

Abstract
Hormone-dependent tumors are characterized by deregulated activity of specific steroid receptors, allowing aberrant expression of many genes involved in cancer initiation, progression and metastasis. In prostate cancer, the androgen receptor (AR) protein has pivotal functions, and over the years it has been the target of different drugs. AR is a nuclear receptor whose activity is regulated by a phosphorylation mechanism controlled by hormone and growth factors. Following phosphorylation, AR interacts with many cofactors that closely control its function. Among such cofactors, Pin1 is a peptidyl-prolyl isomerase that is involved in the control of protein phosphorylation and has a prognostic value in prostate cancer. In the present study, we demonstrate that ARSer81 is involved in the interaction with Pin1, and that this interaction is important for the transcriptional activity of AR. Since Pin1 expression positively correlates with tumor grade, our results suggest that Pin1 can participate in this process by modulating AR function.
Keywords: ARSer81, Pin1, androgen receptor, phosphorylation, prostate cancer
Introduction
The importance of androgen receptor in prostate cancer has been supported for more than 60 y, and androgen-deprivation therapy in advanced prostate cancer is currently used in clinical practice.1 The removal of testicular androgens by castration or by drug treatments results in tumor regression.2 However, recurrent tumors arise within a median of 2–3 y wherein androgen receptor signaling has been inappropriately restored.3 This form of cancer, named castration-resistant prostate cancer (CRPC), is a lethal form of prostate cancer that progresses to form metastatic lesions. Late-stage CRPC retains the expression of AR despite the near absence of circulating androgens, suggesting that receptor activity is subverted but not bypassed.4 Indeed, intracrine androgen production and AR amplification are the principal aberrancies that sustain tumor growth in CRPC patients. For the above-mentioned reasons, secondary hormonal therapies that directly target androgen synthesis or AR have proven successful in clinical trials.5-7 As such, the improvement of these secondary therapies will directly depend on our understanding of the mechanisms of AR function and resistance.8
AR is a ligand-dependent transcription factor that controls the expression of a large number of downstream target genes in organs with reproductive functions, like the prostate. Similar to other steroid hormone receptors, AR becomes activated after binding to a ligand such as dihydrotestosterone (DHT), and the receptor-ligand complex undergoes a conformational change called transformation. During this process, the receptor releases inhibitory heat shock proteins, translocates to the nucleus, binds the androgen responsive elements (ARE) in the genome and regulates the expression of target genes through the recruitment of coactivators, histone acetylases and chromatin remodeling complexes.9-14
The active form of AR is characterized by post-translational modifications such as acetylation, sumoylation and phosphorylation,15,16 which can cooperate with each other.17,18 For instance, AR is heavily phosphorylated in the presence of androgens or after exposure to growth factors.19-23 Each domain of AR has amino acid residues that are phosphorylated and, in turn, regulate AR function. The N-terminal domain, which is a ligand-independent coactivator domain, contains the majority of these sites (13) that are constitutive phosphorylated (Ser94) or phosphorylated after growth factors either androgens stimulation.22 One piece of compelling evidence of AR regulation by phosphorylation is the control of nuclear-cytoplasmic shuttling. AR is constantly shuttling from the nucleus to the cytoplasm, and androgens govern this process. The distribution of kinases and phosphatases between the nucleus and cytoplasm determine in which compartment and at what specific site AR is phosphorylated, thus affecting AR distribution.24,25
Among phosphorylatable amino acids, the serine/threonine residue followed by proline is a key regulatory mechanism for the control of protein activity, with effects on biological processes such as cell proliferation and transformation.26-28 Phospho-serine/threonine-proline (pS/T-P) motifs are potential substrates for the peptidyl prolyl cis/trans isomerase Pin1, which regulates the cis/trans isomerization of target proteins, causing a wide spectrum of effects. Pin1 is a candidate oncogene that is overexpressed in 38 out of 60 different tumor types, including prostate, cervical, brain, ovarian, lung, breast and liver cancer and melanoma.29 The importance of Pin1 in prostate cancer has already been demonstrated. Indeed, Pin1 expression correlates with increased risk of recurrence after radical prostatectomy,30 and it has been established as an independent prognostic marker in PC patients after radical prostatectomy.31 Furthermore, Pin1 depletion significantly suppresses prostate cancer cell growth in nude mice.32
Here we demonstrate that Pin1 forms a protein complex with androgen receptor (AR) and regulates its activity in prostate cancer by changing its phosphorylated status. This interaction could represent the starting point for developing new drugs for patients with prostate cancer.
Results
Pin1 interacts with AR
Over the last decade, much evidence has suggested a relationship between Pin1 and prostate cancer, and an indirect mechanism of AR regulation by Pin1 has been demonstrated.33
From our perspective, since AR contains many pS/P motifs, a direct interaction could be hypothesized (Fig. 1A). To demonstrate the interaction between AR and Pin1, we performed a GST pull-down experiment in LNcaP AR-positive prostate cancer cell line. Figure 1B shows that GSTPin1 interacts with AR. To further demonstrate this interaction, we co-immunoprecipitated the endogenous (E) and overexpressed (OE) Pin1 protein in the presence of androgens. Western blot analysis showed that AR interacts with Pin1 and it is not immunoprecipitated in Pin1 kd cells (Fig. 1C). Co-immunoprecipitation of AR and Pin1 proteins without hormones (FBS charcoal-treated) was unsuccessful (data not shown). These results set up the basis for further investigations.
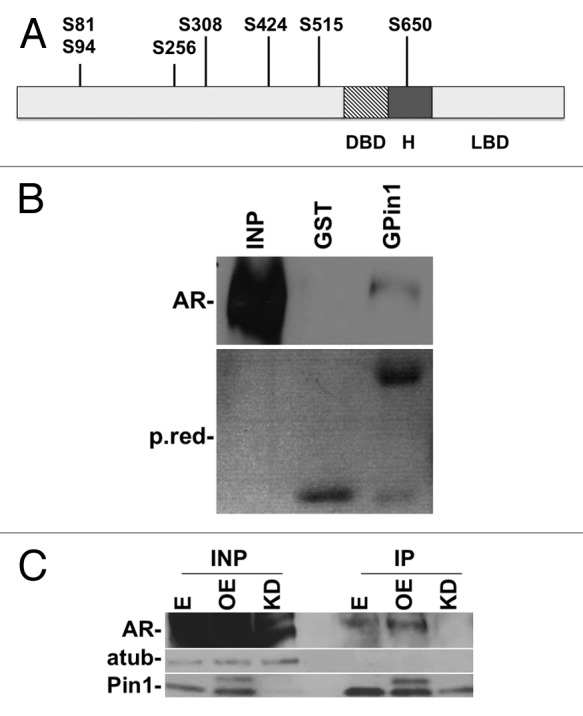
Figure 1. In vitro and in vivo interaction between Pin1 and AR. Schematic representation of AR. (A) Potential Pin1 binding site targets on AR protein. (B) GST-Pin1 interaction with AR. A specific band is detected in the GST-PIN1 lane and no band is detected in GST control lane. (C) Pin1 interacts with AR in vivo. Cells were immunoprecipitated with anti-Pin1 antibody, analyzed by western blot with anti-AR antibody. The membrane was normalized with α-tubulin antibody. E, empty vector; OE, overexpression of Pin1; KD, Pin1 knockdown.
ARSer81 is the target of Pin1
Structural analysis of AR revealed that it contains three major functional domains. The functional domains of AR are conserved with the other members of the “classic” receptor subclass. The N-terminal activation functional domain [(AF-1) residues 1–555] contains different binding sites for transcriptional regulators. Downstream of the AF-1 lies the DNA-binding domain (DBD) (residues 556–624), which mediates sequence-specific binding to DNA regulatory sequences of target genes. The hinge region (residues 625–670) links the DBD to the C-terminal domain. The C-terminal region of AR, the ligand-binding domain (LBD), is responsible for binding hormones and ligand-induced receptor dimerization.34,35
To dissect which functional domain is responsible for AR and Pin1 interaction, we cloned and expressed AR in two different segments: the first one contained the NTD-DBD domains from 1 to 625 a.a., and the second subclone contained the LDB domain from 626 to 919 a.a.. The two domains were overexpressed in 293FT cell lines, and the total protein lysate was pulled-down with GST or GST-Pin1. WB analysis showed that the AR-NTD/DBD domain interacts with Pin1. HSP70 antibody was used to normalize samples (Fig. 2A). Since the the NTD domain contains six different potential Pin1 binding sites, we split the AR NTD domain into three different fragments: NTD-A from 1 to 239 a.a., NTD-B from 240 to 410 a.a., NTD-C from 411 to 560 a.a. GST pull-down experiments narrowed the interaction down to the first 239 a.a. of AR (Fig. 2B).
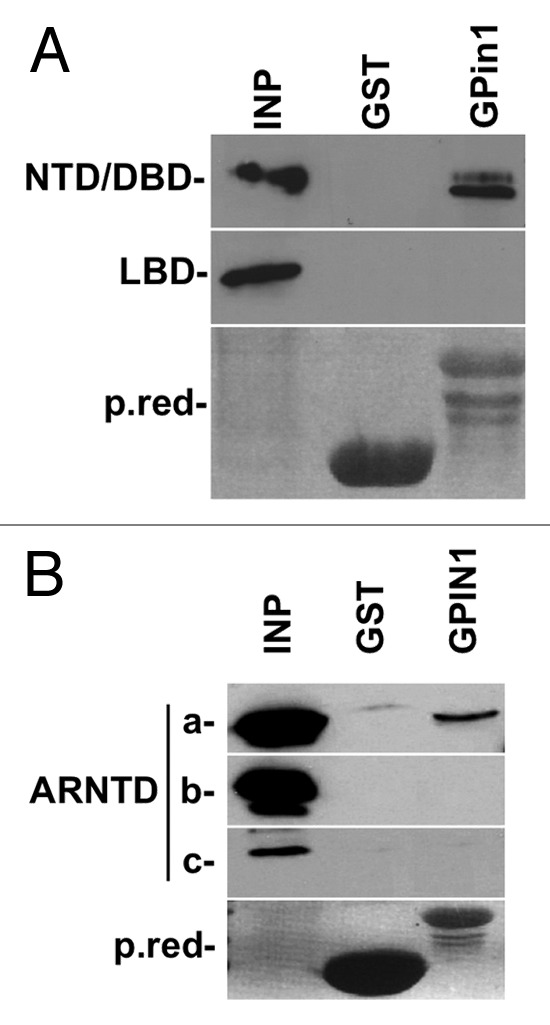
Figure 2. (A) Pin1 interacts with AR-NTD-DBD domain. The AR protein was split into two fragments and pulled down with GST-Pin1. Note no band is detected in GST control line. (B) The N-terminal domain NTD of AR was split into three fragments and pulled down with GST-Pin1. GST alone was used as the negative control. Pin1 interacts with NTD-A corresponding to the first 239 a.a. of AR. NTD, N-terminal domain; DBD, DNA binding domain; LDB, Ligand binding domain.
On the first 239 a.a. of AR, there are two S-P motifs that are potential binding sites of Pin1: Ser 81 and Ser 94. To explore which Ser/Pro motif is bound by Pin1, we mutated ARSer81 and ARSer94 in alanines. The obtained mutant proteins were transfected in 293FT cells and subsequently pulled down with GSTPin1. The immunoblotting assay showed a 70% decrease in interaction with ARSer81Ala compared with the wild-type protein. In contrast, the Ser94Ala mutant did not affect the binding of Pin1 (Fig. 3). We cannot exclude that a non-canonical site in AR protein is a target of Pin1.
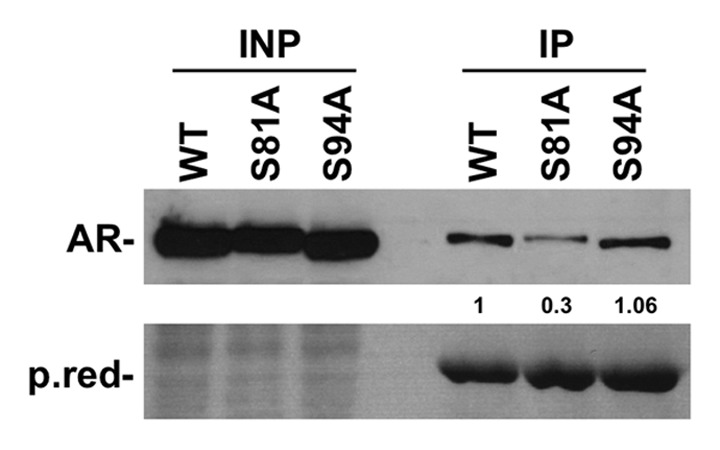
Figure 3. The figure shows a marked decrease in interaction between AR and GST-Pin1 when Ser81 is substituted with Ala, in contrast with the Ser94Ala mutant, which does not show any decrease in interaction compared with the AR wild type.
Pin1 controls the phosphorylation and activity of AR through Ser81
To better clarify the involvement of ARSer81 in AR-dependent transcriptional activity, a luciferase reporter assay was utilized. 293FT cells were transfected with AR, ARSer81Ala and the AR-dependent probasin promoter. Transfected cells were treated with DHT or the ethanol vehicle for 4 or 24 h. Transcriptional activity was measured in relative light unit/second (RLU/s) and normalized to Renilla activity.36 We observed a reduction in ARSer81Ala transcriptional activity compared with AR. The difference was statistically significant (Fig. 4). This experiment showed that ARSer81 is required for transcriptional activation of AR and supports the conclusion that ARSer81 is important for regulating expression of endogenous AR target genes.37
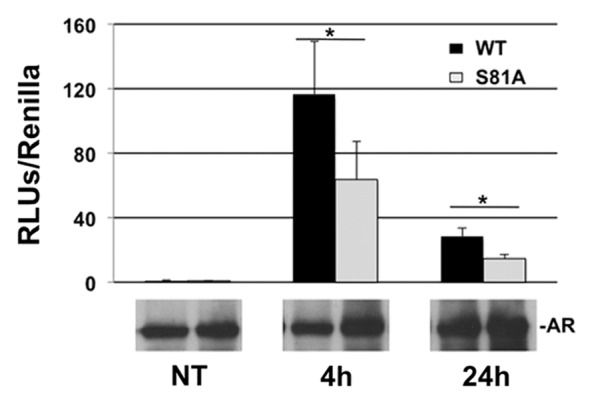
Figure 4. The figure shows that ARSer81 is required for transcriptional activation of AR target gene promoters. 293FT cells were transfected with AR, ARSer81Ala and the AR-dependent probasin promoter and treated with DHT for the indicated time. Transcriptional activity was measured in relative light unit/second (RLU/s) and normalized to Renilla activity. Below the figure is reported the expression of AR protein.
Among CDKs, CDK1 and recently CDK936,37 have been demonstrated to be involved in the phosphorylation of ARSer81. To examine in detail which kinase is responsible for the interaction between Pin1 and AR, we overexpressed the dominant-negative forms of CDK1, CDK2, CDK3, CDK5 and CDK9. Our data show that CDK9 barely affects the interaction between AR and Pin1, but a definitive conclusion is difficult to present (data not shown). These results open up the possibility that further kinases are implicated in ARSer81 phosphorylation.
To understand if Pin1 can control AR phosphorylation, we analyzed the phosphorylation of ARSer650 with the only commercially available antibody. AR and ARSer81Ala were transfected in 293FT cells and treated with DHT for the indicated time. We show that after DHT treatment, the phosphorylation of Ser650 is not dependent on hormone stimulation in the ARSer81Ala mutant protein. This result suggests that Pin1 can control AR phosphorylation after ligand stimulation and further experiments will be necessary to gain additional insight into this mechanism. (Fig. 5)
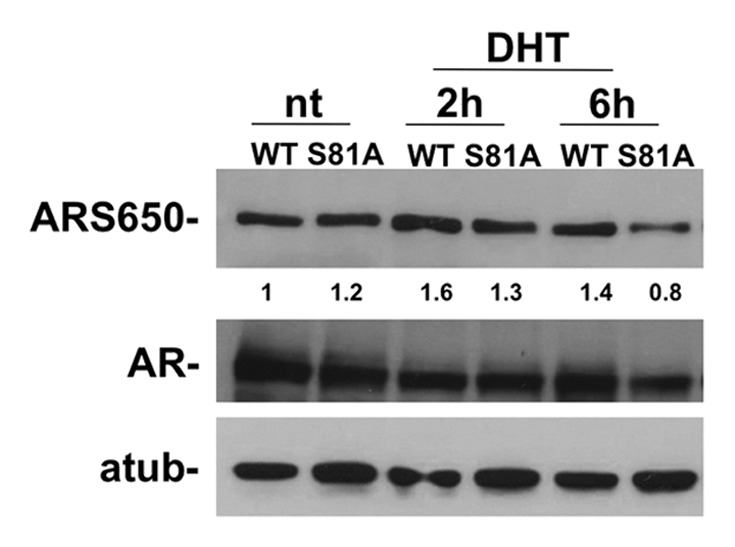
Figure 5. The figure shows ARSer650 phosphorylation after DHT treatment. 293FT cells were grown for three days in media without hormones and then a concentration of 10−8 DHT was added. The time course shows that the phosphorylation of ARSer650 does not increase after DHT treatment in 293FT cells transfected with mutated Ser81Ala.
Discussion
The importance of AR in prostate cancer is demonstrated by the fact that although advanced prostate cancer may be functionally independent of physiological levels of androgen, it is not independent of AR. The increase of serum PSA, a target of AR utilized as a prognostic marker in prostate cancer, indicates that AR function is restored in CRPC. For these reasons, even though androgen ablation is a most commonly prescribed treatment in the first stage of pathology, it is not curative. In-depth knowledge of AR regulation and function will allow for the development of new strategies for the treatment of prostate cancer.5,38,39
During oncogenic transformation, aberrant AR expression, localization and modifications switch the mechanism of control of AR activity in the biological process. In fact, it is hypothesized that the expression of AR in normal stromal cells controls epithelial cell proliferation in a manner that is different from cancer cells in which there is an autonomous epithelial cell mechanism. In this scenario, cofactor proteins play a central role in AR regulation and overexpression of coactivators such as p160 and p300 has long been demonstrated.4 The function of cofactors is complicated by the fact that each cofactor is integrated in a functional network, and more in-depth analyses are necessary to elucidate the mechanism of action. Among them, Pin1 is one of the central players that orchestrate and integrate the intra- and extracellular signals, allowing cancer cells to be highly performing machineries. Recently, it has been reported that Pin1 is overexpressed in human prostate cancer cell lines and prostate cancer tissues with an indirect effect on AR transcriptional activity. Pin1 can block the interaction of AR ligand binding domain with β-catenin. The inhibition of this interaction allows β-catenin and Tcf4 complex formation, and signaling, in turn, contributes to the aggressive behavior of prostate cancer cells.33
We provide evidence that the AR N-terminal domain interacts with Pin1 and clarify the role of this interaction in prostate cancer. We found that Pin1 binds AR at the Ser81 residue and, in turn, it controls the receptor’s transcriptional activity. We suggest that Pin1 can isomerize phosphoSer81 and induces conformational changes in AR. Utilizing a phospho-ARS650 antibody to determine the degree of AR phosphorylation, we demonstrate that ARSer81 is important to modulate the AR phosphorylation following DHT treatment, and further experiments will be done to refine this new model. It seems that the regulation of Pin1 at the N terminus of AR is part of a general mechanism that involves other, different nuclear receptors. In fact, the interaction between Pin1 and the N-terminal region of ERα 40, PPARγ 41 and RARα42,43 has been demonstrated.
The role of ARSer81 has been described with different results.21,33 Recently, the phosphorylation of ARSer81 by CDK9 was demonstrated to be important to regulate the specificity of AR transcriptional activity on the promoter. Up- or downregulation of target genes was observed.44 Overexpression of ARSer81 limits proliferation of prostate cancer cells compared with the wild-type form of AR.37 As for ARSer81, the role of ARSer650 is still controversial. Although early studies have suggested that substitution of Ser650 to Alanine reduces the activity of an MMTV-Luc reporter in CV1 cells at high concentrations of the receptor,44 other laboratories utilizing various cell lines and reporter assays have obtained different results.30 For instance, ARSer650 phosphorylation is involved in AR nuclear export in response to stress kinase signaling,24 and ARSer650 phosphorylation occurs by both hormone-dependent and -independent mechanisms (androgen, protein kinase A, EGF and protein kinase C), thus suggesting a role in AR regulation in response to a variety of physiological stimuli.
All these results indicate that each phosphorylation site on AR must be considered as a highly dynamic integration center of different signals that change over time. On this basis, different AR cofactors such as Pin1 act and balance the equilibrium of intra- and extracellular signals to orchestrate the correct function of AR. By elucidating these mechanisms, we can unravel the complex regulatory network that converges on AR and potentially apply this information to a target therapy in the near future.
Materials and Methods
Cell culture conditions
LNcaP (ATCC) and 293FT (Invitrogen Corp.) prostate cancer cell lines were grown according to the manufacturer’s instructions. For DHT experiments, cells were grown for 3 days in free-phenol red DMEM with 3% charcoal-stripped FBS.
Reagents
Antibodies were purchased as follows: Pin1 (600–401-A20), 6XHis (600–401–382) from Rockland Immunochemicals; AR (sc-7305), HSP70 (SC-24) from Santa Cruz Biotechnology; ARpS650 from Signalway (11120–1); α tubulin (T-6074) from Sigma Inc. shRNA plasmids for Pin1 (SHCLNG-NM_006221) were acquired from Sigma Inc. Scrambled shRNA (17920), psPAX2 packaging plasmid (12260), pMDG.2 envelop plasmid (12259) and PwPI (12254) were obtained from Addgene Inc. To overexpress Pin1, the IMAGE: 3941595 clone was amplified by PCR (Table S1) and cloned in the PwPI lentiviral vector. The plasmid was sequence-verified. The Probasin luciferase plasmid was purchased from Addgene (8392). The AR full length and AR sub-domains were amplified from the clone BC132975 (Table S1). After BamHI/EcoRI or XhoI double digestion, fragments were ligated in pcDNA6 His/Myc vector (Invitrogen Corp). ARSer81 and ARSer94 were generated from plasmid by site-directed mutagenesis using the QuickChange mutagenesis kit (Stratagene) (Table S1).
GST pull-down experiment
GST and GST-Pin1 proteins were produced in BL21 bacteria cells.27 Cells were grown to mid log phase and then induced to express protein by adding 0.25 mM of isopropyl-1-thio-b-D-galactopyranoside (IPTG, Roche Applied Science). The cultures were shaken for 4 h; bacteria were then pelleted and resuspended in NENT buffer [20 mM Tris (pH 8), 100 mM NaCl, 1 mM EDTA (EDTA), 0.5% NP-40]. Cell suspensions were sonicated and pelleted. The supernatant was incubated with glutathione agarose beads (Sigma Inc.) overnight at 4°C. The agarose beads were then pelleted and washed three times in NENT buffer. The GST protein was analyzed by electrophoresis gel and blue coomassie staining. One mg of protein was pulled down with 10 μg of GST or GST-Pin1.
Co-immunoprecipitation assay
Sub-confluent LNcaP cells were harvested and proteins were prepared as follows: the cell pellet was resuspended in lysis buffer (20 mM TRIS-HCl pH 8, 137 mM NaCl, 10% glycerol, 1% NP40, 2 mM EDTA). One mg of proteins was immunoprecipitated, utilizing 4 μg of Pin1 overnight at 4°C. The antibody-protein complex was collected with protein A/G beads (Pierce) for 3 h at 4°C. Immunopellets were washed extensively and subjected to SDS-PAGE followed by immunoblot analyses to detect Pin1 or AR proteins.
Luciferase assay
293FT cells were seeded in 96 multi-plate wells at 2 × 104 cells/well and grown in hormone-free medium. After 3 days, the cells were transfected (Fugene HD, Roche Applied Science) with 50 ng of Probasin luciferase plasmid, 50 ng of renilla plasmid (Promega, E2241) and 100 ng of AR or ARSer81Ala. After 24 h, the cells were treated treated with DHT for 4 or 24 h, then lysed and analyzed with the Dual Luciferase Assay System kit (Promega). The luminescence of each sample was measured in a single tube luminometer (Berthold Technologies, GmbH and CO). Each transfection was performed three times.
Lentiviral production
To generate knock-down cells, lentiviral particles were produced as described (http://www.broadinstitute.org/genome_bio/trc/publicProtocols.html).45 Briefly, 1 × 106 293FT cells (Invitrogen Corp.) were transfected with 2.25 μg of PAX2 packaging plasmid, 0.75 μg of PMD2G envelope plasmid and 3 μg of pLKO.1 hairpin vector utilizing 30 μl of Fugene HD (Roche Applied Science) in 10 cm plates. Polyclonal populations of Pin1 kd or overexpressed and scrambled cells were generated by infection with 1 MOI (multiplicity of infectious) of lentiviral particles. After 3 d post-infection, the cells were selected with 2 µg/ml of puromycin (Sigma-Aldrich) for 1 week.
Statistical analysis
Statistical analyses were performed using GraphPad software by applying unpaired Student’s t-test. Quantification of western blot was performed with Adobe Photoshop.
Supplementary Material
Acknowledgments
We thank Ms. Marie Basso for her assistance in copyediting the manuscript; Associazione Italiana per la Ricerca sul Cancro (AIRC) (A.G.), Special Program Molecular Clinical Oncology, 5 × 1,000, (No. 12214) (G.T.), European Research Council, Programme “Ideas,” Proposal No 269051 (F.R., G.T.), Italian Ministry of Education MIUR [FIRB prot. RBAP11ETKA (G.T.)], Sbarro Health Research Organization (A.G.), Human Health Foundation (A.G.) and Commonwealth of Pennsylvania (A.G.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental material may be found here: www.landesbioscience.com/journals/cc/article/21730
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21730
References
- 1.Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Loblaw DA, Virgo KS, Nam R, Somerfield MR, Ben-Josef E, Mendelson DS, et al. American Society of Clinical Oncology Initial hormonal management of androgen-sensitive metastatic, recurrent, or progressive prostate cancer: 2006 update of an American Society of Clinical Oncology practice guideline. J Clin Oncol. 2007;25:1596–605. doi: 10.1200/JCO.2006.10.1949. [DOI] [PubMed] [Google Scholar]
- 3.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 4.Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. J Carcinog. 2011;10:20. doi: 10.4103/1477-3163.83937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan CJ, Tindall DJ. Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. J Clin Oncol. 2011;29:3651–8. doi: 10.1200/JCO.2011.35.2005. [DOI] [PubMed] [Google Scholar]
- 6.Gioeli DG. The promise of novel androgen receptor antagonists. Cell Cycle. 2010;9:440–1. doi: 10.4161/cc.9.3.11045. [DOI] [PubMed] [Google Scholar]
- 7.Narizhneva NV, Tararova ND, Ryabokon P, Shyshynova I, Prokvolit A, Komarov PG, et al. Small molecule screening reveals a transcription-independent pro-survival function of androgen receptor in castration-resistant prostate cancer. Cell Cycle. 2009;8:4155–67. doi: 10.4161/cc.8.24.10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frezza M, Yang H, Dou QP. Modulation of the tumor cell death pathway by androgen receptor in response to cytotoxic stimuli. J Cell Physiol. 2011;226:2731–9. doi: 10.1002/jcp.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riegman PH, Vlietstra RJ, van der Korput JA, Brinkmann AO, Trapman J. The promoter of the prostate-specific antigen gene contains a functional androgen responsive element. Mol Endocrinol. 1991;5:1921–30. doi: 10.1210/mend-5-12-1921. [DOI] [PubMed] [Google Scholar]
- 10.Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–15. doi: 10.1200/JCO.2002.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Balk SP, Knudsen KEAR. AR, the cell cycle, and prostate cancer. Nucl Recept Signal. 2008;6:e001. doi: 10.1621/nrs.06001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slupianek A, Yerrum S, Safadi FF, Monroy MA. The chromatin remodeling factor SRCAP modulates expression of prostate specific antigen and cellular proliferation in prostate cancer cells. J Cell Physiol. 2010;224:369–75. doi: 10.1002/jcp.22132. [DOI] [PubMed] [Google Scholar]
- 13.Berman BP, Frenkel B, Coetzee GA, Jia L. Androgen receptor responsive enhancers are flanked by consistently-positioned H3-acetylated nucleosomes. Cell Cycle. 2010;9:2249–50. doi: 10.4161/cc.9.11.11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guseva NV, Rokhlin OW, Bair TB, Glover RB, Cohen MB. Inhibition of p53 expression modifies the specificity of chromatin binding by the androgen receptor. Oncotarget. 2012;3:183–94. doi: 10.18632/oncotarget.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu M, Wang C, Wang J, Zhang X, Sakamaki T, Yeung YG, et al. Androgen receptor acetylation governs trans activation and MEKK1-induced apoptosis without affecting in vitro sumoylation and trans-repression function. Mol Cell Biol. 2002;22:3373–88. doi: 10.1128/MCB.22.10.3373-3388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poukka H, Karvonen U, Janne OA, Palvimo JJ. Covalent modification of the androgen receptor by small ubiquitin-like modifier 1 (SUMO-1) Proc Natl Acad Sci USA. 2000;97:14145–50. doi: 10.1073/pnas.97.26.14145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu M, Rao M, Wu K, Wang C, Zhang X, Hessien M, et al. The androgen receptor acetylation site regulates cAMP and AKT but not ERK-induced activity. J Biol Chem. 2004;279:29436–49. doi: 10.1074/jbc.M313466200. [DOI] [PubMed] [Google Scholar]
- 18.Mahajan K, Mahajan NP. Shepherding AKT and androgen receptor by Ack1 tyrosine kinase. J Cell Physiol. 2010;224:327–33. doi: 10.1002/jcp.22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Laar JH, Bolt-de Vries J, Zegers ND, Trapman J, Brinkmann AO. Androgen receptor heterogeneity and phosphorylation in human LNCaP cells. Biochem Biophys Res Commun. 1990;166:193–200. doi: 10.1016/0006-291X(90)91930-Q. [DOI] [PubMed] [Google Scholar]
- 20.van Laar JH, Berrevoets CA, Trapman J, Zegers ND, Brinkmann AO. Hormone-dependent androgen receptor phosphorylation is accompanied by receptor transformation in human lymph node carcinoma of the prostate cells. J Biol Chem. 1991;266:3734–8. [PubMed] [Google Scholar]
- 21.Gioeli D, Ficarro SB, Kwiek JJ, Aaronson D, Hancock M, Catling AD, et al. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J Biol Chem. 2002;277:29304–14. doi: 10.1074/jbc.M204131200. [DOI] [PubMed] [Google Scholar]
- 22.Gioeli D, Paschal BM. Post-translational modification of the androgen receptor. Mol Cell Endocrinol. 2012;352:70–8. doi: 10.1016/j.mce.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Ward RD, Weigel NL. Steroid receptor phosphorylation: Assigning function to site-specific phosphorylation. Biofactors. 2009;35:528–36. doi: 10.1002/biof.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gioeli D, Black BE, Gordon V, Spencer A, Kesler CT, Eblen ST, et al. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol Endocrinol. 2006;20:503–15. doi: 10.1210/me.2005-0351. [DOI] [PubMed] [Google Scholar]
- 25.Kesler CT, Gioeli D, Conaway MR, Weber MJ, Paschal BM. Subcellular localization modulates activation function 1 domain phosphorylation in the androgen receptor. Mol Endocrinol. 2007;21:2071–84. doi: 10.1210/me.2007-0240. [DOI] [PubMed] [Google Scholar]
- 26.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–65. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 27.Rizzolio F, Lucchetti C, Caligiuri I, Marchesi I, Caputo M, Klein-Szanto AJ, et al. Retinoblastoma tumor-suppressor protein phosphorylation and inactivation depend on direct interaction with Pin1. Cell Death Differ. 2012;19:1152–61. doi: 10.1038/cdd.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Napoli M, Girardini JE, Piazza S, Del Sal G. Wiring the oncogenic circuitry: Pin1 unleashes mutant p53. Oncotarget. 2011;2:654–6. doi: 10.18632/oncotarget.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang DG. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol. 2004;164:1727–37. doi: 10.1016/S0002-9440(10)63731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev. 2000;14:2410–34. doi: 10.1101/gad.819500. [DOI] [PubMed] [Google Scholar]
- 31.Ayala G, Wang D, Wulf G, Frolov A, Li R, Sowadski J, et al. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003;63:6244–51. [PubMed] [Google Scholar]
- 32.Ryo A, Uemura H, Ishiguro H, Saitoh T, Yamaguchi A, Perrem K, et al. Stable suppression of tumorigenicity by Pin1-targeted RNA interference in prostate cancer. Clin Cancer Res. 2005;11:7523–31. doi: 10.1158/1078-0432.CCR-05-0457. [DOI] [PubMed] [Google Scholar]
- 33.Chen SY, Wulf G, Zhou XZ, Rubin MA, Lu KP, Balk SP. Activation of beta-catenin signaling in prostate cancer by peptidyl-prolyl isomerase Pin1-mediated abrogation of the androgen receptor-beta-catenin interaction. Mol Cell Biol. 2006;26:929–39. doi: 10.1128/MCB.26.3.929-939.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beato M. Gene regulation by steroid hormones. Cell. 1989;56:335–44. doi: 10.1016/0092-8674(89)90237-7. [DOI] [PubMed] [Google Scholar]
- 35.Centenera MM, Harris JM, Tilley WD, Butler LM. The contribution of different androgen receptor domains to receptor dimerization and signaling. Mol Endocrinol. 2008;22:2373–82. doi: 10.1210/me.2008-0017. [DOI] [PubMed] [Google Scholar]
- 36.Chen S, Xu Y, Yuan X, Bubley GJ, Balk SP. Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc Natl Acad Sci USA. 2006;103:15969–74. doi: 10.1073/pnas.0604193103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gordon V, Bhadel S, Wunderlich W, Zhang J, Ficarro SB, Mollah SA, et al. CDK9 regulates AR promoter selectivity and cell growth through serine 81 phosphorylation. Mol Endocrinol. 2010;24:2267–80. doi: 10.1210/me.2010-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sivanandam A, Murthy S, Chinnakannu K, Bai VU, Kim SH, Barrack ER, et al. Calmodulin protects androgen receptor from calpain-mediated breakdown in prostate cancer cells. J Cell Physiol. 2011;226:1889–96. doi: 10.1002/jcp.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baniwal SK, Little GH, Chimge NO, Frenkel B. Runx2 controls a feed-forward loop between androgen and prolactin-induced protein (PIP) in stimulating T47D cell proliferation. J Cell Physiol. 2012;227:2276–82. doi: 10.1002/jcp.22966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rajbhandari P, Finn G, Solodin NM, Singarapu KK, Sahu SC, Markley JL, et al. Regulation of estrogen receptor α N-terminus conformation and function by peptidyl prolyl isomerase Pin1. Mol Cell Biol. 2012;32:445–57. doi: 10.1128/MCB.06073-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujimoto Y, Shiraki T, Horiuchi Y, Waku T, Shigenaga A, Otaka A, et al. Proline cis/trans-isomerase Pin1 regulates peroxisome proliferator-activated receptor gamma activity through the direct binding to the activation function-1 domain. J Biol Chem. 2010;285:3126–32. doi: 10.1074/jbc.M109.055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brondani V, Schefer Q, Hamy F, Klimkait T. The peptidyl-prolyl isomerase Pin1 regulates phospho-Ser77 retinoic acid receptor alpha stability. Biochem Biophys Res Commun. 2005;328:6–13. doi: 10.1016/j.bbrc.2004.12.130. [DOI] [PubMed] [Google Scholar]
- 43.Gianni’ M, Boldetti A, Guarnaccia V, Rambaldi A, Parrella E, Raska I, Jr., et al. Inhibition of the peptidyl-prolyl-isomerase Pin1 enhances the responses of acute myeloid leukemia cells to retinoic acid via stabilization of RARalpha and PML-RARalpha. Cancer Res. 2009;69:1016–26. doi: 10.1158/0008-5472.CAN-08-2603. [DOI] [PubMed] [Google Scholar]
- 44.Zhou ZX, Kemppainen JA, Wilson EM. Identification of three proline-directed phosphorylation sites in the human androgen receptor. Mol Endocrinol. 1995;9:605–15. doi: 10.1210/me.9.5.605. [DOI] [PubMed] [Google Scholar]
- 45.Rizzolio F, Caligiuri I, Lucchetti C, Fratamico R, Tomei V, Gallo G, et al. Dissecting Pin1 and phospho-pRb regulation. J Cell Physiol. 2012 doi: 10.1002/jcp.24107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.