

Abstract
The essential function of eIF4E-binding protein 1 (4E-BP1) in translation initiation has been well established; however, the role of 4E-BP1 in normal cell cycle progression is coming to attention. Here, we revealed the role of 4E-BP1 on mitotic regulation and chromosomal DNA dynamics during mitosis. First, we have observed the co-localization of the phosphorylated 4E-BP1 at T37/46 with Polo-like kinase 1 (PLK1) at the centrosomes during. Depression of 4E-BP1 by small interfering RNA in HepG2 or HeLa cells resulted in an increased outcome of polyploidy and aberrant mitosis, including chromosomal DNA misaligned and multi-polar spindles or multiple centrosomes. We observed that 4E-BP1 interacted with PLK1 directly in vitro and in vivo in mitotic cells, and the C-terminal aa 77–118 of 4E-BP1 mediates its interaction with PLK1. PLK1 can phosphorylate 4E-BP1 in vitro. Furthermore, the depletion of 4E-BP1 sensitized HepG2 and HeLa cells to the microtubule disruption agent paclitaxel. These results demonstrate that 4E-BP1, beyond its role in translation regulation, can function as a regulator of mitosis via interacting with PLK1, and possibly plays a role in genomic stability maintaining.
Keywords: 4E-BP1, PLK1, cell cycle, centrosome, genomic stability, spindle
Introduction
Translation regulation plays an important role in regulating gene expression in mammalian cells. The principal and primary control point of translation is the formation of eukaryotic translation initiation factor 4F (eIF4F) complex, which is involved in loading ribosomal subunits to the mRNA and starting protein translation. The eIF4F complex is composed of three subunits: scaffold protein eIF4G, ATP-dependent helicase eIF4A and cap-binding protein eIF4E. The eIF4E plays a key role in eIF4F complex assembly by directly binding with scaffold protein eIF4G and the “cap (m7GpppN)” structure. The interaction of eIF4E and eIF4G is negatively regulated by the association of eIF4E-binding proteins (4E-BPs) with eIF4E. 4E-BPs, which share a motif with eIF4G, allowing it to interact with and sequester eIF4E in a competitive fashion, acts as inhibitor of translation initiation.1,2 4E-BP1 is the best-characterized member of 4E-BPs family. The interaction of 4E-BP1 with eIF4E is regulated via the phosphorylation of several serine/threonine residues by mTORC1, which plays an important role in mediating signaling events after exogenous stimuli treatment.3 4E-BP1 phosphorylation is in a stepwise manner in response to agents that activate the translation. The phosphorylation of two N-terminal threonines (T37 and T46) of human 4E-BP1 subsequently promotes the phosphorylation of other sites (T70 and S65), which results in release of eIF4E from the eIF4F complex.4
The rates of protein translation are tightly regulated throughout the cell cycle. Cap-dependent translation initiation is prevalent in G1/S phase and impaired when cells enter mitosis. In contrast to cap-dependent translation, the internal ribosome entry site (IRES)-dependent translation is stimulated immediately during and after the metaphase stage of mitosis.5,6 Hypo-phosphorylated 4E-BP1, through greater sequestrating with eIF4E, can suppress cap-dependent translation and increase IRES-dependent translation during stress condition.7,8 Therefore, 4E-BP1 plays an important role in switching between cap and IRES-dependent translation, and it could be also hypo-phosphorylated during mitosis. However, several articles reported that the phosphorylation of 4E-BP1 increased during mitosis. Recent works reported that the protein rapor, a component of mTORC1, could be phosphorylated by cdk1 and GSK3 at multiple sites during mitosis. Mitotic-specific phosphorylation of raptor altered the activity of mTORC1 and promoted phosphorylation of 4E-BP1 in mitosis.9 Cyclin B1-cdk1 complex has been implicated in the phosphorylation of 4E-BP1 in normal or paclitaxel-induced mitosis phase.10,11 These reports indicated that phosphorylation of 4E-BP1 highly associates with the mitotic regulation. However, the precise functions of 4E-BP1 in this progression haven’t been clearly demonstrated. Transitions through mitosis often require the coordination of cdk1 with another important kinase, PLK1. PLK1 has emerged as a key regulator of mitosis in eukaryotic cells, and it functions in a variety of mitotic events including the G2/M transition, centrosome maturation, spindle formation, chromosome segregation and cytokinesis.12 Normal cell division is exquisitely sensitive to the changes of expression level and activity of PLK1 protein. Cell division is severely impaired in both PLK1 overexpression and depletion cells.13,14 Since 4E-BP1 is a substrate of cdk1/cylcin B1 complex during mitosis, we want to know whether 4E-BP1 regulates mitotic progression and its associated components.
In the present study, we identified that 4E-BP1 is involved in maintaining the spindle stability and chromosomal DNA dynamics during normal mitosis. Suppression of 4E-BP1 led to misaligned chromosome, abnormal centrosomes and polyploidy. We further observed that 4E-BP1 could interact with PLK1. Most importantly, the localization of phosphorylated 4E-BP1 T37/46 at centrosomes during mitosis has also been demonstrated. These observations revealed a new function of 4E-BP1 in cell cycle regulation.
Results
The phosphorylation of 4E-BP1 occurres and localizes at the centrosomes during mitosis
Different phases of synchronized HepG2 cells were harvested at different times after release from the thymidine-induced blockage of G1/S transition; the cell cycle progression was monitored by flow cytometry (Fig. 1A and B). As shown in Figure 1C, an increased expression of PLK1 was observed when the cells entered into G2-M phases by western blot analysis, as expected. Simultaneously, the increased phosphorylated 4E-BP1 at Thr-37/46 and Ser-65 was also detected when the cells entered into G2-M phases using phosphor-specific antibody (Fig. 1C). This phosphorylation effect of 4E-BP1 was also observed by the treatment of nocodazole, a microtubulin-depolymerizing agent that leads to mitotic arrest (data not shown).
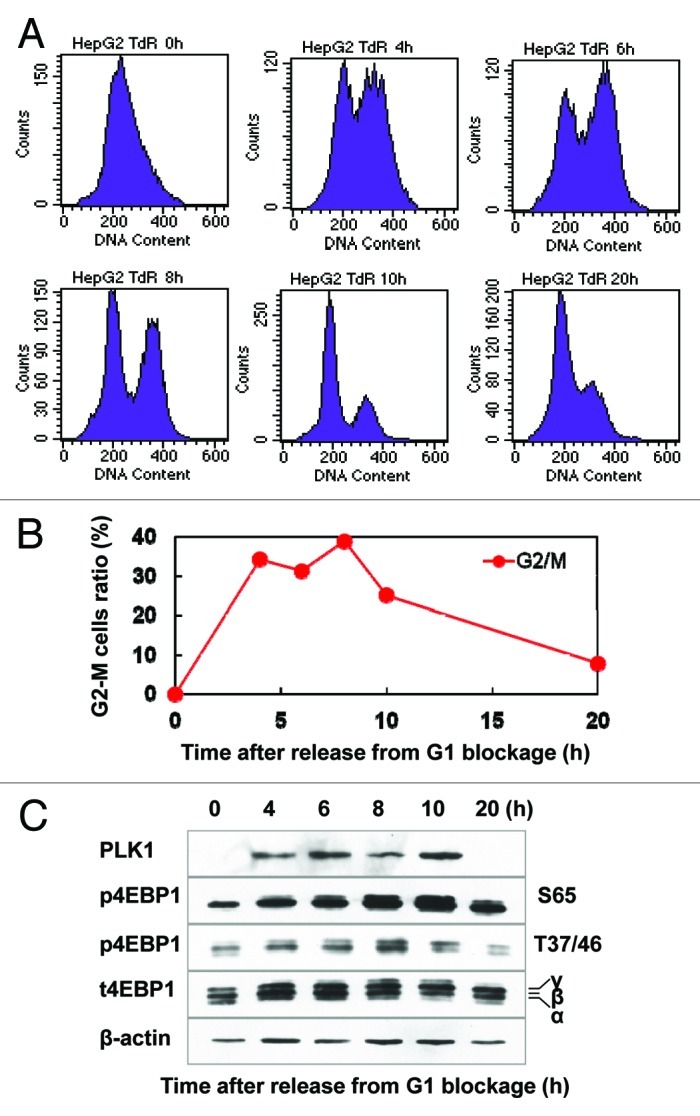
Figure 1A‒C. Phosphorylation of 4E-BP1 is linked with mitotic progression. (A) Representative histograms of flow cytometry analysis for the synchronized HepG2 cells. The synchronized HepG2 cells by thymidine double-blocking method were released by culturing cells in the normal growth medium and were harvested at different time points for flow cytometry analysis. (B) Quantitative data of the proportion of G2-M phases cells at different times after released from G1/S blockage. (C) Phosphorylated and total 4E-BP1 were analyzed by western blotting at indicated times after releasing from G1/S arrest. PLK1 and actin were used as mitotic marker and loading control, respectively.
To further determine the spatial distribution of phosphorylated 4E-BP1 during mitosis, the normally growing HepG2, HeLa (Fig. 1D) and 293T (Fig. S1A) cells were subjected to immunofluorescent staining using anti-4E-BP1 pT37/46-specific antibody. Intriguingly, the Thr-37/46 phosphorylated 4E-BP1 specifically appeared at the centrosomes during metaphase and colocalized with the centrosome molecular marker γ-tubulin, as shown in Figure 1D. To further confirm the specificity of the targeting molecule for the pThr-37/46 4E-BP1 antibody in the immunofluorescence analysis, we knocked down the 4E-BP1 in HepG2 cells by siRNA (Fig. 1F) and found that the immunostaining signal of the anti-pThr-37/46 4E-BP1 antibody was hardly seen at the centrosomes in 4E-BP1-knockdown cells (data not shown), suggesting that this antibody can specifically recognize a phosphorylated form of 4E-BP1 in immunostaining analysis. It was suggested that 4E-BP1 phosphorylation at T37 and T46 is relatively constitutive, and the phosphorylation at S65 is more indicative for its function in mediating cap-dependent translation initiation.15 So we further performed the immunostaining analysis using anti-4E-BP1 pS65-specific antibody. We could also find that Ser 65 phosphorylated 4E-BP1 localizes at centrosomes during mitosis (Fig. S1B). Moreover, we transfected HepG2 cells with the vector of RFP-fused 4E-BP1, and found that this 4E-BP1 fusion protein had a diffuse distribution over the mitotic cell, but was more concentrated at the spindle organelle and also co-localized with γ-tubulin at centrosomes (Fig. 1E).
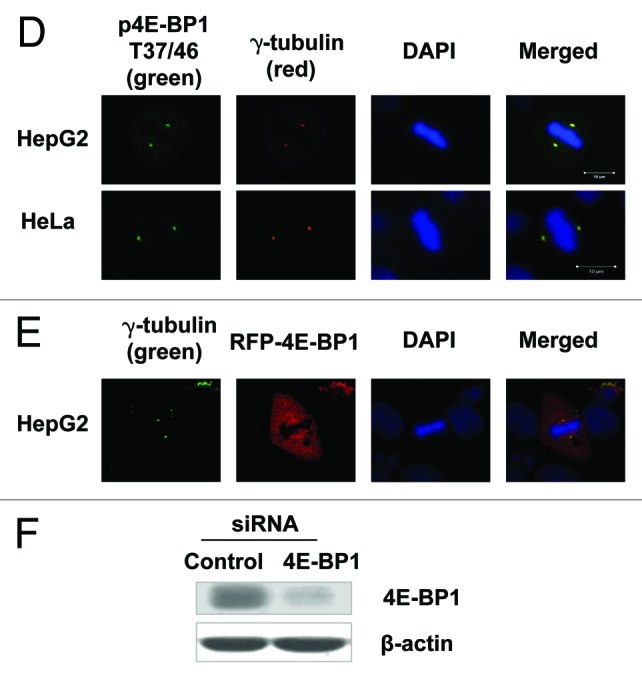
Figure 1D‒F. Phosphorylation of 4E-BP1 is linked with mitotic progression. (D) HepG2 (upper) and HeLa (lower) cells were immunostained with the antibody of phosphorylated 4E-BP1 /pT37/46 and γ-tubulin antibody and were examined by fluorescent microscopy. T37/46-phosphorylated 4E-BP1 overlapped with γ-tubulin at the centrosomes during mitosis. (E) HepG2 cells transfected with a vector of RFP-fused 4E-BP1 were immunostained with anti-γ-tubulin antibodies and subjected to fluorescent microscopy observation. RFP-fused 4E-BP1 protein was shown a diffuse distribution over the mitotic cell, but was more concentrated at the spindle organelle and co-localized with γ-tubulin at centrosomes. (F) The western blotting analysis of 4E-BP1 expression in HepG2 cells transfected with anti-4E-BP1 specific siRNA.
Suppression of 4E-BP1 results in aberrant spindles
The induction of mitotic phosphorylation of 4E-BP1 and its recruitment into the mitotic spindle apparatus strongly indicate an important role of 4E-BP1 in maintaining the normal mitotic progression. Consistent with this hypothesis, we found that 4E-BP1-depressed HepG2 cells showed obvious defects in spindle construction and chromosomal DNA dynamic behaviors during mitosis. For control cells, the highly condensed chromosomes align along the equatorial planes during metaphase. However, depletion of 4E-BP1 results in obviously misaligned chromosomes, and unattached free condensed chromosomal DNA were seen spreading throughout whole nuclei in the metaphase (Fig. 2A and B). A much higher ratio of mitotic cells with multiple centrosomes or multi-polar spindles were shown in 4E-BP1-depleted cells (Fig. 2A and B). Aberrant mitosis spindle construction will lead to aneuploidy, so we further analyzed the DNA content by flow cytometry and observed an increased proportion of aneuploidy cells in the 4E-BP1-knockdown HepG2 cells (Fig. 2C and D). The similar aberrant mitotic appearance has also been observed in 4E-BP1 knockdown HeLa cells (data not shown). These results indicate that 4E-BP1 plays a pivotal role in mitosis progression, and depletion of 4E-BP1 will cause multiple constructional defects and abnormal chromosomal DNA dynamics during mitosis.
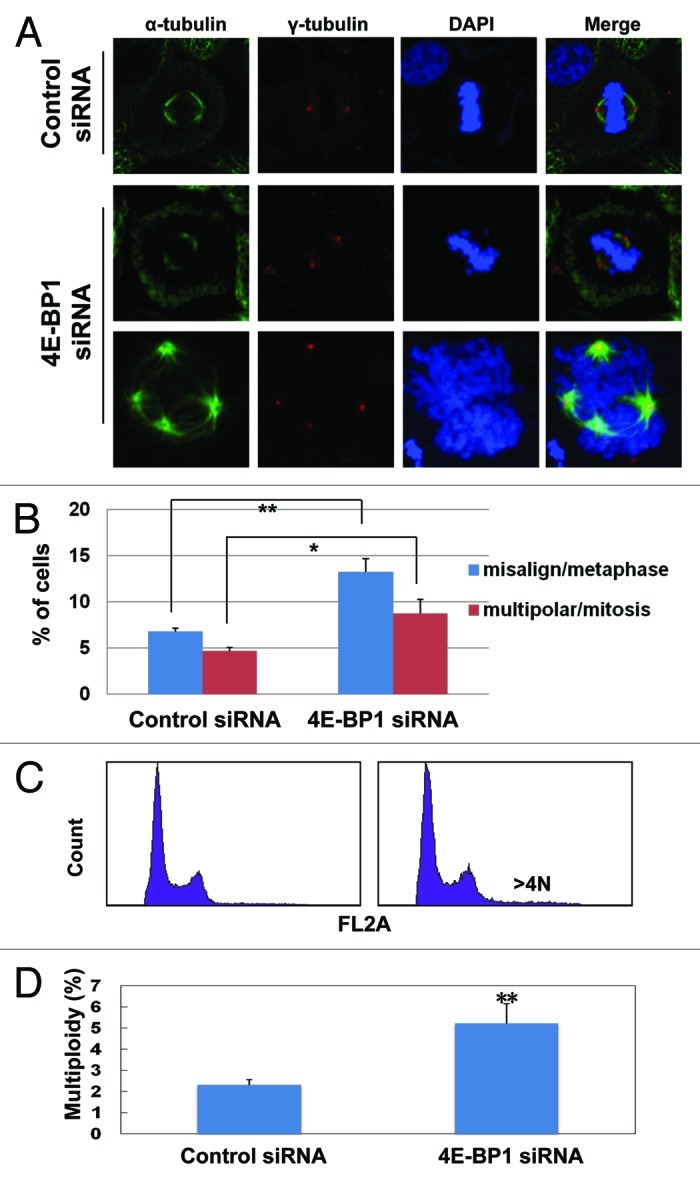
Figure 2. Depletion of 4E-BP1 leads to increased outcome of polyploidy, aberrant spindles and misaligned chromosomal DNA in normal cultured cells. (A) Forty-eight h after transfection with anti-4E-BP1-specific and non-specific siRNA, HepG2 cells were fixed and co-stained with antibodies against α-tubulin and γ-tubulin. Mitotic cells with abnormal spindles were captured. Representative images indicate the misaligned chromosomal DNA (unattached free DNA) and multipolar spindle or multiple centrosomes in 4E-BP1-depleted cells. (B) The polyploidy of the misaligned chromosomal cells to metaphase cells and multipolar spindle cells to mitosis cells were counted (* p < 0.05, ** p < 0.01 as compared with control cells). (C) Forty-eight h after transfected with anti-4E-BP1 specific and non-specific siRNA, HepG2 cells were collected and analyzed by flow cytometry. (D) Quantitative data of the polyploidy cells were presented as the mean and SD from three independent experiments (** p < 0.01 as compared with control cells).
Depletion of 4E-BP1 sensitizes cells to paclitaxel
Our data revealed that 4E-BP1-depleted cells exhibited increased outcome of mitotic catastrophe, which associated with mitotic progression failure, similar to microtubule drug-treated cells. So we next determined whether silencing of 4E-BP1 expression could effectively modulate the sensitivity of HepG2 cells to paclitaxel. Twenty-four h following transfection with 4E-BP1-targeted siRNA, HepG2 cells survival was assessed 72 h after paclitaxel exposure using the MTT assay of cell proliferation. As shown in Figure 3A, 4E-BP1-knockdown HepG2 cells were significantly more sensitive to the treatment of 5 to 100 nM paclitaxel than control siRNA-transfected cells (three asterisks means p < 0.001 compared with control cells). However, depletion of 4E-BP1 had no effect on growth speed in the normal growing condition (Fig. S2A). 4E-BP1-knockdown also sensitizes HeLa cells to paclitaxel (Fig. S2B). We next investigated the effect of 4E-BP1 depletion on the induction of apoptosis using paclitaxel. Twenty-four h after transfected with 4E-BP1-specific or non-specific control siRNA, the cells were treated with 100 nM paclitaxel, and apoptosis was detected by flow cytometry analysis of Annexin V staining 24 h later. 4E-BP1-knockdown HepG2 cells exhibited higher level of Annexin V-positive apoptotic cells compared with control cells in response to paclitaxel treatment. As shown in Figure 3B, after the treatment of 100 nM paclitaxel, the Annexin V-positive apoptotic cells increased from 12.1 ± 1.5% to 15.6 ± 3.1% for control cells and 14.7 ± 1.8% to 22.7 ± 3.9% for 4E-BP1-knockdown cells. These observations suggest that the suppression of 4E-BP1 expression may sensitize HepG2 cells to paclitaxel and provide a new sight, in which 4E-BP1 seems to be an excellent molecular therapeutic target for cancer cells when using combination treatment with paclitaxel. HeLa cells are obviously more resistant to paclitaxel than HepG2 cells; paclitaxel barely induce apoptosis in HeLa cells at the concentration of 100 nM (Fig. S2C). Although depletion of 4E-BP1 sensitizes HeLa cells to paclitaxel (Fig. S2B), which can only slightly increase the induction of apoptosis by 100 nM paclitaxel, this is not statisticaly significant.

Figure 3. Depletion of 4E-BP1 sensitized cells to paclitaxel. (A) Twenty-four h following transfection with 4E-BP1 specific siRNA, HepG2 cells were treated with paclitaxel at various concentrations. Cell growth potency was analyzed after the 72 h treatment of paclitaxel using MTT assay. The data are the means and standard deviations from three independent experiments, ***p < 0.001 as compared with the control siRNA transfected cells under the same treatment of paclitaxel. (B) Twenty-four h following transfection with 4E-BP1-specific siRNA, HepG2 cells were treated with 100 nM paclitaxel or DMSO as control for 24 h. Apoptotic cells were detected by using the annexin V and propidium iodide double staining method. The data are the means and standard deviations from three independent experiments, **p < 0.01 as compared with the control siRNA transfected cells under the same treatment of paclitaxel.
4E-BP1 associates with and is phosphorylated by PLK1
Polo like kinase-1 is a key effector of cell division and has been recently shown to crosstalk with mTOR signal pathway.16 Our data showed that the phosphorylated 4E-BP1 co-localizes with PLK1 at centrosomes during mitosis (Fig. 4A), so we deduced that 4E-BP1 might have an association with PLK1 during mitosis. As we know, PLK1 shows a periodic expression pattern with a markedly increased level in mitotic cells. Indeed, as shown in Figure 4B, an endogenous 4E-BP1 and PLK1 interaction complex was detected by immunoprecipitation (IP) assay in the mitotic cells produced by nocodazole. Importantly, this interaction of 4E-BP1 and PLK1 was hardly detected in the asynchronous cells (Fig. 4B). We further confirmed this interaction using GST pull-down assay. The Flag-fused PLK1 protein, which is expressed in 293T cells, was shown to associate with the GST-fused 4E-BP1 protein, but not with GST control (Fig. 4C), indicating that 4E-BP1 can bind directly to PLK1 in vitro.
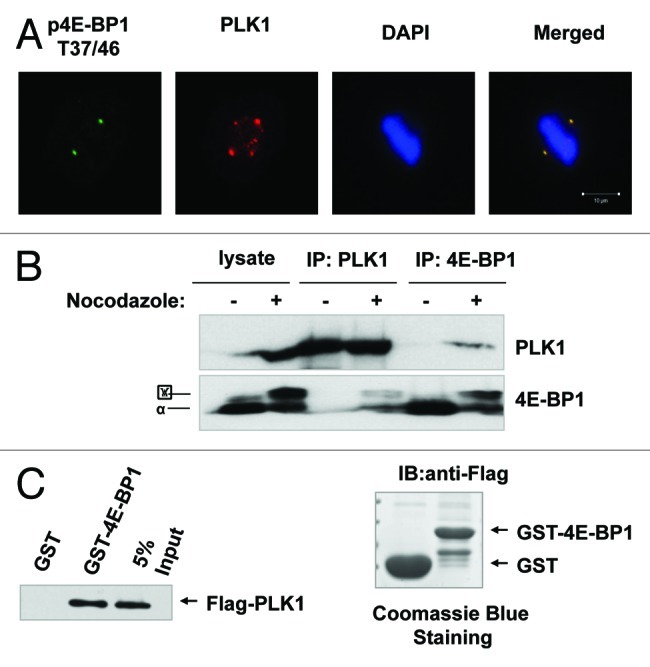
Figure 4A‒C. 4E-BP1 interacts with PLK1. (A) HepG2 cells were immunestained with antibodies against pT37/46 of 4E-BP1 or PLK1, and were examined by fluorescent microscopy. Phosphorylated T37/46 of 4E-BP1 co-localizes with PLK1 at centrosomes. (B) HepG2 cells were treated with nocodazole (200 ng/ml) for 16 h. Mitotic cells were collected by “shake-off.” Asynchronized and mitotic cells lysates were immunoprecipitated using anti-4E-BP1 or PLK1 antibodies. Western blot was performed using anti-PLK1 and 4E-BP1 antibodies. (C) GST and GST-fused 4E-BP1 recombinant bound to glutathione-Sepharose beads. 293T cells transfected with pCMV-2Btag-PLK1 lysates were incubated with the immobilized GST or GST-fusion protein. Bound fractions were analyzed by western blot using anti-Flag antibody.
To further identify the main domain of 4E-BP1 interacting with PLK1, a series of truncated mutants of 4E-BP1 fused with GST were constructed and expressed in E.coli (Fig. 4D and E). It was demonstrated that the mutants of deleted C-terminal aa 77–118 of 4E-BP1 lost their interaction with PLK1 (Fig. 4E). Moreover, the GST-fused 4E-BP1 could pull down the PBD domain of PLK1 protein but not the kinase domain (Fig. 4F and G).

Figure 4D‒G. 4E-BP1 interacts with PLK1. (D) Schematic representation of 4E-BP1 protein deletion mutants. Red domain represents the interaction domain that mediates 4E-BP1 and PLK1 association identified in this study. (E) Recombinant GST and GST fused 4E-BP1 and its mutants bound to glutathione-Sepharose beads were incubated with 293T cells lysates which transfected with pCMV-HA-PLK1 vector. Bound fractions were analyzed by western blotting using anti-HA antibody. (F) Schematic representation of PLK1 protein deletion mutants. Design of these mutants was based on two main function domains of PLK1. Red minus represents the interaction domain. (G) Recombinant GST and GST-fused 4E-BP1 bound to glutathione-Sepharose beads. 293T cells transfected with pCMV-HA-PLK1 KD and PBD domain, respectively; after 48 h, cells were collected and lysates were incubated with the immobilized GST or GST-fusion protein. Bound fractions were analyzed by western blot using anti-HA antibody.
Given that 4E-BP1 interacts with PLK1, and PLK1 is an S/T kinase, we assumed that 4E-BP1 might be a substrate of PLK1. We expressed and purified GST-fused 4E-BP1 and its deletions from bacteria. In vitro phosphorylation assay showed that incubation of PLK1 kinase (commercial) with GST fusion proteins and [γ-32P] ATP led to the incorporation of the radioactive 32P into the recombinant 4E-BP1 protein. Interestingly, the mutants of deleted N-terminal of 4E-BP1 exhibited stronger phosphorylation signal than that of the full-length 4E-BP1 (Fig. 5). This phenomenon suggests that N-terminal of this protein might play a negative role in regulating phosphorylation of 4E-BP1 induced by PLK1.
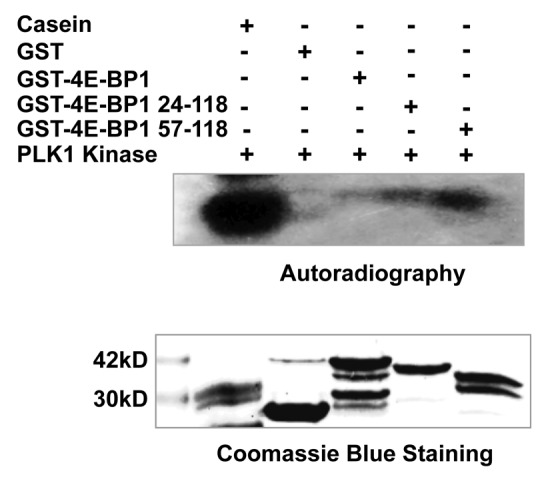
Figure 5. PLK1 phosphorylates 4E-BP1 in vitro. A recombinant GST, GST fused full-length and truncated 4E-BP1 were incubated with a commercial purified PLK1 protein in the presence of [32P]-γ-ATP in the kinase reaction buffer. Proteins were analyzed by SDS–PAGE and visualized by autoradiography (kinase assay) or Coomassie blue staining (staining). Casein was used as positive control.
Discussion
4E-BP1 is reported as an inhibitive factor of tumor cells survival and proliferation because of its role as a negative regulator of cap-dependent translation.17 A previous report showed that human 4E-BP1 gene localizes at chromosome 8p11–12, which is a locus frequently amplified in human cancers specimens.8,18 Immunohistochemistry (IHC) assay using 4E-BP1-specific antibody has revealed that 4E-BP1 is overexpressed in various human cancers, such as advanced breast cancer, colorectal cancer and prostate cancer.8,19-21 The evidence of 4E-BP1 functioning in switching from cap-dependent to cap-independent mRNA translation in response to hypoxia possibly gives one reason why 4E-BP1 keeps a high level in tumor tissues. Here, our study demonstrated that 4E-BP1 participates in maintaining spindle stability and mitotic progression regulation, seemly giving another clue for the association of 4E-BP1 with cancers and explaining why it’s overexpressed in cancer tissues. Cancer cells grow faster than normal cells, so the proteins involved in promotion of mitotic progression are often overexpressed in cancer cells, as is 4E-BP1. In addition to its overexpression behavior in cancers, the phosphorylation of 4E-BP1 was identified as a critical signaling event in cancer resistance to mTOR kinase inhibitors.22,23 Although 4E-BP1 has been showed as a well-characterized substrate of mTORC1,3,22 an mTOR-independent phosphorylation of 4E-BP1 has been also suggested to be associated with the mTOR kinase inhibitor resistance in cancers.23
Spindle integrity, normally aligned chromosomal DNA and precise mitotic progression are important for genomic stability. Azar et al. reported that 4E-BP1 promoter was activated, and 4E-BP1 protein amount increased as culture cells reached confluence, suggesting a critical role for 4E-BP1 in density-mediated cell cycle arrest.24 In the present study, we have provided the direct evidence and partial mechanistic explanation to uncover the fundamental role of 4E-BP1 in mitotic progression control and genomic stability. Our observations showed that inactivation of 4E-BP1 by siRNA resulted in cells with misaligned chromosomal DNA and multipolar spindles/centrosomes. It has long been known that even subtle incorrect execution of mitosis might produce cytokinesis failure and subsequent tetraploidy. Due to extra centrosomes (= 4), tetraploid cells will undergo abnormal mitosis and often become aneuploid.25 Our results also demonstrated that 4E-BP1 deficiency led to increase of aneuploidy. More importantly, the direct evidence of 4E-BP1 involve in mitosis regulation came from the fact that phosphorylated 4E-BP1 at Thr37/46 localized at centrosomes during mitosis. Barnhart has even reported that 4E-BP1 knockdown led to a dramatic increase of aberrant mitosis in Rh30 rhabdomyosarcoma xenografts.26 However, the reason for this phenomenon has not yet been elucidated. Our works here suggest a possible mechanism in which 4E-BP1 locates and functions at the centrosomes and regulates mitotic progression directly.
Although the function of 4E-BP1 in translation regulation has been well investigated, relatively little is known about the precise mechanism of 4E-BP1 in mitotic regulation. Our study has demonstrated that the function of 4E-BP1 in mitosis progression could be related to its direct interaction with PLK1, a critical player through the whole mitotic process.27,28 We identified that the C terminus of 4E-BP1 mediated the interaction between 4E-BP1 and PLK1 PBD domain. PLK1, which plays an essential role in regulation of mitosis, consists of two signature motifs, known as PBD domain at the C terminus and kinase domain localized at N terminus.13 Interaction of the PBD with an exogenous phosphopeptide ligand can cause the release of the kinase domain, which converts the kinase to the active form. We indeed found that PLK1 phosphorylated 4E-BP1 in vitro. Interestingly, N-terminally truncated polypeptide of 4E-BP1 had a much stronger phosphorylation signal. It is reported that depleting the N terminus, which contains the so-called “RAIP” motif (name from the amino acid sequence), results in a low basal level of phosphorylation of 4E-BP1 in response to insulin.29 N terminus-specific cleavage of 4E-BP1 mediated by activated p53 increases the protein stability of 4E-BP1.30 These reports indicated that N terminus domain plays an important role in regulating function of 4E-BP1. We speculate that the N terminus of 4E-BP1 may play a negative role in its phosphorylation by PLK1. Further investigation should be done to reveal the function of 4E-BP1 phosphorylation by PLK1 in mitosis regulation. In addition, 4E-BP1 shares 60% and 57% identity with 4E-BP2 and 4E-BP3, respectively.31 Elucidation of whether 4E-BP2 and 4E-BP3 also contribute to mitosis regulation through PLK1 pathway would be followed up in future studies. PLK1 is a key player in different stages of mitosis, and its protein levels are strictly controlled through protein synthesis and stability. Deletion or mutations of PLK1 cause cells arrested at the early stages of mitosis and, subsequently, a series of mitotic defects. PLK1-overexpressed cells are able to enter mitosis, but mitosis progression is transiently delayed, and cytokinesis appears to be defected.32 Our study indicated that depression of 4E-BP1 resulted in aberrant spindles, multiple centrosomes and chromosomal DNA misalignment, which is similar to the effect of inactivation of PLK1.
Microtubule-disrupting drugs such as microtubule-polymerizing agent paclitaxel have been widely applied for cancer therapy, and the use of these combinations confers some survival advantages in hematological and solid malignancies.33 However, the resistance of cancer cells to microtubule-damaging drugs limits the clinic applications and the success of chemotherapy. A number of reports indicate that targeting the regulators of mitosis is a successful strategy for sensitizing cancer cells to taxane chemotherapy.34,35 4E-BP1 is frequently upregulated in various cancers, and our data indicated that 4E-BP1 is a mitotic regulator. Therefore, targeting 4E-BP1 may sensitize cancer cells to paclitaxel. Actually, 4E-BP1 has already attracted increasing attention in the field of cancer therapy. Knockdown of 4E-BP1 displayed increased baseline level of autophagy and sensitized the MYC-expressing prostate cancer cells to rapamycin.36 Discodermolide is a microtubule-stabilizing agent that induces accelerated cell senescence, and it has recently been shown that reintroduction of 4E-BP1 even a nonphosphorylatable mutant (Thr-37/46 Ala) of 4E-BP1 can revert the resistance of 4E-BP1-defective cancer cells to discodermolide via restoration of induced accelerated senescence.37 Our data here show that depression of 4E-BP1 sensitized HepG2 cells to paclitaxel, which is partially attributed to the increased induction of apoptosis. Although depletion of 4E-BP1 also sensitizes HeLa cells to paclitaxel, no significant apoptosis was induced in response to paclitaxel treatment even with the knock-down of 4E-BP1. Mitotic catastrophe is an effective cell death pathway as the consequence of the mitotic machinery disruption with the typical appearance of multipolar spindle, multiple centrosomes and multiple micronuclei.38 Given the important role of 4E-BP1 in mitotic progression, depletion of 4E-BP1 sensitizing HeLa cells to paclitaxel may be due a mechanism associated with mitotic catastrophe.
Conclusion
The phosphorylated 4E-BP1 localizes at the centrosomes during mitosis and interacts with PLK1. Depression of 4E-BP1 results in a series of defects in mitotic machinery and progression. Our work has shed new light on the function of 4E-BP1 in maintaining spindle integrity and genomic stability and suggests 4E-BP1 as an attractive target for treatment of the cancers with overexpressed 4E-BP1.
Materials and Methods
Cell lines and treatment
The cell lines HepG2 and HeLa were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (HyClone), 10 mM HEPES and 1 mM sodium bicarbonate in a humidified incubator at 37°C with 5% CO2. The sequence of siRNA against 4E-BP1 was described in a previous report.39 Paclitaxel (PTX) was purchased from Sigma; its stock solution was prepared in DMSO. Cells were treated with various concentrations of paclitaxel and harvested 48 h after treatment.
Cells were synchronized using the thymidine (Sigma) double-blocking method. HepG2 cells were plated in 60 mm Petri dishes, and thymidine was added to a final concentration of 2 mM after cell adherence. The cells were treated for 16 h. After removal of the thymidine, cells were cultured for 10 h in the thymidine-free fresh DMEM solution; thymidine was added in the same concentration for an additional 16 h. After removal of thymidine, the synchronized cells were cultured in fresh growth DMEM and harvested at different time points for cell cycle and western blotting analyses.
Antibodies
All of the antibodies were purchased commercial sources: anti-4E-PB1 total, anti-phosphorylated 4E-BP1 (Thr37/46), anti- phosphorylated 4E-BP1 (Ser65) (Cell Signaling), anti-PLK1 and anti-γ-tubulin (Abcam), β-actin. Secondary antibodies were horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (H+L) or HRP-conjugated anti-mouse IgG (H+L) purchased from Zhongshan Golden Bridge Biotechnology.
Flow cytometric analysis of the cell cycle
After washing twice with PBS solution, cells were collected and fixed using 75% ethanol at -20°C for at least 24 h. Then, the cells were washed twice with PBS solution, incubated with 20 µl RNase A (Sigma) (1 mg/ml) for 30 min at 37°C, and stained with 25 µg ml-1 PI (Sigma) for 30 min at room temperature. The cell cycle distribution was then analyzed using flow cytometry, and more than 10,000 cells per sample were counted. All experiments were repeated three times.
Observation of immunofluorescent microscopy
Cells were plated and grew on poly-d-lysine-coated culture slides (BD PharMingen), washed in PBS, fixed in 4% paraformaldehyde/PBS for 30 min, permeabilized in 0.5% Triton X-100/PBS for 15 min and blocked in blocking buffer (1% bovine serum albumin in PBS) for 30 min. Immunostaining was performed using anti-p4E-BP1, anti-γ-tubulin and anti-PLK1 antibodies for 2 h at room temperature in a humidified chamber. After three 10 min washes, the cells were incubated with anti-mouse/rabbit rhodamine-conjugated (1:200) and anti-mouse/rabbit FITC-conjugated (1:200) secondary antibodies. DNA was stained using 4', 6-diamidino-2-phenylindole (DAPI, Sigma) in mounting solution. Confocal immunofluorescence microscopy observation was performed using an LSM 510 laser-scanning confocal microscope (Zeiss). The expression level of pThr37/46 4E-BP1 at centrosomes of HepG2 NC and KD cells during mitosis was quantified using Image-Pro Plus software. Three independent experiments were performed.
Immunoprecipitation and GST pull-down assays
For immunoprecipitation assay, cells were lysed with the lysis buffer [50 mmol/L Tris (pH8.0); 50 mmol/L NaCl; 0.1% Nonidet P-40; protease inhibitor cocktail (Roche)] for 45 min and centrifuged at 12,000 g for 10 min at 4°C. Cell lysates were incubated with anti-4E-BP1 antibody and protein A/G Sepharose overnight, and the Sepharose beads were washed with lysis buffer for three times and resuspended in SDS-PAGE loading buffer for western blotting analysis using anti-PLK1 antibodies.
The GST fused full-length and truncated 4E-BP1 protein were expressed in E. coli BL21 (DE3) cells grown at 37°C in 1 L of LB media to an optical density at 600 nm of 0.5 and then induced with 0.1 mM isopropyl-D-thiogalactopyranoside (IPTG) for 6 h at 25°C. Cells were washed twice with phosphate-buffered saline solution (PBS). Cells were lysed on ice by sonication. The expressions of GST and GST-fusion proteins were purified by GST-Sepharose beads (Pharmacia) according to manufacturer’s guidelines. 293T cells transfected with pCMV-2Btag-PLK1 (or PLK1 PBD and KD domain) lysates were incubated with the immobilized GST or GST-fusion protein at 4°C for 3 h. The beads were washed with lysis buffer for four times and resuspended in SDS-PAGE loading buffer for western blotting analysis using anti-Flag antibodies. The deletions of 4E-BP1 was described in Figure 4D.
Protein kinase production and kinase assays
The purified PLK1 protein was purchased from Cell Signaling Technology. GST-4E-BP1 and its deletion mutants protein were expressed in and purified from E. coli BL21 strain. Kinase and substrates (GST-fused 4E-BP1 and truncated proteins) were incubated in kinase reaction buffer (20 mM Hepes, pH 7.4, 150 mM KCl, 10 nM MgCl2, 1 mM EGTA, 0.5 mM dithiothreitol, 5 mM NaF, 10 μM ATP, 5 µCi γ-32P) for 30 min at 30°C. Reactions were stopped by the addition of 2 × SDS sample buffer. Then samples were heated for 10 min to 95°C before analyses by SDS-PAGE and autoradiography.
Supplementary Material
Acknowledgments
This work was supported by the National Key Basic Research Program (973 Program) of MOST (Grant 2007CB914603), the Chinese National Natural Science Foundation (Grant 81071678, 81272994 and 30970677) and the Distinguished Youth Scientist Foundation of NFSC (Grant 30825011).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental material may be found here: www.landesbioscience.com/journals/cc/article/21770
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21770
Reference
- 1.Jackson RJ, Wickens M. Translational controls impinging on the 5'-untranslated region and initiation factor proteins. Curr Opin Genet Dev. 1997;7:233–41. doi: 10.1016/S0959-437X(97)80133-5. [DOI] [PubMed] [Google Scholar]
- 2.Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–63. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 3.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–18. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 4.Herbert TP, Tee AR, Proud CG. The extracellular signal-regulated kinase pathway regulates the phosphorylation of 4E-BP1 at multiple sites. J Biol Chem. 2002;277:11591–6. doi: 10.1074/jbc.M110367200. [DOI] [PubMed] [Google Scholar]
- 5.Sivan G, Elroy-Stein O. Regulation of mRNA Translation during cellular division. Cell Cycle. 2008;7:741–4. doi: 10.4161/cc.7.6.5596. [DOI] [PubMed] [Google Scholar]
- 6.Wilker EW, van Vugt MA, Artim SA, Huang PH, Petersen CP, Reinhardt HC, et al. 14-3-3sigma controls mitotic translation to facilitate cytokinesis. Nature. 2007;446:329–32. doi: 10.1038/nature05584. [DOI] [PubMed] [Google Scholar]
- 7.Svitkin YV, Herdy B, Costa-Mattioli M, Gingras AC, Raught B, Sonenberg N. Eukaryotic translation initiation factor 4E availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation. Mol Cell Biol. 2005;25:10556–65. doi: 10.1128/MCB.25.23.10556-10565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braunstein S, Karpisheva K, Pola C, Goldberg J, Hochman T, Yee H, et al. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol Cell. 2007;28:501–12. doi: 10.1016/j.molcel.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 9.Ramírez-Valle F, Badura ML, Braunstein S, Narasimhan M, Schneider RJ. Mitotic raptor promotes mTORC1 activity, G(2)/M cell cycle progression, and internal ribosome entry site-mediated mRNA translation. Mol Cell Biol. 2010;30:3151–64. doi: 10.1128/MCB.00322-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heesom KJ, Gampel A, Mellor H, Denton RM. Cell cycle-dependent phosphorylation of the translational repressor eIF-4E binding protein-1 (4E-BP1) Curr Biol. 2001;11:1374–9. doi: 10.1016/S0960-9822(01)00422-5. [DOI] [PubMed] [Google Scholar]
- 11.Greenberg VL, Zimmer SG. Paclitaxel induces the phosphorylation of the eukaryotic translation initiation factor 4E-binding protein 1 through a Cdk1-dependent mechanism. Oncogene. 2005;24:4851–60. doi: 10.1038/sj.onc.1208624. [DOI] [PubMed] [Google Scholar]
- 12.Petronczki M, Lénárt P, Peters JM. Polo on the Rise-from Mitotic Entry to Cytokinesis with Plk1. Dev Cell. 2008;14:646–59. doi: 10.1016/j.devcel.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 13.Barr FA, Silljé HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol. 2004;5:429–40. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 14.McKenzie L, King S, Marcar L, Nicol S, Dias SS, Schumm K, et al. p53-dependent repression of polo-like kinase-1 (PLK1) Cell Cycle. 2010;9:4200–12. doi: 10.4161/cc.9.20.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah OJ, Anthony JC, Kimball SR, Jefferson LS. 4E-BP1 and S6K1: translational integration sites for nutritional and hormonal information in muscle. Am J Physiol Endocrinol Metab. 2000;279:E715–29. doi: 10.1152/ajpendo.2000.279.4.E715. [DOI] [PubMed] [Google Scholar]
- 16.Renner AG, Créancier L, Dos Santos C, Fialin C, Recher C, Bailly C, et al. A functional link between polo-like kinase 1 and the mammalian target-of-rapamycin pathway? Cell Cycle. 2010;9:1690–6. doi: 10.4161/cc.9.9.11295. [DOI] [PubMed] [Google Scholar]
- 17.Avdulov S, Li S, Michalek V, Burrichter D, Peterson M, Perlman DM, et al. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell. 2004;5:553–63. doi: 10.1016/j.ccr.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 18.Ray ME, Yang ZQ, Albertson D, Kleer CG, Washburn JG, Macoska JA, et al. Genomic and expression analysis of the 8p11-12 amplicon in human breast cancer cell lines. Cancer Res. 2004;64:40–7. doi: 10.1158/0008-5472.CAN-03-1022. [DOI] [PubMed] [Google Scholar]
- 19.Rojo F, Najera L, Lirola J, Jiménez J, Guzmán M, Sabadell MD, et al. 4E-binding protein 1, a cell signaling hallmark in breast cancer that correlates with pathologic grade and prognosis. Clin Cancer Res. 2007;13:81–9. doi: 10.1158/1078-0432.CCR-06-1560. [DOI] [PubMed] [Google Scholar]
- 20.Armengol G, Rojo F, Castellví J, Iglesias C, Cuatrecasas M, Pons B, et al. 4E-binding protein 1: a key molecular “funnel factor” in human cancer with clinical implications. Cancer Res. 2007;67:7551–5. doi: 10.1158/0008-5472.CAN-07-0881. [DOI] [PubMed] [Google Scholar]
- 21.Kremer CL, Klein RR, Mendelson J, Browne W, Samadzedeh LK, Vanpatten K, et al. Expression of mTOR signaling pathway markers in prostate cancer progression. Prostate. 2006;66:1203–12. doi: 10.1002/pros.20410. [DOI] [PubMed] [Google Scholar]
- 22.Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8:567–72. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Zheng XF. mTOR-independent 4E-BP1 phosphorylation is associated with cancer resistance to mTOR kinase inhibitors. Cell Cycle. 2012;11:594–603. doi: 10.4161/cc.11.3.19096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Azar R, Susini C, Bousquet C, Pyronnet S. Control of contact-inhibition by 4E-BP1 upregulation. Cell Cycle. 2010;9:1241–5. doi: 10.4161/cc.9.7.11047. [DOI] [PubMed] [Google Scholar]
- 25.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157–62. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 26.Barnhart BC, Lam JC, Young RM, Houghton PJ, Keith B, Simon MC. Effects of 4E-BP1 expression on hypoxic cell cycle inhibition and tumor cell proliferation and survival. Cancer Biol Ther. 2008;7:1441–9. doi: 10.4161/cbt.7.9.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan K, Huang Y, Yao X. Illumination of mitotic orchestra during cell division: a Polo view. Cell Signal. 2011;23:1–5. doi: 10.1016/j.cellsig.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang SM, Song M, Yang TY, Fan R, Liu XD, Zhou PK. HIV-1 Tat impairs cell cycle control by targeting the Tip60, Plk1 and cyclin B1 ternary complex. Cell Cycle. 2012;11:1217–34. doi: 10.4161/cc.11.6.19664. [DOI] [PubMed] [Google Scholar]
- 29.Beugnet A, Wang X, Proud CG. Target of rapamycin (TOR)-signaling and RAIP motifs play distinct roles in the mammalian TOR-dependent phosphorylation of initiation factor 4E-binding protein 1. J Biol Chem. 2003;278:40717–22. doi: 10.1074/jbc.M308573200. [DOI] [PubMed] [Google Scholar]
- 30.Constantinou C, Elia A, Clemens MJ. Activation of p53 stimulates proteasome-dependent truncation of eIF4E-binding protein 1 (4E-BP1) Biol Cell. 2008;100:279–89. doi: 10.1042/BC20070121. [DOI] [PubMed] [Google Scholar]
- 31.Martineau Y, Azar R, Bousquet C, Pyronnet S. Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene. 2012 doi: 10.1038/onc.2012.116. In Press. [DOI] [PubMed] [Google Scholar]
- 32.Mundt KE, Golsteyn RM, Lane HA, Nigg EA. On the regulation and function of human polo-like kinase 1 (PLK1): effects of overexpression on cell cycle progression. Biochem Biophys Res Commun. 1997;239:377–85. doi: 10.1006/bbrc.1997.7378. [DOI] [PubMed] [Google Scholar]
- 33.Martín M, Rodríguez-Lescure A, Ruiz A, Alba E, Calvo L, Ruiz-Borrego M, et al. GEICAM 9906 Study Investigators Randomized phase 3 trial of fluorouracil, epirubicin, and cyclophosphamide alone or followed by Paclitaxel for early breast cancer. J Natl Cancer Inst. 2008;100:805–14. doi: 10.1093/jnci/djn151. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed AA, Lu Z, Jennings NB, Etemadmoghadam D, Capalbo L, Jacamo RO, et al. Australian Ovarian Cancer Study Group SIK2 is a centrosome kinase required for bipolar mitotic spindle formation that provides a potential target for therapy in ovarian cancer. Cancer Cell. 2010;18:109–21. doi: 10.1016/j.ccr.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spänkuch B, Heim S, Kurunci-Csacsko E, Lindenau C, Yuan J, Kaufmann M, et al. Down-regulation of Polo-like kinase 1 elevates drug sensitivity of breast cancer cells in vitro and in vivo. Cancer Res. 2006;66:5836–46. doi: 10.1158/0008-5472.CAN-06-0343. [DOI] [PubMed] [Google Scholar]
- 36.Balakumaran BS, Porrello A, Hsu DS, Glover W, Foye A, Leung JY, et al. MYC activity mitigates response to rapamycin in prostate cancer through eukaryotic initiation factor 4E-binding protein 1-mediated inhibition of autophagy. Cancer Res. 2009;69:7803–10. doi: 10.1158/0008-5472.CAN-09-0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chao SK, Lin J, Brouwer-Visser J, Smith AB, 3rd, Horwitz SB, McDaid HM. Resistance to discodermolide, a microtubule-stabilizing agent and senescence inducer, is 4E-BP1-dependent. Proc Natl Acad Sci USA. 2011;108:391–6. doi: 10.1073/pnas.1016962108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shang ZF, Huang B, Xu QZ, Zhang SM, Fan R, Liu XD, et al. Inactivation of DNA-dependent protein kinase leads to spindle disruption and mitotic catastrophe with attenuated checkpoint protein 2 Phosphorylation in response to DNA damage. Cancer Res. 2010;70:3657–66. doi: 10.1158/0008-5472.CAN-09-3362. [DOI] [PubMed] [Google Scholar]
- 39.Huang S, Shu L, Dilling MB, Easton J, Harwood FC, Ichijo H, et al. Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21(Cip1) Mol Cell. 2003;11:1491–501. doi: 10.1016/S1097-2765(03)00180-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.