

Abstract
The glycogen synthase kinase-3 homolog, Mck1, has been implicated in many cellular functions, from sporulation to calcium stress response in budding yeast. Here, we report a novel function for Mck1 in the inhibition of Clb2-Cdk1 activity post nuclear division. Clb2-Cdk1, the major mitotic cyclin-Cdk complex in yeast, accumulates before anaphase and must be inhibited in telophase for cells to exit mitosis and enter into the next cell cycle. We show that the mck1Δ mutant is highly sensitive to increased Clb2-Cdk1 activity caused either by overexpression of Clb2 or the Cdk1-activating phosphatase Mih1. Deletion of the Cdk1 inhibitory kinase, SWE1, in combination with a mck1Δ mutant results in a synthetic growth defect, suggesting that Mck1 and Swe1 function in parallel pathways to inhibit Clb2-Cdk1. We find that mck1Δ strains have a delay in mitotic exit as well as elevated levels of Clb2-Cdk1 activity post-nuclear division. Using a co-immunoprecipitation assay, we identify a physical interaction between Mck1 and both Clb2 and Mih1. Finally, we demonstrate that phosphorylation of purified Clb2 by Cdk1 is inhibited by catalytically active Mck1 but not catalytically inactive Mck1 in vitro. We propose that Mck1 inhibits the activity of Clb2-Cdk1 via interaction with Clb2. The mammalian glycogen synthase kinase-3 homolog has been implicated in cyclin inhibition, suggesting a conserved cell cycle function for both yeast and mammalian glycogen synthase kinases.
Keywords: cyclin, cyclin-dependent kinase, glycogen synthase kinase, kinase, mitosis
Introduction
Cells possess tightly controlled mechanisms to replicate and segregate chromosomes accurately into daughter cells. The eukaryotic cell cycle is driven by the activity of cyclin-dependent kinases (Cdks). Cdks are serine/threonine protein kinases that consist of a catalytic subunit and an activating subunit called a cyclin.1 In the budding yeast Saccharomyces cerevisiae (S. cerevisiae), the major Cdk that controls cell cycle progression is Cdk1/Cdc28. Cdk1 binds to nine different cyclin subunits, including four mitotic cyclins, of which Clb2 (cyclin B) is the most important for mitotic progression.2,3 The mitotic cyclins (Clb1–4) promote spindle formation and the initiation of mitosis.2,4-8 At the end of mitosis, cells oscillate from a period of high Cdk1 activity to a period of low Cdk1 activity, which is required to exit the cell cycle.9-11 Cdk1 activity is inhibited by degradation of Clb2 via the anaphase-promoting complex (APC) and also by interaction with Sic1.9,12-17 Finally, Cdk1-dependent phosphorylation of substrates is reversed by the release and action of the Cdc14 phosphatase by the Cdc14 early anaphase release (FEAR) and mitotic exit network (MEN) pathways.18,19 High levels of Clb2, or expression of non-degradable forms of Clb2, arrest cells in late telophase, with large buds and divided nuclei.11,15
In fission yeast, Xenopus and human cells, mitotic entry is prevented by inhibitory phosphorylation of Cdk1 tyrosine 15 (Y15) by the Wee1 kinase.20-23 Dephosphorylation of Cdk1-Y15 and subsequent initiation of mitosis is dependent on the Cdc25 phosphatase.24 In S. cerevisiae, the equivalent tyrosine residue, Cdk1-Y19, is phosphorylated in a cell cycle-dependent manner (S/G2); however, unlike other eukaryotes, this phosphorylation is not required to prevent premature mitotic entry or to inhibit Cdk1 activity during DNA damage or replication stress.25,26 Instead, Cdk1-Y19 phosphorylation in S. cerevisiae, which is performed by the Wee1 homolog Swe1, is required for the morphogenesis checkpoint, which inhibits Cdk1 in response to actin depolymerization.27,28 Swe1 preferentially phosphorylates the Clb2-Cdk1 complex but may also target other cyclin-Cdk1 complexes.29 Swe1 also plays a role in the normal progression of mitosis, as a swe1Δ strain enters mitosis prematurely at a smaller cell size.30 In addition to imposing a Cdk1 inhibitory phosphorylation, Swe1 can also inhibit Clb2-Cdk1 by direct binding. This interaction has two consequences: it protects Swe1 from premature degradation and inhibits the activity of Clb2-Cdk1.31,32
The S. cerevisiae Cdc25 phosphatase homolog, Mih1, antagonizes Swe1 Cdk1-Y19 phosphorylation but is not essential for mitotic progression, unlike its counterpart in Schizosaccharomyces pombe.24,33,34 Both Mih1 and Swe1 are phosphorylated by multiple kinases, although the exact role that phosphorylation plays in their regulation is understood only for Swe1. Mih1 is hyperphosphorylated early in the cell cycle and dephosphorylated as cells enter mitosis.33 A number of kinases have been implicated in regulating Mih1 phosphorylation, including Clb2-Cdk1, the casein kinases Yck1/Yck2 as well as the glycogen synthase kinase-3 (GSK-3) kinase Mck1.33 The phosphorylation and subsequent degradation of Swe1 requires the activity and controlled localization of at least four kinases (Cla4, Hsl1, Cdk1, Cdc5) in addition to the localization of Swe1 at the bud neck.35 Mck1 regulates Swe1 during calcium stress response, by delocalization of the bud neck protein kinase Hsl1, which results in accumulation of Swe1 and inhibition of Clb2-Cdk1.36
The S. cerevisiae Mck1 kinase, which is a GSK-3 homolog, has roles in chromosome segregation, genome stability, sporulation, protein kinase A inhibition, transcriptional regulation, protein degradation, RNA pol III inhibition and calcium stress.37-41 The human GSK-3 protein kinase has also been implicated in a wide variety of cellular processes, including cell cycle events, and generally acts as an inhibitory kinase.42-44 GSK-3 directly phosphorylates and promotes cyclin D1 degradation and negatively influences cyclin D1 transcription by inhibiting transcription factors such as c-Jun.45-47 GSK-3 also targets cyclin E and Cdc25 for degradation.48,49
Here, we show, using both genetic and biochemical analyses, that Mck1 has a role in the inhibition of Clb2-Cdk1 activity. Strains carrying a deletion of the MCK1 kinase are unable to tolerate high levels of Clb2-Cdk1 activity caused by overexpression of MIH1, CLB2 or deletion of SWE1. In addition, mck1Δ mutants do not eliminate Clb2-Cdk1 activity as efficiently as wild type cells at the end of mitosis. We also show that Mck1 interacts with both Mih1 and Clb2 but does not promote degradation of these proteins. Finally, we find that Clb2-Cdk1 activity is inhibited in vitro by the Mck1 kinase. Our work demonstrates that the GSK-3/Mck1 kinase has a cell cycle regulatory role by inhibiting Clb2-Cdk1 activity at the end of mitosis.
Results
Increasing the dosage of Cdk1 positive regulators is detrimental to mck1∆ strains
A large-scale genome-wide kinase synthetic dosage lethality (SDL) study was performed, whereby 5380 open reading frames were overexpressed in 92 nonessential kinase deletion mutants, including a mck1∆ strain.50 The premise of SDL screening is that a gene expressed at high levels may not affect wild type cells, but may be detrimental in strains deleted for proteins that interact with, regulate or oppose the function of the overexpressed gene.51-53 The Mih1 phosphatase was identified in the mck1∆ SDL screen.50 Mih1 preferentially dephosphorylates Clb2-Cdk1 over other cyclin-bound Cdk1 complexes.29 To confirm the GAL-GST-MIH1 mck1∆ SDL phenotype, the pGAL-GST-MIH1 inducible plasmid was transformed along with a vector control into wild type and mck1∆ strains. Serial dilutions of the transformants were plated onto GAL-GST-MIH1 non-inducing (dextrose) and inducing (galactose) media. The SDL phenotype was confirmed, as overexpression of pGAL-GST-MIH1 was lethal to a mck1∆ strain but not a wild type strain (Fig. 1A). To determine if the SDL phenotype is specific to a particular cyclin-Cdk complex, we transformed cyclin∆ and mck1∆ double mutants with the pGAL-GST-MIH1 plasmid and performed serial dilution assays. The GAL-GST-MIH1 mck1∆ SDL phenotype was rescued only by the deletion of the mitotic cyclin, CLB2, and not by deletion of any other cyclin (Fig. 1A; Fig. S1). The clb5Δ mck1Δ was not tested, because this strain is synthetic lethal.54 The lethality caused by overexpression of pGAL-GST-MIH1 in mck1∆ cells thus may be due to hyper-active mitotic Clb2-Cdk1. To test this, a pGAL-CLB2-HA plasmid was transformed into a mck1∆ strain, and serial dilution assays were performed. Overexpression of CLB2 from the GAL promoter has been previously shown to increase the activity of Clb2-Cdk1.55 We found that overexpression of CLB2, and thus increasing Clb2-Cdk1 activity, in the absence of MCK1 caused cell lethality (Fig. 1B). The fact that overexpression of two positive regulators of Cdk1 activity (Mih1, Clb2) is detrimental to the MCK1 deletion mutant suggests that high Clb2-Cdk1 activity is not well-tolerated in the absence of MCK1.
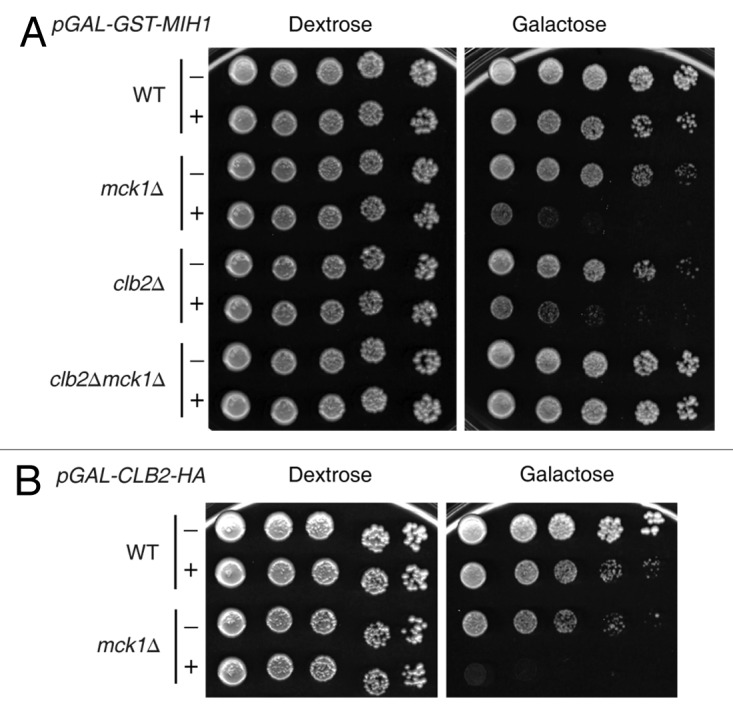
Figure 1. Overexpression of MIH1 is toxic to mck1∆ mutants in a CLB2-dependent fashion. (A) Overexpression of pGAL-GST-MIH1 (+) or vector alone (-) in wild type (WT), mck1∆, clb2∆ and clb2∆mck1∆ strains. (B) Overexpression of pGAL-CLB2-HA (+) or vector alone (-) in a WT or mck1∆ strain. Strains were grown to logarithmic phase, serially diluted onto the indicated media and grown for 4 d at 30°C.
To determine if Mih1 is indeed functional in the absence of MCK1, pGAL-GST-MIH1 was transformed into CLB2-HA and CLB2-HA mck1∆ strains. Clb2-HA was immunoprecipitated (IP’d) and probed for inhibitory Clb2-Cdk1 phosphorylation using a Cdk1-Y19 phosphorylation specific antibody [anti-phospho-Cdc2 (tyr15)]. Overexpression of pGAL-GST-MIH1 caused loss of Cdk1-Y19 phosphorylation with the same timing in the presence or absence of Mck1, indicating that Mih1 is active in mck1Δ strains, and that Mck1 is not required for Mih1 phosphatase activity (Fig. S2). Since Mih1 is functional in the absence of Mck1, we conclude that overexpression of MIH1 increases the abundance of dephosphorylated and, thus, active Clb2-Cdk1, which is detrimental to strains that lack the Mck1 kinase.
The MCK1 deletion mutant has a positive genetic interaction with mih1∆ and conversely has a negative genetic interaction with swe1∆
A complementary approach to analyzing SDL genetic interactions is the analysis of genetic interactions between loss of function double mutants.51 Genetic interactions were examined between MCK1 and MIH1 as well as SWE1 by growth curve analysis performed on both single (mck1∆, mih1∆, swe1∆) and double (swe1∆ mck1∆, mih1∆ mck1∆) mutants. Each strain was grown to log phase and diluted into a 96-well plate in replicates of eight. Optical density (OD600nm) measurements were averaged across the eight replicates (Fig. 2A). To quantify the growth curves, growth rates were determined for the exponential growth phase of each replicate (Table 1A). To determine if the double mutant growth rates are statistically different from the single mutants, p values were calculated (Table 1B). The mck1∆swe1∆ double mutant growth rate is significantly decreased compared with the single swe1∆ or single mck1∆ mutant strains (p < 0.01). Although the mck1∆mih1∆ double mutant growth rate is slower than the mih1∆ strain (p < 0.05), it is faster than the mck1∆ strain (p = 0.05).

Figure 2. The slow growth rate of mck1∆ strains is ameliorated by mih1∆ and exacerbated by swe1∆. (A) The indicated strains were grown in a 96-well plate, and OD600nm measurements were taken every 20.8 min. The average of eight replicate growth curves per strain is plotted. (B) Growth rates relative to WT are plotted with standard deviation. Neutral prediction indicates no genetic interaction. Error bars represent standard deviation. Error bars for neutral predication are too small to observe.
Table 1A. Growth rate of mck1∆, mih1∆, and swe1∆ strains and double mutant combinations.
| Genotype | Mean growth rate1 (107 cells mL−1 hr−1) | STDEV |
|---|---|---|
|
WT |
0.18 |
0.02 |
|
mck1∆ |
0.13 |
0.01 |
|
mih1∆ |
0.16 |
0.01 |
|
mck1∆mih1∆ |
0.15 |
0.02 |
|
swe1∆ |
0.18 |
0.00 |
| mck1∆swe1∆ | 0.11 | 0.01 |
1Average calculated growth rates were determined by the slope of the line of exponential growth with standard deviations.
Table 1B. Calculated p-values for growth rates of double vs. single mutant strains in Table 1A.
| p values1 | WT | mck1∆ | mih1∆ | swe1∆ |
|---|---|---|---|---|
|
WT |
N/A |
N/A |
N/A |
N/A |
|
mck1∆ |
8.68E-05 |
N/A |
N/A |
N/A |
|
mih1∆ |
4.77E-02 |
N/A |
N/A |
N/A |
|
mck1∆mih1∆ |
2.73E-03 |
5.13E-02 |
4.95E-02 |
N/A |
|
swe1∆ |
8.32E-01 |
N/A |
N/A |
N/A |
| mck1∆swe1∆ | 3.13E-06 | 4.68E-03 | N/A | 9.43E-07 |
1Growth rate of strains in rows is compared with those in columns.
In the absence of a genetic interaction, the phenotype of a double mutant is predicted to be the product of the phenotypes (growth rates in this case) of the two individual mutants.51 Using this premise, predictions were made for the growth rate of the mck1∆ double mutants, if no genetic interaction occurs, and compared with what was observed experimentally. Growth rates were calculated as a percentage of the wild type growth rate, and neutral prediction is the product of the growth rates of the two single mutants (Fig. 2B). Our analysis demonstrates that deletion of MIH1 improves the growth rate of mck1∆ strains, and deletion of SWE1 hinders the growth rate of mck1∆ strains when compared with the neutral prediction (Fig. 2B). These results suggest that in the absence of MCK1, Cdk1-Y19 phosphorylation is beneficial.
Mck1 contributes to Mih1 phosphorylation and regulation of Clb2
The fact that removing MIH1 is beneficial to a MCK1 mutant strain suggests that Mih1 may be mis-regulated in the absence of the Mck1 kinase. Similarly, since MCK1 deletion mutants are sensitive to increased levels of Clb2, the deletion mutant might suffer from elevated levels of Clb2-Cdk1. To investigate these possibilities, the protein levels of Mih1, Clb2 and Swe1 were analyzed throughout the cell cycle in a mck1∆ strain (Fig. 3). Cells were arrested in G1 phase, with mating pheromone released and time points taken every 15 min starting 30 min post-pheromone release. Mating factor was added back at 80 min post-release to prevent the subsequent cell cycle. Previous studies have shown that the Clb2 mitotic cyclin appears in G2, and its protein levels reach a maximum just before anaphase and disappear as cells enter anaphase.3,56 We also found that in wild type cells, Clb2 protein levels peaked in G2 at 60 min, whereas in mck1∆ cells, Clb2 protein levels peaked 15 min later at 75 min (Fig. 3A and B). The delay in maximal expression of Clb2 may be because DNA replication is delayed in mck1∆ cells, which we determined by monitoring DNA profiles and confirmed in a separate cell cycle analysis (Fig. S3). Despite the delay in DNA replication and delay in Clb2 expression, mck1Δ mutants enter nuclear division with less Clb2 than wild type cells. When Clb2 levels peaked at 60 min, ~12% of wild type cells had initiated or finished nuclear division (M-phase), whereas when Clb2 levels peaked at 75 min in mck1∆ cells, ~37% of cells had begun or undergone nuclear division (Fig. 3E, graph M). Clb2 levels dropped dramatically at 90 min in wild type strains, whereas at the equivalent time point in mck1∆ strains (105 mins), Clb2 protein was still present (Fig. 3A and B). However, we determined that Mck1 is not required for Clb2 protein turnover by using a Clb2 pulse chase approach (Fig. S4).

Figure 3. Mih1 phosphoforms are reduced and mitotic exit is delayed in mck1∆ strains. Wild type and mck1∆ strains were synchronized in G1 with mating factor. Time points were taken at 30, 45, 60, 75, 90 and 105 min after release and processed for western and microscopy analysis. Cell cycle westerns were probed with (A) anti-Clb2 and anti-PSTAIR (Cdk1, loading control), (C) anti-Mih1. The asterisk (*) indicates a background band. (B) Intensities of Clb2 relative to Cdk1 in (A) is plotted. (D) Intensity of Mih1 top, middle and bottom phosphoforms from (C) relative to Cdk1 in (A) is plotted. (E) Microscopy cell cycle analysis of DAPI stained WT and mck1∆ cells (from experiment in A and C) as a percentage of total cells counted. The experiment was performed three times and shown are representative blots.
Mih1 exists in several phosphoforms that are distinguished by immunoblot analysis as slower migrating bands with anti-Mih1 polyclonal antibodies.33 Mih1 is highly phosphorylated throughout G1 and S phase; phosphorylation levels are reduced as cells enter mitosis; and upon exit of mitosis, Mih1 appears again in its highly phosphorylated form (Fig. 3C).33 In the absence of MCK1, there was a decrease in the slowest migrating Mih1 phosphoform between 45 to 60 min (Fig. 3C and D, top band). An accumulation of the dephosphorylated form (bottom band) of Mih1 was also observed at the end of mitosis in mck1∆ cells (Fig. 3C and D, bottom band, see 90 and 105 min). The accumulation of dephosphorylated Mih1 is transient, as mck1∆ cells enter the cell cycle with high levels of phosphorylated Mih1 similar to wild type cells (Fig. 3C and D). Our data is consistent with a previous report demonstrating an accumulation of dephosphorylated Mih1 in logarithmic phase mck1∆ cells.33 Interestingly, Mih1 dephosphorylation also depends on Clb2-Cdk1.33 Therefore, the observed increase in dephosphorylated Mih1 in mck1∆ cells may be due to higher Clb2-Cdk1 activity or may be because Mih1 is a substrate of the Mck1 kinase. As Swe1 protein levels accumulate in S/G2, Swe1 is phosphorylated on multiple residues by a cascade of kinases starting with Clb2-Cdk1.30,57-59 Early in the mck1∆ mutant cell cycle, Swe1 had increased phosphoforms (S/G2, 30–40 min) compared with wild type cells, but by 60 min Swe1 phosphoforms were equivalent in wild type and mck1∆ cells (Fig. S5). Taken together, our data suggests that Mih1 but not Swe1 phosphoforms are altered, and Clb2 proteolysis is not affected in mck1Δ strains.
Clb2-Cdk1 activity is elevated in mck1∆ cells
To determine if mck1∆ strains have increased levels of Clb2-Cdk1 activity, we performed a cell cycle kinase assay (Fig. 4). Clb2-HA was IPed during a synchronized cell cycle and assayed for activity by incubating with γ-32P-ATP and the Cdk1 substrate histone H1.60 The intensities of the histone H1 phosphorylation bands are displayed relative to Cdk1 levels from whole-cell extracts (Fig. 4B and C). In wild type cells (CLB2-HA), Clb2-Cdk1 activity peaked at 65 min, after the completion of DNA replication (confirmed by FACS analysis, data not shown), and declined at 85 min, when nuclear division occurred (confirmed by microscopy with DAPI stained cells, data not shown). Cells entered the subsequent cell cycle at 105 min, when Clb2-Cdk1 is inactive (Fig. 4A, 105 min). Once wild type cells had nearly completed the second round of replication at 125 min, Clb2-Cdk1 activity increased again (Fig. 4A, 125 min). In the absence of MCK1 (CLB2-HA mck1∆), Clb2-Cdk1 activity is similar to wild type cells up until 65 min but remains active after nuclear division (Fig. 4A, C, 85 and 105 min).

Figure 4. Clb2-Cdk1 remains active post nuclear division in the absence of MCK1. CLB2-HA and CLB2-HA mck1∆ strains were arrested in G1 with mating pheromone. Time points were taken at 25, 45, 65, 85, 105 and 125 min following release and processed for kinase assays (A) and western blots (C). (A) Clb2-HA was IP’d from wild type (CLB2-HA) and mck1∆ (CLB2-HA mck1∆) strains. (C) Western blot analysis of Clb2-HA IP from (A) and WCE probed with the following antibodies as indicated: anti-HA, anti-phospho-cdc2 (Tyr15) and anti-PSTAIR (Cdk1). (B and D) Relative intensities of indicated bands relative to the WCE Cdk1 protein band in (C). The experiment was performed three times, and representative kinase assays and blots are shown.
To determine the level of Cdk1-Y19 phosphorylation, Clb2-HA IPs were also examined by western blot analysis with anti-phospho-cdc2 (tyr15). Surprisingly, there was an increased level of Cdk1-Y19 phosphorylation in mck1∆ vs. wild type cells (Fig. 4B and C). The fact that increased Cdk1-Y19 phosphorylation does not reduce Clb2-Cdk1 activity in the absence of Mck1 suggests that Mck1 is required, along with Cdk1-Y19 phosphorylation, to inhibit Clb2-Cdk1.
Mck1 interacts with Mih1 and Clb2
The inability of mck1∆ mutants to completely inactivate Clb2-Cdk1 and the change in Mih1 phosphoforms in mck1∆ mutants suggests that Mck1 may interact with Clb2-Cdk1 or Mih1. To test this possibility, co-IPs were performed from wild type and Mck1-Myc tagged strains transformed with either a pGAL-CLB2-HA or a pGAL-GST-MIH1 plasmid and grown in galactose media to allow CLB2-HA or GST-MIH1 expression. Mck1 was IPed with anti-Myc beads, and co-purifying Clb2 was detected with an anti-HA antibody, while co-purifying Mih1 was detected by an anti-GST antibody. Clb2-HA was present in the Mck1-Myc IPs but not in the IP from an untagged strain (Fig. 5A, lanes 3 and 4). The interaction between Clb2-HA and Mck1-Myc did not depend on Mih1, since Clb2-HA co-IPed with Mck1-Myc from a mih1∆ strain (Fig. 5B, lane 4). Mih1 also interacted with Mck1-Myc in the presence or absence of Clb2 (Fig. 5C, lanes 4, 6).

Figure 5. Mck1 interacts with Clb2 and Mih1. Myc tagged Mck1 in either wild type (A), wild type and mih1∆ (B) or wild type and clb2∆ (C) strains were grown to mid-log phase in SC-URA 2% raffinose media, washed and resuspended in SC-URA 2% galactose to express either (A) and (B) pGAL-CLB2-HA or (C) pGAL-GST-MIH1 for 1 h. Mck1-myc was IPed and protein interactions with Clb2-HA or GST-Mih1 were detected by probing with anti-HA or anti-GST antibodies, respectively.
Mck1 inhibits Clb2-Cdk1 activity in vitro
Thus far, our genetic and biochemical data suggests that Clb2-Cdk1 activity is elevated in mck1∆ strains, and that Mih1 and Clb2 can both interact with Mck1 from yeast lysates. We tested if Mck1 was able to phosphorylate GST-Mih1 or HA-Clb2 that we purified from yeast lysates but were unable to detect phosphorylation of either substrate (data not shown). Next, we investigated if Mck1 could inhibit Clb2-Cdk1 activity. Clb2-HA was purified from an analog-sensitive Cdk1 strain (cdc28-as1) in the presence (+) or absence (-) of the inhibitor 1NM-PP1 and incubated with γ-32P-ATP. Phosphorylation of Clb2-HA is detected in the absence, but not presence of the inhibitor (Fig. 6, compare lanes 7 and 8). Both inhibited and uninhibited forms of Clb2-HA bound to affinity matrix anti-HA beads that were added to three different kinase reactions. The first kinase reaction consisted of untagged cells incubated with conjugated Myc beads to control for background phosphorylation (empty); the second contained IP’d Mck1-Myc, and the third an IP’d kinase dead allele of MCK1 (mck1D164A-myc). The “no substrate” reactions (lanes 9–11) are Myc bead purifications from the indicated strains that were incubated with γ-32P-ATP but no substrate. Mck1 autophosphorylation (Mck1-P) is detected in the Mck1-Myc purification but not in the kinase-dead mck1D164A-myc purification, as expected61 (Fig. 6, lanes 10–11). When purified Mck1-Myc was incubated with inhibited Clb2-HA (+1NM-PP1) (Fig. 6, lane 2), auto-phosphorylation of Mck1 occured, but no phosphorylation of Clb2-HA was detected, consistent with our previous observation that Mck1 cannot phosphorylate Clb2 when purified in this manner. When uninhibited (-1NM-PP1) Clb2-HA was added to the kinase reactions, Clb2 phosphorylation was observed when incubated with the kinase reaction from the untagged strain (Fig. 6, lane 4), but no Clb2 phosphorylation was observed when incubated with the Mck1-Myc kinase reaction (Fig. 6, lanes 5). In addition, Cdk1 does not appear to be phosphorylated by Mck1. The lack of Clb2 phosphorylation depends on active Mck1, as Clb2 phosphorylation is detected when incubated with the kinase-dead version of Mck1 (Fig. 6, lane 6). Our data suggest that Mck1 kinase activity may prevent Clb2 phosphorylation via inhibition of Clb2-Cdk1 activity.

Figure 6. The Mck1 kinase inhibits Clb2 phosphorylation. Clb2-HA was IP’d from the cdc28-as1 strain. The IP was split in half and incubated with either 1NM-PP1 or solvent control (DMSO). Mck1-myc and Mck1D164A-myc were IP’d and equivalent amounts of either inhibited Clb2-HA cdc28as1 (+1NM-PP1, lanes 1–3) or uninhibited Clb2-HA cdc28as1 (-1NM-PP1, lanes 4–6) were added to Mck1 IPs. Inhibited and uninhibited purified Clb2-HA cdc28as1 is shown in lanes 7 and 8 without addition of the Mck1 kinase. Mck1 kinase purifications with no added substrate are shown in lanes 9–11. Mck1 autophosphorylation (Mck1-P), Clb2 phosphorylation (Clb2-P) and Cdk1 phosphorylation (Cdk1-P) bands are indicated.
Overexpression of MCK1 causes bud elongation, consistent with Clb2-Cdk1 inhibition
Activation of Cdk1 by G1 cyclins triggers polarized growth whereas activation of Cdk1 by G2 cyclins triggers depolarized growth.62 If Cdk1 activity is inhibited in budded cells, hyperpolarized growth is triggered, and the bud elongates.62 To determine if increased levels of Mck1 alter cell morphology, wild type cells were transformed with an overexpression plasmid (pGAL-GST-MCK1) and a vector control and grown in galactose for a total of 16 h. Logarithmic growth was maintained throughout the time course. After 8 hours of MCK1 overexpression, cells exhibited elongated buds (Table 2; Fig. S6A) and a greater proportion of 2N cells than the vector control (Fig. S6B). The elongated bud morphology suggests that an increased dosage of Mck1 can inhibit Clb2-Cdk1 activity in vivo. Removal of SWE1 partially rescues the bud elongation detected upon overexpression of MCK1 (Table 2; Fig. S6C). Therefore, Mck1 may be able to inhibit Clb2-Cdk1 activity by both Swe1-dependent and Swe1-independent mechanisms.
Table 2. Increased levels of Mck1 induces bud elongation.
| Round Bud | Elongated Bud1 | |
|---|---|---|
|
WT vector |
100.00 |
0.00 |
|
WT pGAL-GST-MCK1 |
72.40 |
27.60 |
|
swe1∆ vector |
100.00 |
0.00 |
| swe1∆ pGAL-GST-MCK1 | 84.85 | 15.15 |
1Bud elongation defined as having a longer axis of growth vs. width was counted for 200 cells. Percentages are displayed.
Discussion
In this study, we have uncovered a cell cycle role for the Mck1 GSK-3 kinase in budding yeast. The following evidence strongly suggests that Mck1 is an inhibitor of Clb2-Cdk1 activity post-nuclear division. (1) Cells devoid of MCK1 do not tolerate an increase in Clb2-Cdk1 activity, either by removal of SWE1 or by overexpression of either MIH1 or CLB2 (Figs. 1 and 2). (2) mck1∆ cells have a mitotic exit delay (Fig. 3; Fig. S3). (3) Clb2-Cdk1 activity is not eliminated in mck1Δ cells post-nuclear division (Fig. 4). (4) Mck1, Clb2 and Mih1 physically interact (Fig. 5). (5) Mck1 kinase activity inhibits Clb2 phosphorylation in vitro (Fig. 6). (6) Overexpression of GAL-GST-MCK1 results in bud elongation, which is a hallmark sign of Clb2-Cdk1 inhibition (Table 2; Fig. S6).
Potential mechanisms by which Mck1 inhibits Clb2-Cdk1 activity
We have demonstrated that overexpression of either CLB2 or MIH1 results in slow growth to mck1Δ strains, and that both Clb2 and Mih1 can interact with Mck1 independently of each other, suggesting that either of these proteins may be targets for inhibition by Mck1 (Figs. 1 and 5). As well, we detected changes in Mih1 phosphoforms in mck1∆ strains (Fig. 3C). However, we were unable to detect phosphorylation of Mih1 in our Mck1 in vitro kinase assay (data not shown). GSK-3 kinases, including Mck1, often require a preceding or “priming” phosphorylation of their target proteins.39,41,43,63 In an effort to preserve any priming signals, we purified Mih1 from yeast extracts; however, the required priming phosphorylation may have been removed during purification. The phosphorylation of Mih1 in vivo is partially dependent on the casein kinase I homologs Yck1/2, which are known to prime the Ubr1 ubiquitin ligase for phosphorylation by Mck1.33,39 In addition, casein kinase I primes β-catenin for phosphorylation by GSK-3 in mammalian cells.43 Future experiments will determine if Yck1/2 indeed primes Mih1 for phosphorylation by Mck1.
We also tested if Clb2-Cdk1 purified from an analog-sensitive Cdk1 (cdc28-as1) strain in the presence or absence of inhibitor was phosphorylated by Mck1 in vitro. We did not detect phosphorylation of Clb2 purified in the absence of Cdk1 activity or increased phosphorylation of Clb2 purified in the presence of Cdk1 activity when incubated with purified Mck1 (Fig. 6). On the contrary, we detected a dramatic reduction of phosphorylated Clb2 when incubated with Mck1 but not a catalytically inactive form of Mck1 (Fig. 6). This data suggests that the kinase active form of Mck1 is capable of inhibiting Clb2 phosphorylation by Cdk1 in vitro. Mck1 was previously shown to bind and inhibit, but not phosphorylate, the Tpk1 catalytic subunit of protein kinase A.64 Similar to our findings with Clb2-Cdk1, the ability of Mck1 to inhibit Tpk1 requires Mck1 catalytic activity, suggesting that Mck1 may inhibit Tpk1 and Clb2-Cdk1 in a similar manner—by direct binding, but not transfer, of a phosphate.64 Mck1 is autophosphorylated on both serine and tyrosine residues61,65 Therefore, it is plausible that Mck1 kinase activity is required for autophosphorylation, and that phosphorylated Mck1 inhibits Clb2-Cdk1 (Fig. 7).

Figure 7. Model of Clb2-Cdk1 regulation. Inhibitory phosphorylation of Cdk1 on tyrosine 19 is controlled by the Mih1 phosphatase and the Swe1 kinase prior to mitotic entry. Clb2 is targeted for degradation during mitotic exit by the APCCdc20/ Cdh1, while the kinase activity of Cdk1 is inhibited by Sic1 and counteracted by the Cdc14 phosphatase (FEAR/MEN pathways). Catalytically active Mck1 inhibits Cdk1 activity most likely via interaction with Clb2.
Could Mck1 function in a known Clb-Cdk1 inhibitory pathway?
The destruction of the Clb2-Cdk1 mitotic kinase is required for mitotic exit.11 There are three documented mechanisms for Clb2-Cdk1 inhibition upon mitotic exit (Fig. 7): (1) the anaphase-promoting complex (APCCDC20/CDH1) degrades the mitotic cyclin, Clb266; (2) Sic1 is a potent inhibitor of Clb-Cdk1 kinase complexes14,16; (3) release of the Cdc14 phosphatase, which antagonizes the kinase activity of Clb2-Cdk1 and is controlled by the FEAR pathway in early anaphase, and the MEN pathway in late anaphase.67 It is unlikely that Mck1 inhibits Clb2-Cdk1 activity via one of these pathways. Since mck1∆ mutants do not arrest in G2, nor do they have difficulties degrading Clb2, Mck1 does not likely promote APC activity (Fig. S4). A strain deleted for SIC1 prematurely replicates its DNA, whereas we show that mck1∆ mutants are slightly delayed in DNA replication, suggesting that Mck1 does not share a pathway with Sic1 (Fig. S3).68 As well, mck1 sic1 double mutants have synthetic growth defects, suggesting that they function in parallel pathways to inhibit Clb-Cdk1 activity.69-71 Mck1 could possibly act via the FEAR or MEN pathway, and large-scale protein interaction studies have found that Mck1 binds to the FEAR components Cdc14 and Net1.72 However, since Mck1inhibits Clb2-Cdk1 activity in vitro, where FEAR and MEN pathway components are likely not present, it is unlikely that Mck1 acts through any of the mechanisms previously described to inhibit Clb2-Cdk1.
The inhibition of Clb2-Cdk1 phosphorylation by purified Mck1 in our in vitro kinase assay (Fig. 6) suggests that Mck1 may directly inhibit Clb2-Cdk1 activity either by binding Clb2, thus protecting it from Cdk1 phosphorylation or by altering Cdk1 kinase activity (Fig. 7). We did not detect phosphate transfer to either Clb2 or Cdk1 by Mck1, suggesting that Mck1 inhibition is most likely through binding. We observe a direct interaction between Mck1 and Clb2 but not Cdk1 (data not shown); therefore, it is most plausible that Mck1 inhibits Clb2-Cdk1 activity through binding of Clb2. It is unknown at this time what role Clb2 phosphorylation plays in Clb2 regulation.
Mammalian GSK-3 regulates cell cycle progression by targeting cyclin D1, cyclin E and Cdc25A for proteolysis.45,48,49 Although we have no evidence that Mck1 targets Clb2 or Mih1 for degradation, it is clear that both yeast and mammalian GSK-3 homologs interact with cyclins and the Cdk1 activating phosphatase to regulate cell cycle transitions. Further elucidation of the mechanism by which Mck1 inhibits Clb2-Cdk1 will contribute to our understanding of mammalian GSK-3 function.
Materials and Methods
Strains, plasmids and media
Yeast strains and plasmids used in this study are described in Table S1 and Table S2, respectively. The S288C- BY4742 strain background was used for experiments that required galactose induction. The kinase reactions were done with the W303 strain background. Genes were deleted or epitope tagged by using standard PCR-based yeast homologous recombination methods.73 To construct the mck1D164Amyc strain the pmck1D164A plasmid was transformed into a mck1∆ strain and was epitope tagged with MYC:TRP using standard methods. The tagged plasmid was checked by both PCR and epitope tag expression. The plasmid was subsequently digested to excise the mck1D164Amyc:TRP insert and transformed into a mck1∆KanMX6 strain. Media for yeast growth was either yeast peptone with 2% dextrose (YPD) or supplemental minimal media (SC) lacking the amino acid uracil (SC-URA) with either dextrose, raffinose or galactose as carbon sources.74
Spot assays
Cultures were grown in minimal SC-URA media (2% raffinose, 0.1% dextrose) overnight. Cell concentration was measured as optical density (OD600nm) by spectrometer and cultures diluted to allow for doubling at least twice in SC-URA (2% raffinose). Serial dilutions were performed by first diluting the cultures to an OD600nm of 106 cells mL−1 (OD600nm 0.1), then subsequently diluting 5-fold. Cultures (4 μL) were spotted on both SC-URA (2% dextrose) and SC-URA (2% galactose) plates.
Growth curves
All strains were grown overnight in YPD. Cultures were diluted to an OD600nm of 0.05 to allow for two doublings. Cultures were then diluted to an OD600nm of 0.1 and 200 μL was added to each well of a BD Falcon 96-well flat bottom plate (BD Biosciences) with eight technical replicates. OD600nm measurements were taken on an Infinite® Pro microplate reader (Magellan v7.0™ Software; Tecan Group Ltd.) every 20.8 min for 16 h. Every 14 min, the plate underwent orbital rotation for 5 min, rested for 30 sec, and then the OD600nm was measured. Each time point was averaged among replicates, and the mean values were plotted vs. time (Fig. 2A). Separately, the mean growth rate was calculated for each replicate. The growth rate was taken as the slope of the line at exponential growth. Exponential growth was defined as the time between 50% OD600nm max and 75% OD600nm max. OD600nm max was the average of the last three time points. The growth rate was averaged for the eight replicates, and the mean value and standard deviation calculated. Two-sample t-tests were performed on growth rates, and p-values recorded. Growth rate was calculated as a percentage of WT for each of the strains. Neutral predictions for the double mutants were calculated as the product of the two single mutants; their standard deviation was the product of the single mutants’ standard deviation (Fig. 2B). All calculations were done with Numbers ’09 [Apple Inc. Version 2.1 (436)].
Cell synchronization
Cells were grown to early logarithmic phase in YPD at 30°C, arrested with 5 μg/ mL α-mating factor (BioVectra) for 1 h followed by a second dose for an additional hour. Cells were washed twice and resuspended in media lacking α-mating factor. In order to prevent subsequent cell cycles, α-mating factor was added back to cultures at 80 min, except in Figure S5 and Figure 4.
Flow cytometry
Cells were fixed in 70% ethanol (EtOH), 30% 1 M sorbitol overnight at 4°C. Fixed cells were pelleted and resuspended in 1 mg/mL RNase A, 0.2 M TRIS-HCl pH 7.5 and incubated overnight at 37°C. Proteinase K (0.5 mg/mL) was added and incubated for at least 1 h at 50°C. Cells were pelleted and re-suspended in 400 μL of 0.2 M Tris pH7.5. Cell clumps were disrupted by sonication and stained with 600 μL of 1 μg/μL propidium iodide, 0.2 M Tris pH 7.5 for at least 1 hour. Flow cytometry was performed by a BD FACScan (BD Biosciences) with BD Cell Quest Pro software (BD Biosciences) and analyzed using FlowJo software (Version 9.4.10). Data was gated for 1N, 2N and > 2N populations.
Microscopy
Cells were fixed in 3.7% formaldehyde for 10 min. To avoid microtubule destruction, cells were centrifuged at 2,500 rpm, washed and resuspended in 1X PBS. Membranes were permeabilized by addition of 70% EtOH and incubated at room temperature for 30 min. Following resuspension in PBS, cells were sonicated, pelleted and re-suspended in a 50 ng/mL DAPI solution. Microscopy analysis of samples was performed on a Zeiss Colibri LED illuminator with a Zeis Axiocam Ultra High Resolution Monochrome Digital Camera Rev3.0. Images were analyzed using ImageJ software (version 1.45b, http://imagej.nih.gov.ij). Graphs were produced using Numbers ’09 Apple Inc. [Version 2.1 (436)].
Western blot
In Figure 3, 2 mL samples were taken for each time point. Cells were pelleted and supernatant removed. Cold sample buffer (100 μL) (0.065 M TRIS-HCl pH 6.8, 3% SDS, 10% glycerol, 5% β-mercaptoethanol, 1X Phosphatase Cocktail 2 SIGMA-Aldrich, 2 mM PMSF, 0.01% bromophenol blue) was added to each pellet with 100 μL glass beads prior to freezing in liquid nitrogen. Cell pellets were lysed by vortexing (4 × 30 sec at 5 m/s) in the FastPrep Homogenizer (MP Biomedicals). Samples were centrifuged (20 sec at 10,000 rpm) to reduce suds, boiled for 5 min, centrifuged (5 min at 10,000 rpm), and 10 μL was loaded onto a poly-acrylamide gel (12% Acrylamide/Bis, 78:1). Proteins were transferred onto a nitrocellulose membrane by wet transfer. Membranes were blocked for 1 h in 1X PBS 5% dry milk at room temperature.
Endogenous untagged Clb2 was detected using a rabbit polyclonal antibody (Y-180 Santa Cruz, sc-9071) (1:2000); Cdk1 was detected using monoclonal mouse anti-PSTAIR (Sigma Aldrich P 7962) (1:7500). Myc-tagged proteins were detected with anti-myc antibody (Roche) (1:5000). All primary antibodies were incubated with blocking buffer overnight at 4°C. To detect Mih1, blots were blocked in Mih1 block (1X PBS, 0.5 M NaCl, 0.1% Tween 20, 3% dry milk) for 1 hour. Polyclonal rabbit anti-Mih1 antibody (gift from Doug Kellogg) was diluted to a working concentration of 4 μg/mL in Mih1 block and incubated at 4°C for at least 20 h. Detection of phosphorylated tyrosine 19 Cdk1 was performed by first blocking the membrane in 1X TBS, 0.1% Tween 20, 5% dry milk for 1 hour at room temperature, then incubating with the phospho-specific antibody P-cdc2 Y15 (Cell signaling 9111S) (1:1000) dilution in 1X TBS, 0.1% Tween 20, 5% BSA overnight at 4°C. Epitope-tagged Clb2-HA was detected using mouse monoclonal anti-HA antibody (Roche) (1:2500 in either 1X PBS 5% dry milk or 1X TBS 5% BSA) incubated overnight at 4°C. Epitope-tagged Mih1-GST was detected using anti-GST antibodies (GE Healthcare RPN1236) at a dilution of 1:5000 in either 1X PBS 5% dry milk or 1X TBS 5% BSA, incubated overnight at 4°C.
Horseradish peroxidase (HRP)-conjugated secondary antibodies were used to visualize and amplify the primary signal. Secondary antibodies were goat-anti-mouse (Roche) (1:5000) or goat anti-rabbit (Roche) (1:5000) and were incubated in blocking buffer with the membranes for 1 h at room temperature. All proteins were imaged by chemiluminesence on film. Band intensity analysis was done by measuring pixel intensity in ImageJ (version 1.45b, http://imagej.nih.gov.ij). Relative intensities were calculated using Numbers ’09 Apple Inc. [Version 2.1 (436)].
Clb2 cell cycle kinase assay
For the Clb2-HA/Cdk1 kinase assay, following synchronization, 100 mL samples were pelleted and frozen on dry ice. Samples were lysed in 600 μL of kinase lysis buffer (0.25 M NaCl, 0.05 M Tris pH7.5, 2.5 mM NaF, 12 μM 6DMAP, 0.1% NP-40, 5 μM EDTA, 1 mM PMSF, 1μg/mL Leupeptin, 10 μg/ mL TLCK, 0.98 μg/ mL Pepstatin, 1 mM DTT, 10 μg/mL TPCK, 10 μg/mL SBTI, 1 μg/mL Aprotinin) and 100 μL glass beads with vortexing for 5 × 1 min in a FastPrep homogenizer (MP Biomedicals) beads. Protein lysates (18 mg) were incubated with affinity matrix HA beads (Covance, 40 μL of a 50% solution) for 1 hour at 4°C before proceeding with the washes. Half the beads were prepared for kinase reactions, while the other half were prepared for western blot analysis. WCE (160 μg) was also used for western blot analysis. Beads with bound protein were washed four times in 1 mL wash buffer (0.25 M NaCl, 0.05 M Tris pH 7.5, 0.1% NP-40, 5 μM EDTA and 1 mM DTT) and twice in 1 mL kinase buffer (10 mM MgCl2, 5 mM MnCl2, 0.05 M Tris pH7.5, 0.5 μM ATP, 1 mM DTT). Kinase reactions were incubated with the addition of 4 μL γ-32P-ATP in kinase buffer. Histone H1 (Roche), 1 μg, was added as a substrate. Clb2-HA kinase reactions were incubated for 30 min at 30°C until addition of 2X SDS loading buffer, run on polyacrylamide gels, dried on a gel dryer and imaged using film.
Clb2-Cdk1 and Mck1 kinase assay
Cultures were lysed in kinase lysis buffer as above. Protein lysate (8 mg) from CLB2-HA cdc28as1 strain was incubated with 20 μL of 50% slurry of anti-HA affinity matrix (Covance). Mck1-myc and mck1D164A-myc were IP’d from 20 mg protein lysate per 20 μL of 50% affinity-matrix anti-myc beads. Beads were washed in wash buffer and kinase buffer as described above. The kinase reaction was run with the addition of 1 μL of γ-32P-ATP. The inhibitor 1 NM-PP1 (Toronto Research Chemicals) was prepared according to manufacturer’s instructions in DMSO. Inhibitor or control (DMSO) was incubated with Clb2-HA preparations for 10 min at 30°C. Clb2-HA kinases were incubated for 45 min at 30°C until addition of 2X SDS loading buffer.
Galactose induced overexpression
All strains were grown in SC-URA (2% raffinose, 0.1% dextrose) overnight. OD600nm was measured and strains diluted in SC-URA (2% raffinose) to obtain logarithmically growing cultures of sufficient quantity for experiments. To express genes under the control of the GAL1 promoter, cultures were pelleted and washed once and re-suspended in SC-URA (2% galactose) for the indicated amount of time.
Supplementary Material
Acknowledgments
We would like to thank Brenda Andrews’ lab for performing SDL screens, Nina Piggott for technical assistance, Gayatri Pal and Doug Kellogg for the generous gift of Mih1 antibodies and discussions on the project, Amy Ikui for the W303 mck1Δ strain and discussions on the project and Jeremy Thorner for the mck1D164A plasmid. J.M. was supported by a Canadian Institutes of Health Research (CIHR) Frederick Banting and Charles Best Canada Graduate Scholarship. This work was supported by CIHR operating grant MOP-84242 to V.M. V.M. is a Canada Research Chair in Enology and Yeast Genomics.
Glossary
Abbreviations:
- APC
anaphase-promoting complex
- Clb2
cyclin B
- Cdk1
cyclin-dependent kinase -1
- GSK-3
glycogen synthase kinase-3
- IP
immunoprecipitation
- Mck1
meiotic and centromere regulatory ser, tyr-kinase
- SDL
synthetic dosage lethal
- WCE
whole-cell extract
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental material may be found here: www.landesbioscience.com/journals/cc/article/21731
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21731
References
- 1.Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol. 2007;8:149–60. doi: 10.1038/nrm2105. [DOI] [PubMed] [Google Scholar]
- 2.Richardson H, Lew DJ, Henze M, Sugimoto K, Reed SI. Cyclin-B homologs in Saccharomyces cerevisiae function in S phase and in G2. Genes Dev. 1992;6:2021–34. doi: 10.1101/gad.6.11.2021. [DOI] [PubMed] [Google Scholar]
- 3.Surana U, Robitsch H, Price C, Schuster T, Fitch I, Futcher AB, et al. The role of CDC28 and cyclins during mitosis in the budding yeast S. cerevisiae. Cell. 1991;65:145–61. doi: 10.1016/0092-8674(91)90416-V. [DOI] [PubMed] [Google Scholar]
- 4.Amon A, Tyers M, Futcher B, Nasmyth K. Mechanisms that help the yeast cell cycle clock tick: G2 cyclins transcriptionally activate G2 cyclins and repress G1 cyclins. Cell. 1993;74:993–1007. doi: 10.1016/0092-8674(93)90722-3. [DOI] [PubMed] [Google Scholar]
- 5.Fitch I, Dahmann C, Surana U, Amon A, Nasmyth K, Goetsch L, et al. Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol Biol Cell. 1992;3:805–18. doi: 10.1091/mbc.3.7.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuczera T, Bayram O, Sari F, Braus GH, Irniger S. Dissection of mitotic functions of the yeast cyclin Clb2. Cell Cycle. 2010;9:2611–9. doi: 10.4161/cc.9.13.12082. [DOI] [PubMed] [Google Scholar]
- 7.Schwob E, Nasmyth K. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev. 1993;7(7A):1160–75. doi: 10.1101/gad.7.7a.1160. [DOI] [PubMed] [Google Scholar]
- 8.Signon L. New insights into the regulation of anaphase by mitotic cyclins in budding yeast. Cell Cycle. 2011;10:1655–68. doi: 10.4161/cc.10.10.15632. [DOI] [PubMed] [Google Scholar]
- 9.Irniger S, Piatti S, Michaelis C, Nasmyth K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 1995;81:269–78. doi: 10.1016/0092-8674(95)90337-2. [DOI] [PubMed] [Google Scholar]
- 10.Nasmyth K. At the heart of the budding yeast cell cycle. Trends Genet. 1996;12:405–12. doi: 10.1016/0168-9525(96)10041-X. [DOI] [PubMed] [Google Scholar]
- 11.Surana U, Amon A, Dowzer C, McGrew J, Byers B, Nasmyth K. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO J. 1993;12:1969–78. doi: 10.1002/j.1460-2075.1993.tb05846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amon A, Irniger S, Nasmyth K. Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell. 1994;77:1037–50. doi: 10.1016/0092-8674(94)90443-X. [DOI] [PubMed] [Google Scholar]
- 13.Irniger S, Nasmyth K. The anaphase-promoting complex is required in G1 arrested yeast cells to inhibit B-type cyclin accumulation and to prevent uncontrolled entry into S-phase. J Cell Sci. 1997;110:1523–31. doi: 10.1242/jcs.110.13.1523. [DOI] [PubMed] [Google Scholar]
- 14.Mendenhall MD. An inhibitor of p34CDC28 protein kinase activity from Saccharomyces cerevisiae. Science. 1993;259:216–9. doi: 10.1126/science.8421781. [DOI] [PubMed] [Google Scholar]
- 15.Schwab M, Lutum AS, Seufert W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 1997;90:683–93. doi: 10.1016/S0092-8674(00)80529-2. [DOI] [PubMed] [Google Scholar]
- 16.Schwob E, Böhm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–44. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 17.Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase-promoting complex. Science. 1998;282:1721–4. doi: 10.1126/science.282.5394.1721. [DOI] [PubMed] [Google Scholar]
- 18.Bardin AJ, Amon A. Men and sin: what’s the difference? Nat Rev Mol Cell Biol. 2001;2:815–26. doi: 10.1038/35099020. [DOI] [PubMed] [Google Scholar]
- 19.D’Amours D, Amon A. At the interface between signaling and executing anaphase--Cdc14 and the FEAR network. Genes Dev. 2004;18:2581–95. doi: 10.1101/gad.1247304. [DOI] [PubMed] [Google Scholar]
- 20.Draetta G, Beach D. Activation of cdc2 protein kinase during mitosis in human cells: cell cycle-dependent phosphorylation and subunit rearrangement. Cell. 1988;54:17–26. doi: 10.1016/0092-8674(88)90175-4. [DOI] [PubMed] [Google Scholar]
- 21.Dunphy WG, Newport JW. Fission yeast p13 blocks mitotic activation and tyrosine dephosphorylation of the Xenopus cdc2 protein kinase. Cell. 1989;58:181–91. doi: 10.1016/0092-8674(89)90414-5. [DOI] [PubMed] [Google Scholar]
- 22.Gautier J, Matsukawa T, Nurse P, Maller J. Dephosphorylation and activation of Xenopus p34cdc2 protein kinase during the cell cycle. Nature. 1989;339:626–9. doi: 10.1038/339626a0. [DOI] [PubMed] [Google Scholar]
- 23.Morla AO, Draetta G, Beach D, Wang JY. Reversible tyrosine phosphorylation of cdc2: dephosphorylation accompanies activation during entry into mitosis. Cell. 1989;58:193–203. doi: 10.1016/0092-8674(89)90415-7. [DOI] [PubMed] [Google Scholar]
- 24.Russell P, Nurse P. cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell. 1986;45:145–53. doi: 10.1016/0092-8674(86)90546-5. [DOI] [PubMed] [Google Scholar]
- 25.Amon A, Surana U, Muroff I, Nasmyth K. Regulation of p34CDC28 tyrosine phosphorylation is not required for entry into mitosis in S. cerevisiae. Nature. 1992;355:368–71. doi: 10.1038/355368a0. [DOI] [PubMed] [Google Scholar]
- 26.Sorger PK, Murray AW. S-phase feedback control in budding yeast independent of tyrosine phosphorylation of p34cdc28. Nature. 1992;355:365–8. doi: 10.1038/355365a0. [DOI] [PubMed] [Google Scholar]
- 27.McMillan JN, Sia RA, Lew DJ. A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J Cell Biol. 1998;142:1487–99. doi: 10.1083/jcb.142.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sia RA, Herald HA, Lew DJ. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol Biol Cell. 1996;7:1657–66. doi: 10.1091/mbc.7.11.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keaton MA, Bardes ES, Marquitz AR, Freel CD, Zyla TR, Rudolph J, et al. Differential susceptibility of yeast S and M phase CDK complexes to inhibitory tyrosine phosphorylation. Curr Biol. 2007;17:1181–9. doi: 10.1016/j.cub.2007.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harvey SL, Kellogg DR. Conservation of mechanisms controlling entry into mitosis: budding yeast wee1 delays entry into mitosis and is required for cell size control. Curr Biol. 2003;13:264–75. doi: 10.1016/S0960-9822(03)00049-6. [DOI] [PubMed] [Google Scholar]
- 31.Hu F, Gan Y, Aparicio OM. Identification of Clb2 residues required for Swe1 regulation of Clb2-Cdc28 in Saccharomyces cerevisiae. Genetics. 2008;179:863–74. doi: 10.1534/genetics.108.086611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simpson-Lavy KJ, Brandeis M. Clb2 and the APC/C(Cdh1) regulate Swe1 stability. Cell Cycle. 2010;9:3046–53. doi: 10.4161/cc.9.15.12457. [DOI] [PubMed] [Google Scholar]
- 33.Pal G, Paraz MT, Kellogg DR. Regulation of Mih1/Cdc25 by protein phosphatase 2A and casein kinase 1. J Cell Biol. 2008;180:931–45. doi: 10.1083/jcb.200711014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russell P, Moreno S, Reed SI. Conservation of mitotic controls in fission and budding yeasts. Cell. 1989;57:295–303. doi: 10.1016/0092-8674(89)90967-7. [DOI] [PubMed] [Google Scholar]
- 35.Lee KS, Asano S, Park JE, Sakchaisri K, Erikson RL. Monitoring the cell cycle by multi-kinase-dependent regulation of Swe1/Wee1 in budding yeast. Cell Cycle. 2005;4:1346–9. doi: 10.4161/cc.4.10.2049. [DOI] [PubMed] [Google Scholar]
- 36.Mizunuma M, Hirata D, Miyaoka R, Miyakawa T. GSK-3 kinase Mck1 and calcineurin coordinately mediate Hsl1 down-regulation by Ca2+ in budding yeast. EMBO J. 2001;20:1074–85. doi: 10.1093/emboj/20.5.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Archambault V, Ikui AE, Drapkin BJ, Cross FR. Disruption of mechanisms that prevent rereplication triggers a DNA damage response. Mol Cell Biol. 2005;25:6707–21. doi: 10.1128/MCB.25.15.6707-6721.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 1998;12:2956–70. doi: 10.1101/gad.12.18.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hwang CS, Varshavsky A. Regulation of peptide import through phosphorylation of Ubr1, the ubiquitin ligase of the N-end rule pathway. Proc Natl Acad Sci USA . 2008;105:19188–93. doi: 10.1073/pnas.0808891105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kassir Y, Rubin-Bejerano I, Mandel-Gutfreund Y. The Saccharomyces cerevisiae GSK-3 beta homologs. Curr Drug Targets. 2006;7:1455–65. doi: 10.2174/1389450110607011455. [DOI] [PubMed] [Google Scholar]
- 41.Lee J, Moir RD, McIntosh KB, Willis IM. TOR signaling regulates ribosome and tRNA synthesis via LAMMER/Clk and GSK-3 family kinases. Mol Cell. 2012;45:836–43. doi: 10.1016/j.molcel.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–76. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 43.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–86. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu C, Kim NG, Gumbiner BM. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle. 2009;8:4032–9. doi: 10.4161/cc.8.24.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tullai JW, Graham JR, Cooper GMA. A GSK-3-mediated transcriptional network maintains repression of immediate early genes in quiescent cells. Cell Cycle. 2011;10:3072–7. doi: 10.4161/cc.10.18.17321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tullai JW, Tacheva S, Owens LJ, Graham JR, Cooper GM. AP-1 is a component of the transcriptional network regulated by GSK-3 in quiescent cells. PLoS ONE. 2011;6:e20150. doi: 10.1371/journal.pone.0020150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, et al. GSK-3 beta targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3 beta inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008;13:36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, et al. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell. 2003;12:381–92. doi: 10.1016/S1097-2765(03)00287-9. [DOI] [PubMed] [Google Scholar]
- 50.Sharifpoor S, van Dyk D, Costanzo M, Baryshnikova A, Friesen H, Douglas AC, et al. Functional wiring of the yeast kinome revealed by global analysis of genetic network motifs. Genome Res. 2012;22:791–801. doi: 10.1101/gr.129213.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boone C, Bussey H, Andrews BJ. Exploring genetic interactions and networks with yeast. Nat Rev Genet. 2007;8:437–49. doi: 10.1038/nrg2085. [DOI] [PubMed] [Google Scholar]
- 52.Measday V, Hieter P. Synthetic dosage lethality. Methods Enzymol. 2002;350:316–26. doi: 10.1016/S0076-6879(02)50971-X. [DOI] [PubMed] [Google Scholar]
- 53.Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell. 2006;21:319–30. doi: 10.1016/j.molcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 54.Ikui AE, Cross FR. Specific genetic interactions between spindle assembly checkpoint proteins and B-Type cyclins in Saccharomyces cerevisiae. Genetics. 2009;183:51–61. doi: 10.1534/genetics.109.105148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mui MZ, Roopchand DE, Gentry MS, Hallberg RL, Vogel J, Branton PE. Adenovirus protein E4orf4 induces premature APCCdc20 activation in Saccharomyces cerevisiae by a protein phosphatase 2A-dependent mechanism. J Virol. 2010;84:4798–809. doi: 10.1128/JVI.02434-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grandin N, Reed SI. Differential function and expression of Saccharomyces cerevisiae B-type cyclins in mitosis and meiosis. Mol Cell Biol. 1993;13:2113–25. doi: 10.1128/mcb.13.4.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Asano S, Park JE, Sakchaisri K, Yu LR, Song S, Supavilai P, et al. Concerted mechanism of Swe1/Wee1 regulation by multiple kinases in budding yeast. EMBO J. 2005;24:2194–204. doi: 10.1038/sj.emboj.7600683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McMillan JN, Longtine MS, Sia RA, Theesfeld CL, Bardes ES, Pringle JR, et al. The morphogenesis checkpoint in Saccharomyces cerevisiae: cell cycle control of Swe1p degradation by Hsl1p and Hsl7p. Mol Cell Biol. 1999;19:6929–39. doi: 10.1128/mcb.19.10.6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shulewitz MJ, Inouye CJ, Thorner J. Hsl7 localizes to a septin ring and serves as an adapter in a regulatory pathway that relieves tyrosine phosphorylation of Cdc28 protein kinase in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:7123–37. doi: 10.1128/mcb.19.10.7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wittenberg C, Reed SI. Control of the yeast cell cycle is associated with assembly/disassembly of the Cdc28 protein kinase complex. Cell. 1988;54:1061–72. doi: 10.1016/0092-8674(88)90121-3. [DOI] [PubMed] [Google Scholar]
- 61.Lim MY, Dailey D, Martin GS, Thorner J. Yeast MCK1 protein kinase autophosphorylates at tyrosine and serine but phosphorylates exogenous substrates at serine and threonine. J Biol Chem. 1993;268:21155–64. [PubMed] [Google Scholar]
- 62.Lew DJ, Reed SI. Morphogenesis in the yeast cell cycle: regulation by Cdc28 and cyclins. J Cell Biol. 1993;120:1305–20. doi: 10.1083/jcb.120.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hilioti Z, Gallagher DA, Low-Nam ST, Ramaswamy P, Gajer P, Kingsbury TJ, et al. GSK-3 kinases enhance calcineurin signaling by phosphorylation of RCNs. Genes Dev. 2004;18:35–47. doi: 10.1101/gad.1159204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rayner TF, Gray JV, Thorner JW. Direct and novel regulation of cAMP-dependent protein kinase by Mck1p, a yeast glycogen synthase kinase-3. J Biol Chem. 2002;277:16814–22. doi: 10.1074/jbc.M112349200. [DOI] [PubMed] [Google Scholar]
- 65.Dailey D, Schieven GL, Lim MY, Marquardt H, Gilmore T, Thorner J, et al. Novel yeast protein kinase (YPK1 gene product) is a 40-kilodalton phosphotyrosyl protein associated with protein-tyrosine kinase activity. Mol Cell Biol. 1990;10:6244–56. doi: 10.1128/mcb.10.12.6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morgan DO. Regulation of the APC and the exit from mitosis. Nat Cell Biol. 1999;1:E47–53. doi: 10.1038/10039. [DOI] [PubMed] [Google Scholar]
- 67.Stegmeier F, Amon A. Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu Rev Genet. 2004;38:203–32. doi: 10.1146/annurev.genet.38.072902.093051. [DOI] [PubMed] [Google Scholar]
- 68.Lengronne A, Schwob E. The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G(1) Mol Cell. 2002;9:1067–78. doi: 10.1016/S1097-2765(02)00513-0. [DOI] [PubMed] [Google Scholar]
- 69.Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature. 2007;446:806–10. doi: 10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- 70.Fiedler D, Braberg H, Mehta M, Chechik G, Cagney G, Mukherjee P, et al. Functional organization of the S. cerevisiae phosphorylation network. Cell. 2009;136:952–63. doi: 10.1016/j.cell.2008.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pan X, Ye P, Yuan DS, Wang X, Bader JS, Boeke JD. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell. 2006;124:1069–81. doi: 10.1016/j.cell.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 72.Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, et al. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–6. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–61. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 74.Kaiser C, Michaelis S, Mitchell A. Methods in yeast genetics. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press, 1994. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.