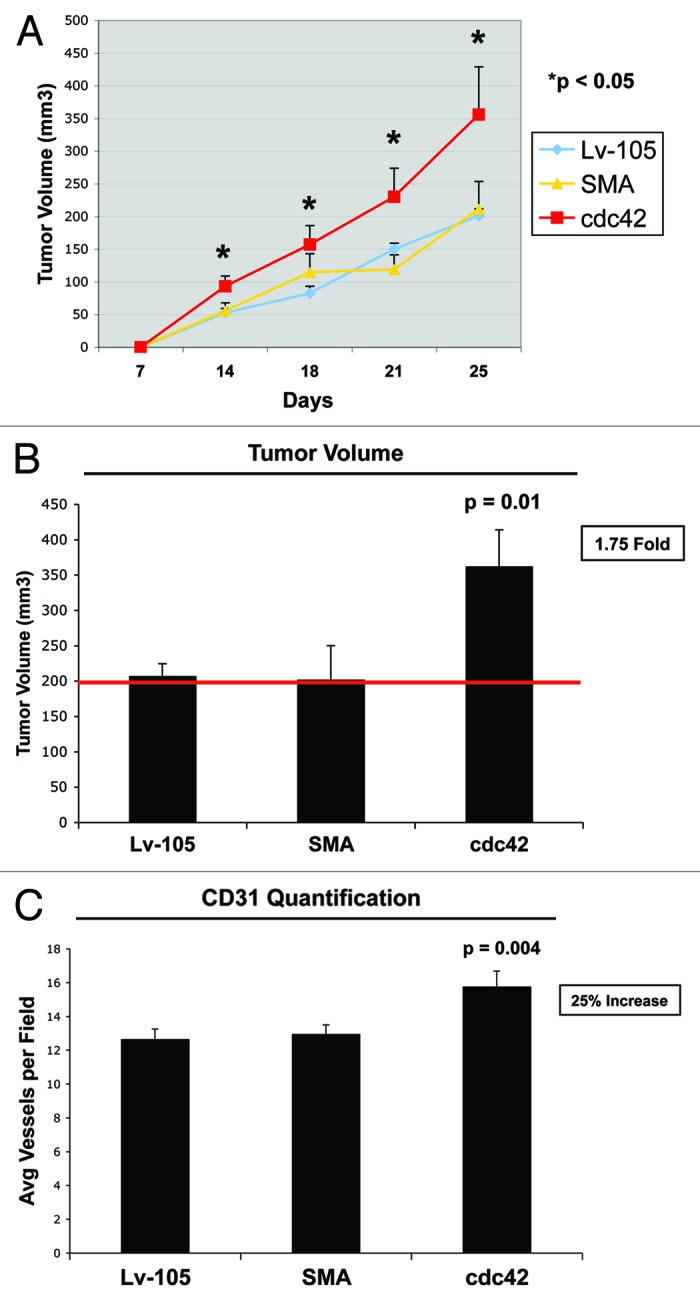
Figure 9. Cdc42 overexpressing fibroblasts promote tumor growth in vivo. We used a xenograft model employing MDA-MB-231 breast cancer cells injected into the flanks of athymic nude mice. MDA-MB-231 breast cancer cells were co-injected with either the empty vector (Lv-105), SMA- or Cdc42- overexpressing fibroblasts. (A) Comparative trend measurements for tumor growth (days 7–25 post-injection). Tumor volumes were measured with callipers about twice a week and mean tumor volume is plotted vs. time, for each experimental group. (B) Tumor growth. Tumor volumes were also measured at 4 weeks post-injection. Note that fibroblasts overexpressing Cdc42 significantly promote tumor growth, resulting in a 1.75-fold increase in tumor volume. p = 0.01; n = 10 tumors per experimental group. (C) Tumor angiogenesis. Tumor frozen sections were cut and immunostained with anti-CD31 antibodies. Then, vascular density (number of vessels per field) was quantified. The observed 25% increase in tumor angiogenesis in Cdc42 tumors is not sufficient to account for a near 2-fold increase in tumor growth. Instead, metabolic reprogramming of the tumor microenvironment, toward L-lactate production, is a more likely mechanism.
Finally, to determine the role of neo-vascularization in Cdc42-mediated tumor growth, we quantified neo-vascularization via immunostaining with CD31 (Fig. 9C). However, a 25% increase of tumor angiogenesis in Cdc42 tumors is not sufficient to account for a near 2-fold increase in tumor growth. Instead, metabolic reprogramming of the tumor microenvironment toward L-lactate production is a more likely mechanism (Fig. 10).

Figure 10. MSF expression in cancer-associated fibroblasts drives tumor growth. MSF leads to the upregulation of SMA and a number of other myo fibroblast marker proteins, conferring a myo fibroblast phenotype. The fibroblast activation by MSF is probably induced by TGF-β signaling and the increased expression of the small GTPase Cdc42, driving the activation of NFκB that, in turn, induces autophagy, mitophagy and aerobic glycolysis, thereby promoting tumor growth.
Discussion
The role of the host stromal microenvironment in promoting tumor initiation and progression is now well-established.1-4 However, the exact molecular mechanism(s) of how cancer-associated fibroblasts promote tumor growth remain unknown. Here, we highlight that MSF (migration-stimulating factor) functions to metabolically reprogram stromal fibroblasts toward glycolytic metabolism, resulting in the generation of a catabolic tumor microenvironment that actively “fuels” anabolic tumor growth.
More specifically, MSF-overexpressing fibroblasts were used to mimic the “activated microenvironment” that is now widely known to support tumor growth. We demonstrated that MSF fibroblasts show many characteristics of differentiated myo fibroblasts, including the expression of smooth muscle-specific proteins.
Transforming growth factor-β (TGF-β) is a potent inducer of myo fibroblast differentiation that has been implicated in conferring the tumor-associated fibroblast phenotype.3,18,19,49-52 Here, we have demonstrated that MSF overexpression in stromal fibroblasts leads to the increased production of TGF-β and is associated with a reduction in the expression of its receptor, TGF-β-RI. The expression of TGF-β, specifically TGF-β1, is upregulated in most tumors and seems to play a key role in cancer progression.3,18,19,49-52
Increased TGF-β expression in fibroblasts benefits cancer progression, likely via paracrine effects on tumor cells.18,19,49-52 In particular, the release of TGF-β in the vicinity of cancer cells may result in a more hospitable microenvironment, facilitating tumor growth. Several authors have shown that TGF-β overexpression leads to an increased metabolic rate, due to enhanced glycolysis.53,54 MSF may induce glycolysis in stromal fibroblasts via increased endogenous production of TGF-β. The observed increase in glycolytic metabolism may be due to the autophagic destruction of mitochondria in MSF-overexpressing fibroblasts. This assertion is consistent with our previous observations that autophagy in cancer-associated fibroblasts is able to generate a catabolic tumor stroma that drives the anabolic growth of cancer cells.8,9 Regardless of the exact mechanism activating glycolysis, MSF is able to produce a catabolic, energy-rich microenvironment that favors tumor growth.
Small GTPases, such as Rac1 and Cdc42, are known to play a critical role in cell migration and invasion.55,56 However, their potential roles in myo fibroblast differentiation, autophagy and cellular metabolism are underappreciated. In the current study, we demonstrated that MSF-overexpressing fibroblasts have increased expression both Rac1 and Cdc42. To determine whether increased Rac1 and/or Cdc42 expression influences the activation of tumor microenvironment, we generated Rac1- and Cdc42-overexpressing fibroblasts.
Our results demonstrate that both Rac1 and Cdc42 fibroblasts undergo myo fibroblast differentiation, with characteristic re-organization of the actin cytoskeleton. However, only Cdc42 fibroblasts show activation of NFκB, with the onset of autophagy and a shift toward predominantly glycolytic metabolism in the tumor stroma resulting in the promotion of tumor growth. Therefore, overexpression and/or activation of Cdc42 is a likely mechanism by which MSF induces NFκB-activation, leading to increased autophagy and glycolysis due to reduced mitochondrial function. As such, glycolytic/catabolic MSF fibroblasts create a favorable metabolic microenvironment to support tumor growth.
In conclusion, our results highlight the critical functional role of MSF as a driver of cancer progression. This is consistent with its ability to stimulate the migration/invasion in both stromal and tumor cells and with its effects on the metabolic remodeling of the tumor microenvironment.
Materials and Methods
Materials
Reagents were purchased as follows: the specific and cell-permeable proteasome inhibitor (MG132) was from Calbiochem (used at a final concentration of 10 µM for 16 h); Metformin (1.1-dimethylbiguanide hydrochloride) was from Sigma (D150959); Alexa Fluor 633 Phalloidin (A222284) was from Invitrogen. Antibodies to the following target proteins were also used: Fibronectin N-terminal (Chemicon International, MAB1936; to recognize MSF); Fibronectin (Abcam, ab23750); Vimentin (BD PharMingen, 550513); Calponin 1/2/3 (Santa Cruz Biotech, sc-28545); Smooth Muscle Actin (Dako, M0851); Beclin (Novus Biologicals, NBP1–00085); BNIP-3 (Abcam, ab10433); LC3 (Abcam, ab48394); β-actin (Sigma-Aldrich, A5441); TGF-β (Cell Signaling, 3711); TGF-β-RI (Santa Cruz Biotech, sc-398); phospho-Akt (Cell Signaling, 9271); Akt (Cell Signaling, 2967); phospho-mTOR (Cell Signaling, 2971); mTOR (Cell Signaling, 2972); phospho-p70 S6 kinase (Cell Signaling, 9234); p70 S6 kinase (Cell Signaling, 9202); CD31 (BD Biosciences, 550274); Rac1 (Santa Cruz, sc-95); Cdc42 (Santa Cruz, sc-8401); p-NFκB (Cell Signaling, 3037); NFκB (Cell Signaling, 3034); p14ARF (Santa Cruz, sc-53639); p16 (Santa Cruz, sc-759); p21 (Santa Cruz, sc-6246); LAMP1 (Santa Cruz, sc-17768); cathepsin B (Santa Cruz, sc-13985).
Cell culture and stable transfection
Human immortalized fibroblasts (hTERT-BJ1) were used to generate the cell lines overexpressing migration-stimulatory factor (MSF), SMA, Rac1 and Cdc42. Lentiviral plasmids [EX-NEG-Lv105 (empty vector), Ex- Z4998-Lv105 (human MSF), Ex-D0101-Lv105 (human SMA), Ex-A0247-Lv105 (human Rac1), Ex-X0047-Lv105 (human cdc42) (obtained from GeneCopoeia, Inc.)] were used to transfect GeneCopoeia 293Ta lentiviral packaging cells using Lenti-PacTM HIV Expression Packaging Kit following the manufacturer’s instructions. After 48 h, lentivirus containing culture medium was added to human fibroblasts in the presence of 5 μg/ml Polybrene. Infected fibroblasts were selected with puromycin (1.5 μg/ml).
All cell lines used in the following experiments were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum in a 37°C humidified atmosphere (5% CO2) unless otherwise noted.
Immunoblot analysis
For immunoblotting, cultured cells were harvested in lysis buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, and 60 mM n-octyl-glucoside) or RIPA lysis buffer containing protease inhibitors (Roche, 11836153001) and phosphatase inhibitors (Thermoscientific, 78420). The pooled cells were rotated for 40 min at 4°C, centrifuged at 10.000 × g for 15 min at 4°C, and the protein concentration of the supernatant was determined using the BCA reagent (Pierce). Protein samples (30–50 μg total proteins per lane) were then subjected to 12% or 15% SDS-PAGE, and the proteins were then electrophoretically transferred to a nitrocellulose membrane. After blocking for 1 h at room temperature with TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.05% Tween 20) supplemented with 5% nonfat dry milk and 1% BSA, membranes were incubated for 1 h at room temperature with primary antibodies and then for 1 h at RT with specific (HRP)-conjugate secondary antibodies [anti-mouse, 1:6,000 dilution (Pierce) or anti-rabbit, 1:5,000 dilution (BD PharMingen)]. HRP activity was visualized by enhanced chemiluminescent substrate (Thermo Scientific) followed by exposure of the membrane to X-ray film.
Migration assay
The effects of MSF overexpression on fibroblast migration and the effects of MSF fibroblasts on the migratory potential of MDA-MB-231 cells were measured in vitro using a modified Boyden chamber assay. Briefly, fibroblasts [EV (empty vector) or MSF overexpressing] in 0.5 ml of serum-free Dulbecco's modified Eagle's medium were added to the wells of 8 µm pore uncoated membrane of modified Boyden chambers. The lower chambers contained 10% fetal bovine serum in Dulbecco's modified Eagle's medium to serve as a chemo-attractant. Cells were incubated at 37°C and allowed to migrate throughout the course of 6 h. To assess the effect of fibroblast overexpressing MSF protein on the migratory capacity of MDA-MB-231 cells, fibroblasts were seeded in the lower chambers in DMEM supplemented with 10% NuSerum and used as chemo-attractant. MDA-MB-231 cells were added to the wells of 8 µm pore uncoated membrane of modified Boyden chambers and allowed to migrate throughout the course of 4 h at 37°C. In both cases, cells were removed from the upper surface of the membrane by scrubbing with cotton swabs. Chambers were stained in 0.5% crystal violet diluted in 100% methanol for 30 min, rinsed in water and examined under a bright-field microscope. Values for migration were obtained by counting five fields per membrane (X20 objective) and represent the average of three independent experiments.
L-lactate assay
Cells (empty vector or MSF-, SMA-, Cdc42- overexpressing fibroblasts) were seeded in quadruplicate (100,000 cells per well) in 12-well plates in 1 ml of complete media. After 18 h, the media was changed to DMEM containing 2% FBS and incubated under hypoxic conditions (0.5% O2). SMA and cdc42 overexpressing fibroblasts were also with or without metformin (1 mM). After 48 h, the media of each well was collected and the concentration of L-lactate was measured using the EnzyChrom™ L-Lactate Assay Kit (ECLC-100, BioAssay Systems, Inc.). After removing the media, cells were tyrpsinized, spun down and resuspended in 1 mL of media for quantification. Cells were counted in 4–6 fields, using a 40× objective lens and a hemocytometer. Cells were then lysed, and the protein concentration was determined using the BCA reagent (Pierce). The amount of L-lactate in the media was normalized to total cell number or to total cell protein content.
Mitochondrial staining
To evaluate mitochondrial activity, cells were stained with MitoTracker Orange (CMTMRos; M7510, Invitrogen, Inc.). Lyophilized MitoTracker was dissolved in DMSO to generate a 1 mM stock solution that was then diluted into serum-free DMEM at a final concentration of 25 nM. Briefly, control or MSF-, SMA-, Cdc42-overexpressing fibroblasts (90,000 cells per well) were cultured for 48 h in normoxia or under hypoxic conditions. Then, they were incubated with pre-warmed MitoTracker staining solution for 12 min at 37°C in the dark. Cells were then washed in PBS Ca2+/Mg2+, three times and fixed with 2% PFA 30 min a RT. Cell were washed again with PBS Ca2+/Mg2+, incubated with the nuclear stain DAPI and mounted.
Murine xenograft studies
All animals were housed and maintained in a barrier facility at the Kimmel Cancer Center at Thomas Jefferson University under National Institutes of Health (NIH) guidelines. Mice were kept on a 12-h light/dark cycle with ad libitum access to food and water. Animal protocols used for this study were pre-approved by the Institutional Animal Care and Use Committee (IACUC). Briefly, MDA-MB-231-GFP human breast cancer cells (1 × 106 cells) were co-injected with control (empty vector) or MSF-, SMA-, Cdc42- overexpressing fibroblasts (3 × 105 cells) in 100 µl of sterile PBS into the flanks of athymic NCr nude mice (NCRNU; Taconic Farms; 6–8 weeks of age). Mice were then sacrificed at 4 weeks post-injection; tumors were dissected to determine their weight and size using calipers. Tumor volume was calculated using the formula (X2Y)/2, where X and Y are the short and long dimensions, respectively, of the tumor. After the dissection, tumors were fixed with 10% formalin or flash-frozen in liquid nitrogen-cooled isopentane.
Quantitation of tumor angiogenesis
Immunohistochemical staining for CD31 was performed on frozen tumor sections using a three step biotin-streptavidin-horseradish peroxidase method. Frozen tissue sections (6 µm) were fixed in 4% paraformaldehyde in PBS for 10 min and washed with PBS. After blocking with 10% rabbit serum, the sections were incubated overnight at 4°C with rat anti-mouse CD31 antibody (BS Biosciences) at a dilution of 1:200, followed by biotinylated rabbit anti-rat IgG (Vector Labs, 1:200) antibody and streptavidin-HRP (Daki, 1:1000). Immunoreactivity was revealed with 3.3'-diaminobenzidine (DAB).
Phalloidin staining
Cell monolayers were stained with Alexa Fluor 633-phalloidin to examine the structure of filamentous F-actin. Washed cells were fixed with paraformaldehyde 2%, washed again with PBS Ca2+/Mg2+ and permeabilized for 10 min with TBP buffer (0.1% Triton X-100, 0.2% BSA in PBS Ca2+/Mg2+. The cells were stained with Phalloidin Staining Solution (diluted 1:40 in PBS 1% BSA) 30 min at RT in dark conditions. Stained F-actin was visualized using a Zeiss LSM510 meta-confocal system. Images were acquired with a 20× objective.
Statistical analysis
Statistical significance was examined using the Student's t-test. Values of p ≤ 0.05 were considered significant. Values were expressed as means ± SEM.
Acknowledgments
F.S. and her laboratory were supported by grants from the Breast Cancer Alliance (BCA) and the American Cancer Society (ACS). U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660), and the Susan G. Komen Breast Cancer Foundation. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072, and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L. and U.E.M.-O.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21701
References
- 1.Petersen OW, Nielsen HL, Gudjonsson T, Villadsen R, Rank F, Niebuhr E, et al. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol. 2003;162:391–402. doi: 10.1016/S0002-9440(10)63834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rønnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev. 1996;76:69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- 3.Sieweke MH, Bissell MJ. The tumor-promoting effect of wounding: a possible role for TGF-beta-induced stromal alterations. Crit Rev Oncog. 1994;5:297–311. doi: 10.1615/CritRevOncog.v5.i2-3.90. [DOI] [PubMed] [Google Scholar]
- 4.Sieweke MH, Thompson NL, Sporn MB, Bissell MJ. Mediation of wound-related Rous sarcoma virus tumorigenesis by TGF-beta. Science. 1990;248:1656–60. doi: 10.1126/science.2163544. [DOI] [PubMed] [Google Scholar]
- 5.Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64:8492–5. doi: 10.1158/0008-5472.CAN-04-1708. [DOI] [PubMed] [Google Scholar]
- 6.Santos AM, Jung J, Aziz N, Kissil JL, Puré E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest. 2009;119:3613–25. doi: 10.1172/JCI38988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123–8. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- 8.Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab. 2012;15:4–5. doi: 10.1016/j.cmet.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 9.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–67. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 10.Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011;43:1045–51. doi: 10.1016/j.biocel.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez-Outschoorn UE, Pavlides S, Sotgia F, Lisanti MP. Mitochondrial biogenesis drives tumor cell proliferation. Am J Pathol. 2011;178:1949–52. doi: 10.1016/j.ajpath.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011;10:4047–64. doi: 10.4161/cc.10.23.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772–83. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal. 2012;16:1264–84. doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 16.Casey TM, Eneman J, Crocker A, White J, Tessitore J, Stanley M, et al. Cancer associated fibroblasts stimulated by transforming growth factor beta1 (TGF-beta 1) increase invasion rate of tumor cells: a population study. Breast Cancer Res Treat. 2008;110:39–49. doi: 10.1007/s10549-007-9684-7. [DOI] [PubMed] [Google Scholar]
- 17.Desmoulière A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–11. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci USA. 2010;107:20009–14. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–33. doi: 10.4161/cc.9.12.12048. [DOI] [PubMed] [Google Scholar]
- 20.Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331–9. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rønnov-Jessen L, Petersen OW. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Invest. 1993;68:696–707. [PubMed] [Google Scholar]
- 22.Surowiak P, Murawa D, Materna V, Maciejczyk A, Pudelko M, Ciesla S, et al. Occurence of stromal myofibroblasts in the invasive ductal breast cancer tissue is an unfavourable prognostic factor. Anticancer Res. 2007;27(4C):2917–24. [PubMed] [Google Scholar]
- 23.Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol Med. 2010;2:211–30. doi: 10.1002/emmm.201000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsujino T, Seshimo I, Yamamoto H, Ngan CY, Ezumi K, Takemasa I, et al. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin Cancer Res. 2007;13:2082–90. doi: 10.1158/1078-0432.CCR-06-2191. [DOI] [PubMed] [Google Scholar]
- 25.Vozenin-Brotons MC, Sivan V, Gault N, Renard C, Geffrotin C, Delanian S, et al. Antifibrotic action of Cu/Zn SOD is mediated by TGF-beta1 repression and phenotypic reversion of myofibroblasts. Free Radic Biol Med. 2001;30:30–42. doi: 10.1016/S0891-5849(00)00431-7. [DOI] [PubMed] [Google Scholar]
- 26.Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, et al. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 2005;19:854–6. doi: 10.1096/fj.04-2882fje. [DOI] [PubMed] [Google Scholar]
- 27.Schor SL, Haggie JA, Durning P, Howell A, Smith L, Sellwood RA, et al. Occurrence of a fetal fibroblast phenotype in familial breast cancer. Int J Cancer. 1986;37:831–6. doi: 10.1002/ijc.2910370606. [DOI] [PubMed] [Google Scholar]
- 28.Schor SL, Schor AM, Howell A, Crowther D. Hypothesis: persistent expression of fetal phenotypic characteristics by fibroblasts is associated with an increased susceptibility to neoplastic disease. Exp Cell Biol. 1987;55:11–7. doi: 10.1159/000163389. [DOI] [PubMed] [Google Scholar]
- 29.Haggie JA, Sellwood RA, Howell A, Birch JM, Schor SL. Fibroblasts from relatives of patients with hereditary breast cancer show fetal-like behaviour in vitro. Lancet. 1987;1:1455–7. doi: 10.1016/S0140-6736(87)92206-9. [DOI] [PubMed] [Google Scholar]
- 30.Colletta AA, Wakefield LM, Howell FV, van Roozendaal KE, Danielpour D, Ebbs SR, et al. Anti-oestrogens induce the secretion of active transforming growth factor beta from human fetal fibroblasts. Br J Cancer. 1990;62:405–9. doi: 10.1038/bjc.1990.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schor SL, Grey AM, Picardo M, Schor AM, Howell A, Ellis I, et al. Heterogeneity amongst fibroblasts in the production of migration stimulating factor (MSF): implications for cancer pathogenesis. EXS. 1991;59:127–46. doi: 10.1007/978-3-0348-7494-6_9. [DOI] [PubMed] [Google Scholar]
- 32.Picardo M, Schor SL, Grey AM, Howell A, Laidlaw I, Redford J, et al. Migration stimulating activity in serum of breast cancer patients. Lancet. 1991;337:130–3. doi: 10.1016/0140-6736(91)90798-T. [DOI] [PubMed] [Google Scholar]
- 33.Schor SL, Grey AM, Ellis I, Schor AM, Howell A, Sloan P, et al. Fetal-like fibroblasts: their production of migration-stimulating factor and role in tumor progression. Cancer Treat Res. 1994;71:277–98. doi: 10.1007/978-1-4615-2592-9_15. [DOI] [PubMed] [Google Scholar]
- 34.Schor AM, Rushton G, Ferguson JE, Howell A, Redford J, Schor SL. Phenotypic heterogeneity in breast fibroblasts: functional anomaly in fibroblasts from histologically normal tissue adjacent to carcinoma. Int J Cancer. 1994;59:25–32. doi: 10.1002/ijc.2910590107. [DOI] [PubMed] [Google Scholar]
- 35.Schor SL, Ellis IR, Jones SJ, Baillie R, Seneviratne K, Clausen J, et al. Migration-stimulating factor: a genetically truncated onco-fetal fibronectin isoform expressed by carcinoma and tumor-associated stromal cells. Cancer Res. 2003;63:8827–36. [PubMed] [Google Scholar]
- 36.Kay RA, Ellis IR, Jones SJ, Perrier S, Florence MM, Schor AM, et al. The expression of migration stimulating factor, a potent oncofetal cytokine, is uniquely controlled by 3′-untranslated region-dependent nuclear sequestration of its precursor messenger RNA. Cancer Res. 2005;65:10742–9. doi: 10.1158/0008-5472.CAN-05-2038. [DOI] [PubMed] [Google Scholar]
- 37.Schor SL, Schor AM, Rushton G. Fibroblasts from cancer patients display a mixture of both foetal and adult-like phenotypic characteristics. J Cell Sci. 1988;90:401–7. doi: 10.1242/jcs.90.3.401. [DOI] [PubMed] [Google Scholar]
- 38.Schor SL, Schor AM, Grey AM, Rushton G. Foetal and cancer patient fibroblasts produce an autocrine migration-stimulating factor not made by normal adult cells. J Cell Sci. 1988;90:391–9. doi: 10.1242/jcs.90.3.391. [DOI] [PubMed] [Google Scholar]
- 39.Schor SL, Schor AM. Foetal-to-adult transitions in fibroblast phenotype: their possible relevance to the pathogenesis of cancer. J Cell Sci Suppl. 1987;8:165–80. doi: 10.1242/jcs.1987.supplement_8.9. [DOI] [PubMed] [Google Scholar]
- 40.Grey AM, Schor AM, Rushton G, Ellis I, Schor SL. Purification of the migration stimulating factor produced by fetal and breast cancer patient fibroblasts. Proc Natl Acad Sci USA. 1989;86:2438–42. doi: 10.1073/pnas.86.7.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schor SL, Schor AM, Grey AM, Chen J, Rushton G, Grant ME, et al. Mechanism of action of the migration stimulating factor produced by fetal and cancer patient fibroblasts: effect on hyaluronic and synthesis. In Vitro Cell Dev Biol. 1989;25:737–46. doi: 10.1007/BF02623727. [DOI] [PubMed] [Google Scholar]
- 42.Schor SL, Schor AM. Characterization of migration-stimulating factor (MSF): evidence for its role in cancer pathogenesis. Cancer Invest. 1990;8:665–7. doi: 10.3109/07357909009018941. [DOI] [PubMed] [Google Scholar]
- 43.Ellis I, Grey AM, Schor AM, Schor SL. Antagonistic effects of TGF-beta 1 and MSF on fibroblast migration and hyaluronic acid synthesis. Possible implications for dermal wound healing. J Cell Sci. 1992;102:447–56. doi: 10.1242/jcs.102.3.447. [DOI] [PubMed] [Google Scholar]
- 44.Schor SL, Grey AM, Ellis I, Schor AM, Coles B, Murphy R. Migration stimulating factor (MSF): its structure, mode of action and possible function in health and disease. Symp Soc Exp Biol. 1993;47:235–51. [PubMed] [Google Scholar]
- 45.Schor SL. Fibroblast subpopulations as accelerators of tumor progression: the role of migration stimulating factor. EXS. 1995;74:273–96. doi: 10.1007/978-3-0348-9070-0_14. [DOI] [PubMed] [Google Scholar]
- 46.Schor SL, Schor AM. Phenotypic and genetic alterations in mammary stroma: implications for tumour progression. Breast Cancer Res. 2001;3:373–9. doi: 10.1186/bcr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perona R, Montaner S, Saniger L, Sánchez-Pérez I, Bravo R, Lacal JC. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11:463–75. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- 48.Cammarano MS, Minden A. Dbl and the Rho GTPases activate NF kappa B by I kappa B kinase (IKK)-dependent and IKK-independent pathways. J Biol Chem. 2001;276:25876–82. doi: 10.1074/jbc.M011345200. [DOI] [PubMed] [Google Scholar]
- 49.Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 50.Löhr M, Schmidt C, Ringel J, Kluth M, Müller P, Nizze H, et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001;61:550–5. [PubMed] [Google Scholar]
- 51.Massagué J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Massagué J. TGF-β signaling in development and disease. FEBS Lett. 2012;586:1833. doi: 10.1016/j.febslet.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 53.Racker E, Resnick RJ, Feldman R. Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc Natl Acad Sci USA. 1985;82:3535–8. doi: 10.1073/pnas.82.11.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nowak G, Schnellmann RG. Autocrine production and TGF-beta 1-mediated effects on metabolism and viability in renal cells. Am J Physiol. 1996;271:F689–97. doi: 10.1152/ajprenal.1996.271.3.F689. [DOI] [PubMed] [Google Scholar]
- 55.Monypenny J, Zicha D, Higashida C, Oceguera-Yanez F, Narumiya S, Watanabe N. Cdc42 and Rac family GTPases regulate mode and speed but not direction of primary fibroblast migration during platelet-derived growth factor-dependent chemotaxis. Mol Cell Biol. 2009;29:2730–47. doi: 10.1128/MCB.01285-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]