

Abstract
The circadian clock is a global regulatory mechanism that confers daily rhythmicity on many biochemical and physiological functions, including DNA excision repair in mammalian organisms. Here, we investigated the effect of the circadian clock on the major DNA damage response pathways by using mouse cell lines mutated in genes encoding proteins in the positive (Bmal1, CLOCK) or negative (Cry 1/2, Per 1/2) arms of the transcription-translation feedback loop that generates the circadian clock. We find that cells mutated in these genes are indistinguishable from wild-type in their response to UV, ionizing radiation and mitomycin C. We conclude that either the majority of DNA damage response reactions are not controlled by the circadian clock or that, even if such a control exists at the organism level, it is supplanted by homeostatic control mechanisms at the cellular level in tissue culture. We suggest that caution must be exercised in extrapolating from experiments in tissue culture to whole animals with respect to the effect of the circadian clock on cellular response to DNA damaging agents.
Keywords: Cryptochrome, Period, Clock, BMal1, DNA repair, checkpoint, apoptosis
Introduction
Circadian rhythm is the oscillation of biochemical, physiological and behavioral functions of organisms with daily periodicity. In mammals, the rhythm is generated by an endogenous self-sustaining molecular clock consisting of positive and negative transcription factors that operate on the basis of a transcription-translational feedback loop (TTFL).1 The positive factors are BMAL1 and CLOCK, and the negative factors are CRY 1/2 and PER 1/2. The core clock, either through regulating other genes or interacting with proteins in other pathways, interfaces with all major cellular biochemical pathways.2 As a consequence, disruption of the circadian clock has been reported to affect various physiopathological states, ranging from sleep to metabolism to cancer.2
With regard to the DNA damage response, it has been reported that the ATR-mediated DNA damage checkpoint directly interfaces with the core clock mechanism through interactions with the clock proteins timeless and cryptochrome.3 Similarly, it was reported that the ATM-mediated DNA damage checkpoint interfaces with the clock through interaction of PER1 with ATM and CHK2.4 Finally, recently we reported that the expression of a key protein in mammalian nucleotide excision repair, XPA, is controlled by the circadian clock in mouse brain, liver and skin.5-7 In addition, in a recent siRNA screen for genes whose downregulation causes sensitivity to genotoxic agents, it was found that CLOCK downregulation rendered cells sensitive to IR, which causes double-strand breaks, and to mitomycin C, which produces interstrand cross-links.8 Moreover, when cells were focally irradiated with laser, CLOCK accumulated at the damage site along with other proteins known to be involved in processing double-strand breaks, consistent with the notion that CLOCK may directly participate in double-strand break (dsb) repair or dsb-intitiated checkpoint signaling. In addition to these studies at cellular and biochemical levels, a number of epidemiological and clinical studies, and a few studies with animal model systems, have also indicated that the circadian clock might be an important determinant in cellular/organismal response to genotoxic agents.2
With these considerations, we then decided to systematically analyze the circadian clock-DNA damage response connection, specifically, because conflicting data exist from cellular, animal and clinical studies regarding the magnitude of the clock effect on DNA damage response using cells or animals with a particular clock gene mutation.9 We reasoned that if cell lines with mutations in all individual clock genes were compared under, to the extent possible, identical conditions, a better insight may be gained about the clock-DNA damage response connection. Hence, in this study we have used cell lines mutated in each of the clock genes, and, moreover, we have tested three groups of DNA damaging agents that produce bulky adducts, double-strand breaks and interstrand crosslinks that are repaired by three major repair pathways: nucleotide excision repair, double-strand break repair and interstrand crosslink repair.10 We find that cell lines with mutations in the positive arm (Bmal1 and Clock) of the core clock and mutations in the negative arm (Cry1/2 and Per1/2) are indistinguishable from their wild-type counterparts for all the DNA damage response endpoints we tested, including cell survival, repair, checkpoint activation and apoptosis. We conclude that, at least at cell culture level, either the clock does not affect cellular response to DNA damage, or homeostatic mechanisms in tissue culture override any circadian effect that might exist on DNA damage response at the organism level. Hence, we suggest that caution must be exercised in extrapolating from cell culture studies with clock mutants to genotoxic responses at the animal level.
Results
Effect of clock gene mutations on cellular survival to UV, ionizing radiation and mitomycin C
Three separate studies have linked the clock to responses to UV-, ionizing radiation- and mitomycin C-induced DNA damage by using one of the clock mutant animals or cell lines.5,8,11 To address this question comprehensively, we used Clock−/−, Bmal1−/−, Cry1/2−/−, Per1/2-/- mouse cell lines, along with isogenic wild-type cell lines in clonogenic survival assays.
Figure 1A and B show the UV survival of the mutants in the positive and negative arms of the clock, respectively. As apparent from the figure, mutations in clock genes do not affect clonogenic UV survival. Because it has been shown that in mouse brain, liver and skin, the XPA protein and nucleotide excision repair exhibit robust circadian rhythmicity5-7 and that excision repair is the major determinant of UV survival,10 these results were somewhat unexpected. Hence, we performed in vivo repair assays in the mutant cell lines to determine if, despite altered repair activity, the clock mutants retained the same survival properties. Figures 2 and 3 show that the rates of repair of the two major UV photoproducts, cyclobutane pyrimidine dimers and (6–4) photoproducts are not affected by mutations in either the positive arm (Fig. 2) or the negative arm (Fig. 3) of the circadian clock, which explains the lack of clock effect on survival. We reasoned that the lack of effect of clock gene mutations on repair may be due to loss of clock control of XPA and, hence, excision repair in tissue culture in which homeostatic gene control might be dominating. Hence, we tested the level of Xpa transcripts in wild-type and Cry1/2−/− mutant cell lines. In contrast to the finding in Cry1/2−/− mouse tissues, where XPA levels are constitutively high at all circadian time points,5 in the cell lines the levels of XPA in Cry1/2−/− mutant are indistinguishable from the wild-type (Fig. S1). This finding is contrary to expectations that removal of a negative regulator would lead to constitutively high Xpa transcription and XPA protein level, but is in agreement with the recent realization that in tissue culture, the circadian control of genes other than core clock genes disappear and circadian effect on cell physiology is blunted.12

Figure 1. Mouse cells deficient in clock genes are not sensitive to UV irradiation. Cells were plated at a low density, allowed to attach and then UV irradiated; after colony formation, cells were stained with Giemsa, and percent survival was determined as described in the text. Each data point represents the average of at least three independent experiments and bars signify the standard deviation. (A) Positive arm of the transcription-translational feedback loop (TTFL). Mouse embryonic fibroblasts were derived from mice with the following genotypes: wild-type (squares), Clock−/− (circles) and Bma1l−/− (triangles). (B) Negative arm of the TTFL. Mouse skin fibroblasts were derived from mice with the following genotypes: wild-type (squares), Cry1−/−Cry2−/− (circles) and Per1−/−Per2−/− (triangles).

Figure 2. Excision repair capacity of UV-induced photoproducts in mouse cells deficient in positive feedback loop of the core molecular clock. Exponentially growing, primary mouse embryonic fibroblast cells were irradiated with 5 J/m2 UVC, allowed to repair from 0–24 h, lysed and genomic DNA collected for immuno slot blot analysis. (A) An immuno slot blot assay was used to measure (6–4) PP repair at various time points post-irradiation with UV light. The image shows the (6–4) PP signal detected with an anti-(6–4) PP antibody and SYBR gold staining to show equal loading of total genomic DNA. (B) Quantitative analysis of the repair assay from (A) and experiments conducted under identical conditions. (C) An immuno slot blot assay was used to measure CPD repair at various time points, post-irradiation with UV light. The image shows the CPD signal detected with an anti-CPD antibody and SYBR gold staining to show equal loading of total genomic DNA. (D) Quantitative analysis of the repair assay from (C) and experiments conducted under identical conditions. The averages and standard deviations are from at least three independent experiments.

Figure 3. Excision repair capacity of UV-induced photoproducts in mouse cells deficient in negative feedback loop of the core molecular clock. Exponentially growing primary mouse skin fibroblast cells were irradiated with 5 J/m2 UVC, allowed to repair from 0–24 h, lysed and genomic DNA collected for immuno slot blot analysis. (A) Immuno slot blot assay for measuring (6–4) PP repair at various time points post-irradiation with UV light. The image shows the (6–4) PP signal detected with an anti-(6–4) PP antibody and SYBR gold staining to show equal loading of total genomic DNA. (B) Quantitative analysis of the repair assay from (A) and two additional assays conducted under identical conditions. (C) Immuno slot blot assay for measuring CPD repair at various time points, post-irradiation with UV light. The image shows the CPD signal detected with an anti-CPD antibody and SYBR gold staining to show equal loading of total genomic DNA. (D) Quantitative analysis of the repair assay from (C) and two additional assays conducted under identical conditions. The averages and standard deviations are from at least three independent experiments.
Similar to UV damage, survival following DNA damage caused by IR11 and mitomycin C8 are reported to be modulated by the circadian clock. Hence, we tested the four core clock gene mutants for their sensitivity to IR, which kills cells mainly by introducing double-strand breaks, and mitomycin C, which kills cells mainly by introducing interstrand crosslinks as evidenced by the sensitivity of XP-F mutant cells (Fig. S2), which are known to be defective in crosslink repair.10 The results with Clock mutants are shown in Figures 4 and 5. As is apparent, clock gene mutations do not affect the sensitivity of cell lines to these genotoxic agents, consistent with previous reports on the effect of Cry1/2 −/− mutation on cellular survival to UV and IR.13,14 These results are also consistent with the view that the effect of the circadian clock on clock-controlled genes in tissue culture is limited and supplanted by homeostatic regulatory systems.12
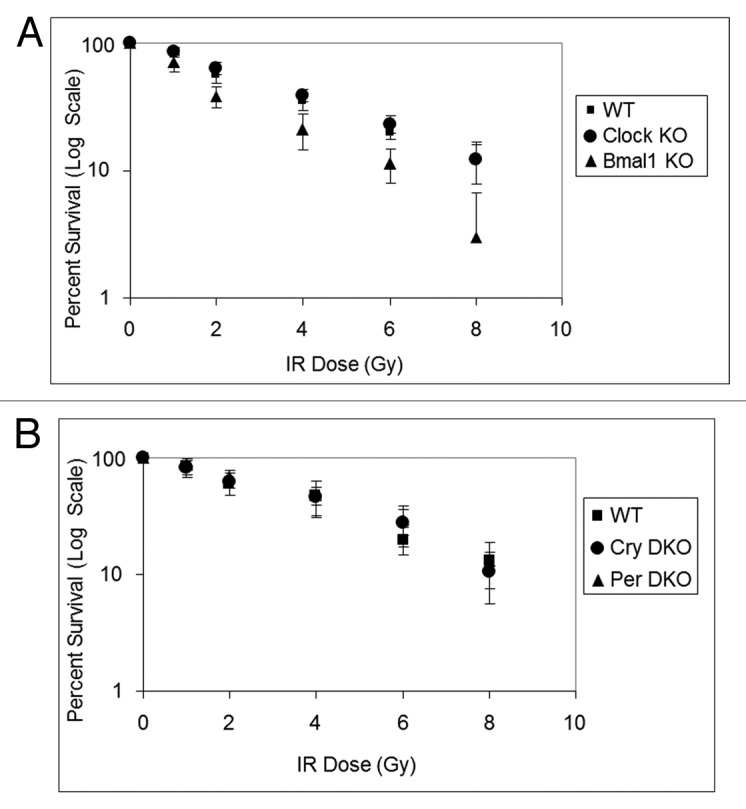
Figure 4. Mouse cells deficient in clock genes are not sensitive to IR irradiation. Cells were plated at low density, allowed to attach and then IR irradiated; after colony formation, cells were stained with Giemsa and percent survival was determined as described in the text. Each data point represents the average of at least four independent experiments and bars signify the standard deviation. (A) Mouse embryonic fibroblasts were derived from mice with the following genotypes: wild-type (squares), Clock−/− (circles) and Bmal1−/− (triangles). (B) Mouse skin fibroblasts were derived from mice with the following genotypes: wild-type (squares), Cry1−/−Cry2−/− (circles) and Per1−/−Per2−/− (triangles).
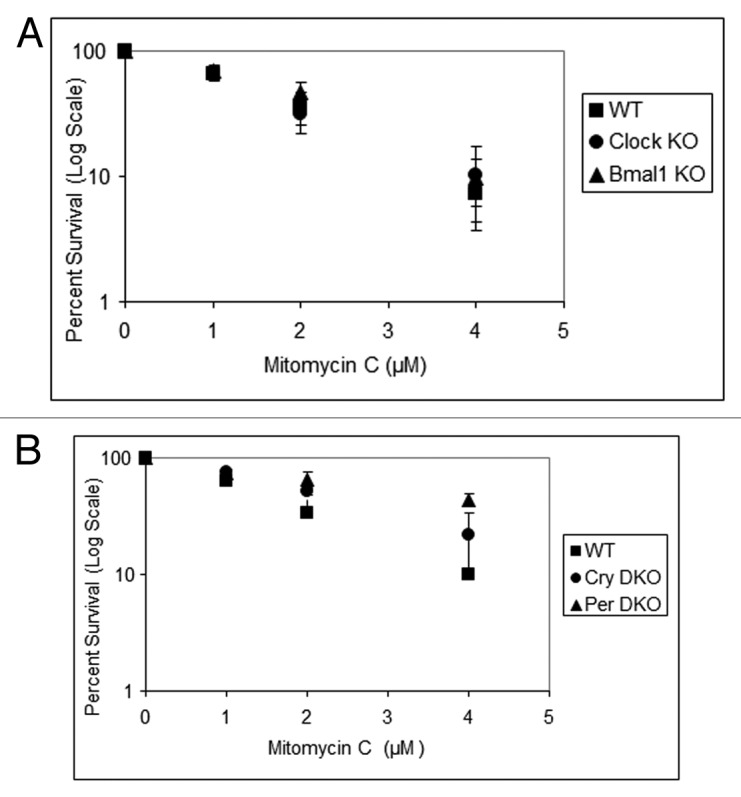
Figure 5. Mouse cells deficient in clock genes are not sensitive to mitomycin C. Cells were plated at a low density, allowed to attach and then treated with mitomycin C (MMC); after colony formation, cells were stained with Giemsa, and percent survival was determined as described in the text. Each data point represents the average of at least three independent experiments and bars signify the standard deviation. (A) Mouse embryonic fibroblasts were derived from mice with the following genotypes: wild-type (squares), Clock−/− (circles) and Bmal1−/− (triangles). (B) Mouse skin fibroblasts were derived from mice with the following genotypes: wild-type (squares), Cry1−/−Cry2−/− (circles) and Per1−/−Per2−/− (triangles).
Effect of clock disruption on DNA damage checkpoint response
In addition to DNA repair, DNA damage checkpoint and apoptosis are the two major cellular responses to DNA damage that dictate cellular fate after genotoxic stress.10 Therefore, as part of our efforts to determine the effect of the clock on cellular responses to DNA damage, we also analyzed checkpoints and apoptosis in clock-disrupted cells. We found that various mutant cell lines isolated in different labs expressed different basal levels of checkpoint and apoptosis proteins that made comparison with the wild-type problematic after genotoxic stress. Therefore, we analyzed the contribution of the circadian clock to these responses by using primary mouse skin fibroblasts and downregulating clock protein expression by siRNA. The results for the UV-induced checkpoint are shown in Figure 6. It is evident that downregulation of any of these clock proteins (Fig. 6A and C) does not affect UV-induced Chk1 phosphorylation (Fig. 6A–D). Interestingly, and in agreement with the “standard model” for the circadian clock,1 downregulation of CRY1 leads to overexpression of CRY2 and PER2, while downregulation of PER1 leads to upregulation of PER2 and a moderate upregulation of CRY1. While these findings are of some relevance for the “standard model,” from the perspective of this study, it is clear that downregulation of clock genes either in the positive arm or the negative arm of the core clock circuitry does not affect the UV-induced checkpoint as determined by the kinetics of Chk1 phosphorylation in primary mouse skin fibroblasts.

Figure 6. Effect of UV induced Chk1 activation by reduced levels of circadian clock genes in mouse cells. siRNA transfected, primary mouse skin fibroblast cells 72 h later were irradiated with 5 J/m2 UVC, allowed to grow for 0, 0.5 or 1 h and proteins lysates were prepared for immunoblot analysis. (A and C) indicated proteins were analyzed by immunoblotting with appropriate antibodies at various times after UV damage. (B and D) quantitative analysis of the immunoblots. The averages and standard deviations are from at least three independent experiments.
Effect of clock gene downregulation on apoptosis
Next, we tested the effect of downregulating clock proteins on apoptosis (Fig. 7). We found that CLOCK and BMAL1 downregulation moderately enhances apoptosis at low UV dose but not at the high dose (Fig. 7A and B). The significance of this finding is not apparent at present. When we downregulated CRY1/2, the expression level of PER1/2 was elevated, but there was no significant effect on apoptosis as measured by PARP and caspase-3 cleavage (Fig. 7C and D). PER1/2 downregulation caused reduced levels of CRY1 and, similarly, reduced PARP-1 and caspase-3 cleavage at all UV doses tested (Fig. 7C and D). Taken together, the results lead us to conclude that PER1/2 downregulation reduces apoptosis, but the significant PER1/2 upregulation observed in CRY1/2-downregulated cells does not lead to enhanced apoptosis.

Figure 7. Effect of UV-induced apoptosis by reduced levels of circadian clock genes in mouse cells. siRNA transfected primary mouse skin fibroblast cells were irradiated with 0, 5, 10 or 25 J/m2 UVC, allowed to grow for 24 h and protein lysates were prepared for immunoblot analysis. (A) Protein levels were analyzed by immunoblotting 24 h after UV damage with the indicated antibodies. (B) Quantitative analysis of the immunoblot from three experiments. (C) Protein levels were analyzed by immunoblotting 24 h after UV damage with the indicated antibodies. (D) Quantitative analysis of the immunoblots. Averages and standard deviations are from at least three independent experiments. C-PARP and C-Caspase-3 stand for cleaved PARP and cleaved capsase-3, respectively.
Effect of PER1 downregulation or overexpression on ATM-Chk2 mediated DNA damage checkpoints and apoptosis
Both PER1 and PER2 proteins have been implicated in IR-induced apoptosis. In particular, it has been reported that PER1 interacts with ATM and Chk2 proteins and directly participates in the double-strand break activated ATM→Chk2 signaling pathway in human cell lines.4 Further, it was reported that downregulation of PER1 conferred resistance to apoptosis by IR and radiomimetic agents, and that overexpression conferred sensitivity to IR in a number of human cancer cell lines.4 We wished to reassess these findings in light of our other data reported in this study.
First, we tested the effect of PER1 downregulation and overexpression in the NCI-H460 human cancer cell line that was used in the previous study.4 The results of the effect of PER1 downregulation on apoptosis are shown in Figure 8A. In contrast to the findings reported in the previous study,4 we find that downregulation of PER1 does not affect IR-induced ATM signaling as measured by Chk2 and p53 phosphorylation, nor does it affect apoptosis as measured by PARP-1 cleavage. (Fig. 8A and B).

Figure 8. PER1 expression neither sensitizes human lung cancer cells to apoptosis nor affects Chk2 phosphorylation following ionizing radiation (IR). siRNA transfected NCI-H460 human lung cancer cells were irradiated with 0, 10 or 30 Gy of IR and allowed to grow for 48 h, protein lysates were prepared for immunoblot analysis. (A) Protein levels were analyzed by immunoblotting 48 h after irradiation with IR with the indicated antibodies from NCI-H460 cells transfected with either negative control (non-target, NT) or Per1 siRNAs. (B) Quantitative analysis of the immunoblots. (C) Protein levels were analyzed by immunoblotting 48 h after irradiation with IR with the indicated antibodies from NCI-H460 cells transfected with either empty vector (pCDNA3) or a mPer1-overexpressing vector for 72 h. (D) Quantitative analysis of the immunoblots. Averages and standard deviations are from at least three independent experiments. T-PARP and C-PARP stand for total PARP and cleaved PARP, respectively.
Next, we tested the effect of PER1 overexpression on ATM-mediated checkpoint and apoptosis by transfecting cells with a PER1-expressing vector and subjecting them to ionizing radiation followed by analysis by western blotting for target proteins to assess checkpoint activation and apoptosis. The results are shown in Figure 8C and D. As is apparent from Figure 8C, under our experimental conditions, transfection of NCI-H460 cells with Per1 expression vector leads to significant overproduction of PER1. However, when these cells were exposed to IR and analyzed for checkpoint activation as measured by Chk2 phosphorylation and for apoptosis as measured by PARP1 cleavage, they were indistinguishable from the control, vector-transfected cells at two different IR doses used (Fig. 8C and D). Thus, we conclude that neither downregulation, nor overexpression of PER1, affect the IR-induced ATM→Chk2 pathway or apoptosis.
Effect of PER1 levels on clonogenic survival to IR of human cancer cell lines
To further reassess the effect of PER1 expression level on the IR effect on human cancer cell lines, we performed clonogenic survival assays. Both the NCI-H460 human lung cancer cell line and the HCT-116 human colorectal cancer cell line that were used in the previous study4 were transfected with Per1 siRNA, which reduced PER1 levels, or transfected with PER1 expression vector, which overexpressed the PER1 protein, plated and irradiated with increasing doses of IR. Figure 9 shows the results of this colony formation assay, which reveals that neither downregulation nor overexpression of PER1 affects the survival of these two human cancer cell lines against IR.

Figure 9. PER1 expression does not sensitize human cancer cells to ionizing radiation in a colony formation assay. Cells were transfected with indicated siRNAs or expression vectors and exposed to IR, and IR survival was measured by colony formation assay. (A) Colony formation assay results for NCI-H460 human lung cancer cells. After 48 h of siRNA treatment, cells were trypsinized and plated at low density (500 cells per dish) in 6-well culture dish plates. After allowing cells to attach for 8 h, they were irradiated with the indicated fluence of IR and allowed to grow in fresh growth media for 7–10 d. Colonies (> 50 cells) were counted using a digital counter after staining with Giemsa and the surviving cell fraction was calculated. (B) Colony-formation assay results for HCT-116 human colon cancer cells. After 48 h of siRNA treatment, cells were trypsinized and plated at low density in 6-well plates. The results are the average of three biological experiments and error bars represent the standard deviation of the mean.
Discussion
The circadian clock system is so intimately integrated into all the metabolic and signaling networks that even in pathways where a clear link with the clock has not been established, it is intuitive to think that the clock would affect and be affected by all physiological functions. As a corollary, it is commonly thought that clock disruption would be a factor in essentially all pathophysiological phenomena, including sleep disorders, obesity, cardiovascular diseases and cancer. Because cancer is caused by abnormal DNA damage response and cell cycle disregulation, attempts have been made to correlate circadian clock disruption with increased cancer risk. In support of this notion, three types of studies have been performed: epidemiology of human populations, carcinogenesis in animal models and analysis of DNA damage response in cell lines with mutations in clock genes. In the following section we will briefly summarize these previous studies to put our current work in context.
Epidemiological studies
Several studies have reported that night shift or rotating shift workers and flight attendants on international/transcontinental flights have increased risk of various cancers, including breast and colon cancer, and correlations were reported between the length of rotating shift work and cancer.15-19 While these studies are important from a public health perspective, they do not establish a cause and effect relationship, nor do they provide a mechanistic insight into the clock disruption and cancer. Indeed, it has been argued that night shift/rotating shift work and cancer are two independent outcomes of certain personality traits.20 In support of this argument, it was reported that in a large study on Chinese women, the incidence of breast cancer in night shift/rotating shift workers was actually lower than the breast cancer incidence in women with conventional working schedules.21 Hence, when all factors are taken into account, the epidemiological data fall short of establishing causality between circadian clock disruption and elevated risk of cancer, and the epidemiology must be supported by cellular and molecular data to link clock disruption to cancer.
Animal model studies
Two types of animal studies have been performed to investigate the effect of circadian clock disruption on carcinogenesis. In one study, the suprachiasmatic nucleus (SCN), which is the master circadian clock that coordinates all peripheral clocks, was surgically destroyed, and then tumor implants were placed into these arrhythmic mice, and growth of the implants was monitored.22 In the second group of experiments, the circadian clock was disrupted by mutating one of the core clock genes, and the incidence of spontaneous and DNA damage-induced cancers was monitored.11,13,23-25
In mice in which the circadian clock was disrupted by SCN lesioning, both the pancreatic carcinoma and Glasgow osteosarcoma xenografts grew at a faster rate than the xenografts in mice with intact SCN.22 This was taken as evidence that clock disruption promotes cancer growth. However, as the clock was disrupted only in the SCN of the host mice, it is not known whether the implanted tumors had an intrinsic clock, and whether that clock was influenced by the host circadian clock. Hence, based on all these uncertainties, the implications of this study of clock disruption-carcinogenesis are problematic. In a related study, when Clock gene expression was downregulated in Lewis lung carcinoma (LLC1) cells by siRNA and then the cells were inoculated subcutaneously into mice, the resulting tumor tissue grew at a slower rate than the control, suggesting that in this experimental system, clock disruption actually interfered with tumor growth.25 However, it could be argued that clock disruption by CLOCK mutation stops the circadian clock at around midnight (when CLOCK-BMAL1 activity is at its minimum), and hence, stopping the clock by other means and at other times of the biological time may differentially affect tumor growth. This qualification notwithstanding, based on this evidence alone, it cannot be generalized that clock disruption, by whatever means, promotes tumorigenesis, and whether clock disruption facilitates tumorigenesis may depend on the means by which the clock is stopped and at what point in the circadian cycle the clock is stopped.
Genetic models
In the second group of animal studies, one (or both isoforms when they exist) of the clock genes was knocked out genetically, and in these genetically engineered mice, the incidence of spontaneous and DNA damage-induced cancers was monitored. In the first such experiment it was reported that Per2-knockout mice were predisposed to spontaneous and IR induced cancers.11 This was ascribed to elevated c-Myc expression and reduced p53 levels, which together were presumed to promote cell proliferation and prevent apoptosis. In view of these findings then, the results came as a surprise when Cry1/2−/− mice were tested for tumorigenesis13: CRY and PER proteins comprise the negative arm of the TTFL that constitutes the core circadian clock, and hence, to a first approximation, Per and Cry mutants are expected to behave similarly. Contrary to this expectation, Cry1/2−/− mutant mice were indistinguishable from wild-type mice with regard to incidence of spontaneous and IR-induced cancers.13 It was concluded that although PERs and CRYs collaborate in inhibiting BMAL1-CLOCK activated transcription, they may also participate in reactions unique to each protein. However, there was also the possibility that Cry mutation, for whatever reason, had a weaker phenotype than the Per2 mutant, and that in an appropriate background, Cry mutation may also enhance the cancer incidence. With these considerations in mind then, Cry1/2−/−p53−/− mutant mice were generated and tested.26 The p53−/− mutant mice had high incidence of lymphomas and essentially all animals died within 5 mo.27 When these mice are crossed with a weakly penetrant cancer promoting mutation, such as RasG12D, the rate of cancer incidence accelerates.28 Hence, it was suspected that Cry1/2−/− mutation had a weakly penetrant cancer-promoting phenotype that by itself is not evident, but would become apparent when combined with p53−/− mutation. In fact, the results were quite the opposite: the Cry1/2−/−p53−/− mutants develop cancer later and live about 1.5-fold longer than the p53−/− mutants.26 At the cellular level, as will be elaborated below, it was found that Cry1/2−/−p53−/− cells were more sensitive to apoptosis by UV-mimetic agents than p53−/− cells, and therefore, it was concluded that the lack of CRY prevented/delayed cancer, not by preventing malignant transformation, but by actually promoting p53-independent apoptosis of cancerous cells (immortalized and transformed). Although this conclusion was questioned in a subsequent study23 that claimed mutation in any of the core clock genes (Cry, Per, Clock and Bmal1) predisposed mice to cancer, that study itself has been questioned on grounds of employing mice of different genetic background for comparison,9 and therefore, it will not be further elaborated on here.
In follow-up work, to understand the late onset of cancer in p53−/−Cry1/2−/− mice compared with p53−/− mice, it was found that p53−/−Cry1/2−/− cells were more sensitive to both intrinsic and extrinsic apoptosis than p53−/− cells, making cancer cells with the triple mutation more sensitive to both genotoxic agents29 and cytokines30 compared with p53−/− cells. To summarize, Cry mutation does not predispose mice to cancer, and, in fact, in the appropriate genetic background, Cry mutation may reduce cancer risk.
Finally, analyses of Clock and Bmal1 mutants, which constitute the positive arm of the TTFL that make up the core clock, has also revealed that these mutations do not predispose animals to cancer.24,25 Bmal1, in fact, among all clock genes, exhibits the strongest non-circadian phenotype in the form of early aging, but even this aging phenotype is not associated with an increased incidence of cancer.24
Cell biology and in vivo biochemistry
The first biochemical study on the link between DNA damage checkpoint and the clock was performed to examine the contribution of timeless to the ATR→Chk1 signaling pathway.3 Although timeless is a core clock gene in Drosophila,1 it has been difficult to establish its clock role in mice, because in mammals it appears to be an essential gene.31 Nevertheless, studies from multiple directions have indicated that timeless in mammals plays a role both in the circadian clock3,31 and in replication/S-phase checkpoint.32,33 In an attempt to understand the mechanism of the timeless checkpoint function, its interactions with checkpoint proteins were analyzed. It was found that cryptochrome 1 bound to timeless, which, in turn, bound to ATR and Chk1.3 Most importantly, it was also found that downregulation of timeless disrupted both the circadian clock in SCN slices31 and the ATR→Chk1 signal transduction pathway in tissue culture cells.3
In a parallel set of experiments, it was reported that Per1 interacted with both ATM and Chk2, and that this interaction was enhanced by ionizing radiation and was essential for IR-induced ATM→Chk2 signaling.4 In support of this, it was reported that downregulation of Per1 interfered with ATM→Chk2 signaling and IR-induced apoptosis, and that overproduction of Per1 sensitized a variety of human cancer cell lines to apoptosis.4 However, these seemingly rather strong links between Per1 and checkpoint and apoptosis have not been reproduced.34,35 In fact, it was reported that in the human gingival cancer cell line CA9–22, pancreatic cancer lines MTA PCa-2 and PANC-1 and the hepatocellular carcinoma cell line, HepG2, Per1 acts as an anti-apoptotic factor, as evidenced by the fact that Per1 siRNA reduced both spontaneous and cisplatin-induced apoptosis and, depending on the cell line, either marginally decreased or increased the sensitivity of the cell line to cisplatin induced killing.34-36 Consistent with these findings, in this paper, we find that neither upregulation nor downregulation of Per1 affects IR- induced survival, apoptosis or ATM→Chk2 checkpoint signaling in human cancer cell lines used in the study reporting a Per1-checkpoint connection4 nor in mouse Per1- or Per2-knockout cell lines.
In contrast to these reports, which have not yielded a consistent model for a Per1/2-DNA damage response connection, studies with Cry mutants have produced a consistent model regarding the clock and DNA repair, apoptosis, carcinogenesis and cancer treatment. First, it is now well established that nucleotide excision repair in mice is under strong circadian control, reaching a maximum in subjective dusk and a minimum in subjective dawn.5-7 Second, Cry-null mutations of both Cry isoforms in otherwise “normal” immortalized or primary mouse fibroblasts does not affect any of the DNA damage responses, including repair, checkpoint signaling, apoptosis or clonogenic survival.26 In contrast, in Ras-transformed, p53-null mutant cell lines, Cry downregulation or Cry-null mutation makes cells sensitive to oxaliplatin-induced apoptosis and clonogenic killing by activating p73 expression through clock-controlled Egr1 transcription factor.29 Similarly, these cells are sensitive to TNFα-induced apoptosis and clonogenic death because of clock-controlled downregulation of anti-apoptotic activity of NFκB.30
In summary, while it is intuitive to suspect that a global regulatory system such as the circadian clock would interface with cellular responses to intrinsic and extrinsic carcinogenic stimuli, at present there is no compelling evidence to presume that clock disruption predisposes mice or humans to cancer. However, it is also evident that DNA repair and at least skin carcinogenesis have a strong circadian component,5-7 and that in the appropriate genetic background, the clock does affect cellular responses to DNA damage.26,29,30 Finally, we wish to note that while cell-based in vitro circadian systems have been quite useful for elaborating the basic clockworks in mammalian cells,37 these systems have serious limitations with regard to studying outputs that are clock-controlled at animal level but not at tissue culture, and hence, results based on tissue culture must be interpreted in this light.
Materials and Methods
Cell culture, siRNA, pCDNA3 and mPer1 transfections
Primary mouse skin fibroblasts (MSFs) were isolated as described26 using skin biopsies from wild-type, Cry1−/−Cry2−/− and Per1−/−Per2−/− mice.38,39 Primary mouse embryonic fibroblasts (MEFs) from wild-type, Bmal1−/− and Clock−/− mice24,25 were a gift from M.P. Antoch (Roswell Park Cancer Institute). Cells were cultured in a humidified incubator at 37°C under 5% CO2. Mouse fibroblast cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM-high glucose) supplemented with 10% fetal bovine serum, penicillin at 50 U/ml and streptomycin at 50μg/ml. NCI-H460 and HCT-116 (obtained from ATCC) cell lines were grown in RPMI-1640 medium or McCoy’s 5A medium (GIBCO) supplemented with 10% FBS. For siRNA transfections, exponentially growing cells were transfected either with human Per1, Per2, mouse Per1, Per2, Cry1, Cry2, Clock and Bmal1, and Cyclophilin B siRNAs (Santa Cruz) or non-target siRNA (Dharmacon) using the Lipofectamine RNAiMax (Invitrogen) transfection reagent for 48 h according to the manufacturer’s directions before UV or IR irradiation. pCDNA3 or V5- tagged mouse Per1 genes were transfected using the Lipofectamine 2000 (Invitrogen) transfection reagent for 48 h according to the manufacturer’s directions as described previously.40
Clonogenic UV survival assays
Trypsinized cells were plated such that there would be 150–200 viable cells per 10 cm2 dish. For each experiment, cells were plated in triplicate dishes for each UV dose (0, 2, 4, 6, 8 J/m2). Cells were allowed to attach for 4–5 h; media was removed, and cells were washed once with phosphate buffered saline or Hank’s balanced salt solution (Gibco). Wash buffer was removed, and cells were irradiated with a germicidal lamp emitting primarily at 254 nm. The UV fluence was determined with a UVX digital radiometer, and cells were irradiated at a dose rate of 1.2–1.3 J/m2/s (120–130 μW/cm2). Conditioned media was added back to the cells and the cells were incubated at 37°C in growth media until colonies were visible (7–10 d). Then, the media was removed, cells were fixed for 10 min with methanol:acetic acid (3:1, v/v), air-dried, stained 60–90 min with Giemsa (Ricca), gently washed with tap water and air-dried. Colonies containing > 50 cells were counted and percent survival at each dose was determined relative to the number of colonies on control plates (no UV).
Clonogenic mitomycin C survival assays
MMC was obtained from Sigma, resuspended in sterile water, and the concentration was determined by absorbance at 367 nm using a molar extinction coefficient of 21,800 M−1 cm−1. The stock solution of MMC was stored at -20°C and diluted in water, and then DMEM just prior to use. Trypsinized cells were plated such that there were 150–200 viable cells per 10 cm2 dish. For each experiment, cells were plated in triplicate dishes for each Mitomycin C (MMC) dose (0, 1, 2, 4 μM) and allowed to attach for 3–4 h prior to adding drug. After 1.5–2 h treatment with the drug, medium was removed, and cells were washed once with Hank’s balanced salt solution. Fresh medium was added to the cells, which were then incubated at 37°C until colonies were visible (7–10 d). Fixing, staining and quantitation were as described for UV survival assays.
Clonogenic IR survival assays
Trypsinized cells were plated such that there would be 75–100 viable cells per 6-well plate dish. For each experiment, cells were plated in triplicate dishes for each IR dose (0, 2, 2.5, 4, 5, 6, 7.5, 8 and 10 Gy). Cells were allowed to attach for 6–8 h, IR irradiated with the indicated Grays using an RS2000 X-ray Biological Irradiator (RadSource). The medium was changed 3 d post-irradiation, and the cells were incubated at 37°C in growth media until colonies were visible (7–10 d). Fixing, staining and counting of the colonies were as described for UV survival assays.
Slot blot assay for measurement of CPD and (6–4) photo product repair rate in vivo
Repair of CPDs and (6–4) PPs was quantified as reported previously.41,42 Briefly, exponentially growing cells were irradiated with UV-C (254 nm) light with a dose of 5 J/m2. Cells were harvested at the indicated time points, and cell pellets were stored at -80°C until processing. RNA-free genomic DNA was isolated using QIAamp DNA Mini kit (Qiagen) with RNase A treatment. Quality and quantity of genomic DNA was assessed using the NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific) and electrophoresis in an ethidium bromide-stained agarose gel. Genomic DNA was denatured with 0.4 M NaOH, 10 mM EDTA for CPD repair assays or with water instead of alkali for (6–4) PP repair measurements, boiled for 10 min and then neutralized by addition of cold ammonium acetate to a final concentration of 1 M. DNA was then loaded onto a nitrocellulose membrane pre-wetted with 6X SSC buffer using a Bio-Dot SF Slot-Blot apparatus (Bio-Rad). The membrane was baked for 2 h at 80°C in a vacuum oven and then blocked in PBS/T (1X PBS with 0.1% Tween-20) containing 5% nonfat dry milk for 1 h at room temperature with gentle shaking. For immunodetection of CPDs and (6–4) PPs, membranes were incubated for 12–16 h at 4°C with anti-CPD (Kamiya Biomedical Company) or anti-(6–4) PP (Cosmo Bio Co. LTD) antibodies, respectively. After washing with PBS/T, membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody and chemiluminescent signals were detected with ECL-Plus Reagent (GE). Scanned images were quantified with Adobe Photoshop CS5 software. SYBR Gold (Invitrogen) was used to confirm equal loading of DNA.
Immunoblot analyses
Protein lysates from whole-cell lysates were prepared as described previously,42 and the protein expression levels were determined by immunoblot assay.42 The following antibodies were used to detect the respective proteins: XPA, Cyclophilin-B, p53, Chk1, Chk2 and actin (Santa Cruz Biotechnology); cleaved PARP, cleaved caspase-3, P-chk1, P-chk2 and GAPDH (Cell Signaling Technology); Cry2, Per2, Clock and Bmal1 (Bethyl Labs); Per1 (Kerafast) and Cry1.38
RNA isolation and real-time PCR
Total RNA from mouse liver and mouse skin fibroblasts, isolated using TRIZOL reagent (Invitrogen), was subjected to reverse transcription using ImProm-II Reverse Transcription System (Promega). Real-time PCR experiments were performed using an ABI 7300 system (Applied Biosystems) and MaximaTM SYBR Green/ROX qPCR Master Mix (Fermentas). RT-PCR cycle thresholds of individual genes were normalized to the corresponding actin mRNA expression values obtained with the primer set. The following primers were used for PCR (for Xpa 5′:CAAAGGTGGCTTCATTTTAG and 3′:GGTACATGTCATCTTCTAAG; for Cry1 5′:CGATGGTGAACCATGCTGAG and 3′:GTACTGACTTTCCCACCAAC; and for Actin 5′:GTTCCGATGCCCTGAGGCTC and 3′:CACTTGCGGTGCACGATGGA).
Supplementary Material
Acknowledgments
We thank Dr. M.P. Antoch and Dr. C.C. Lee for providing the Primary MEFs from wild-type, Bmal−/− and Clock−/− mice and Per1/2−/− mouse, respectively. This work was supported by National Institutes of Health grants (GM31082 and GM32833) to A.S.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21771
References
- 1.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–41. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 2.Hastings MH, Reddy AB, Maywood ES. A clockwork web: circadian timing in brain and periphery, in health and disease. Nat Rev Neurosci. 2003;4:649–61. doi: 10.1038/nrn1177. [DOI] [PubMed] [Google Scholar]
- 3.Unsal-Kaçmaz K, Mullen TE, Kaufmann WK, Sancar A. Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol. 2005;25:3109–16. doi: 10.1128/MCB.25.8.3109-3116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gery S, Komatsu N, Baldjyan L, Yu A, Koo D, Koeffler HP. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol Cell. 2006;22:375–82. doi: 10.1016/j.molcel.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 5.Kang TH, Reardon JT, Kemp M, Sancar A. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc Natl Acad Sci USA. 2009;106:2864–7. doi: 10.1073/pnas.0812638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang TH, Lindsey-Boltz LA, Reardon JT, Sancar A. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc Natl Acad Sci USA. 2010;107:4890–5. doi: 10.1073/pnas.0915085107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaddameedhi S, Selby CP, Kaufmann WK, Smart RC, Sancar A. Control of skin cancer by the circadian rhythm. Proc Natl Acad Sci USA. 2011;108:18790–5. doi: 10.1073/pnas.1115249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotta-Ramusino C, McDonald ER, 3rd, Hurov K, Sowa ME, Harper JW, Elledge SJ. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science. 2011;332:1313–7. doi: 10.1126/science.1203430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu EA, Weaver DR. Disrupting the circadian clock: gene-specific effects on aging, cancer, and other phenotypes. Aging (Albany NY) 2011;3:479–93. doi: 10.18632/aging.100323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 11.Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50. doi: 10.1016/S0092-8674(02)00961-3. [DOI] [PubMed] [Google Scholar]
- 12.Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, et al. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009;5:e1000442. doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gauger MA, Sancar A. Cryptochrome, circadian cycle, cell cycle checkpoints, and cancer. Cancer Res. 2005;65:6828–34. doi: 10.1158/0008-5472.CAN-05-1119. [DOI] [PubMed] [Google Scholar]
- 14.Destici E, Oklejewicz M, Saito S, van der Horst GT. Mammalian cryptochromes impinge on cell cycle progression in a circadian clock-independent manner. Cell Cycle. 2011;10:3788–97. doi: 10.4161/cc.10.21.17974. [DOI] [PubMed] [Google Scholar]
- 15.Stevens RG. Light-at-night, circadian disruption and breast cancer: assessment of existing evidence. Int J Epidemiol. 2009;38:963–70. doi: 10.1093/ije/dyp178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stevens RG. Circadian disruption and breast cancer: from melatonin to clock genes. Epidemiology. 2005;16:254–8. doi: 10.1097/01.ede.0000152525.21924.54. [DOI] [PubMed] [Google Scholar]
- 17.Schernhammer ES, Razavi P, Li TY, Qureshi AA, Han J. Rotating night shifts and risk of skin cancer in the nurses’ health study. J Natl Cancer Inst. 2011;103:602–6. doi: 10.1093/jnci/djr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schernhammer ES, Kroenke CH, Laden F, Hankinson SE. Night work and risk of breast cancer. Epidemiology. 2006;17:108–11. doi: 10.1097/01.ede.0000190539.03500.c1. [DOI] [PubMed] [Google Scholar]
- 19.Poole EM, Schernhammer ES, Tworoger SS. Rotating night shift work and risk of ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2011;20:934–8. doi: 10.1158/1055-9965.EPI-11-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kantermann T, Roenneberg T. Is light-at-night a health risk factor or a health risk predictor? Chronobiol Int. 2009;26:1069–74. doi: 10.3109/07420520903223984. [DOI] [PubMed] [Google Scholar]
- 21.Pronk A, Ji BT, Shu XO, Xue S, Yang G, Li HL, et al. Night-shift work and breast cancer risk in a cohort of Chinese women. Am J Epidemiol. 2010;171:953–9. doi: 10.1093/aje/kwq029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filipski E, King VM, Li X, Granda TG, Mormont MC, Liu X, et al. Host circadian clock as a control point in tumor progression. J Natl Cancer Inst. 2002;94:690–7. doi: 10.1093/jnci/94.9.690. [DOI] [PubMed] [Google Scholar]
- 23.Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS One. 2010;5:e10995. doi: 10.1371/journal.pone.0010995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006;20:1868–73. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antoch MP, Gorbacheva VY, Vykhovanets O, Toshkov IA, Kondratov RV, Kondratova AA, et al. Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle. 2008;7:1197–204. doi: 10.4161/cc.7.9.5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozturk N, Lee JH, Gaddameedhi S, Sancar A. Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc Natl Acad Sci USA. 2009;106:2841–6. doi: 10.1073/pnas.0813028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr., Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 28.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–87. doi: 10.1016/S1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 29.Lee JH, Sancar A. Circadian clock disruption improves the efficacy of chemotherapy through p73-mediated apoptosis. Proc Natl Acad Sci USA. 2011;108:10668–72. doi: 10.1073/pnas.1106284108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JH, Sancar A. Regulation of apoptosis by the circadian clock through NF-kappaB signaling. Proc Natl Acad Sci USA. 2011;108:12036–41. doi: 10.1073/pnas.1108125108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnes JW, Tischkau SA, Barnes JA, Mitchell JW, Burgoon PW, Hickok JR, et al. Requirement of mammalian Timeless for circadian rhythmicity. Science. 2003;302:439–42. doi: 10.1126/science.1086593. [DOI] [PubMed] [Google Scholar]
- 32.Unsal-Kaçmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro-Stone M, Sancar A, et al. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol. 2007;27:3131–42. doi: 10.1128/MCB.02190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gotter AL, Suppa C, Emanuel BS. Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork-associated factors. J Mol Biol. 2007;366:36–52. doi: 10.1016/j.jmb.2006.10.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sato F, Wu Y, Bhawal UK, Liu Y, Imaizumi T, Morohashi S, et al. PERIOD1 (PER1) has anti-apoptotic effects, and PER3 has pro-apoptotic effects during cisplatin (CDDP) treatment in human gingival cancer CA9-22 cells. Eur J Cancer. 2011;47:1747–58. doi: 10.1016/j.ejca.2011.02.025. [DOI] [PubMed] [Google Scholar]
- 35.Sato F, Nagata C, Liu Y, Suzuki T, Kondo J, Morohashi S, et al. PERIOD1 is an anti-apoptotic factor in human pancreatic and hepatic cancer cells. J Biochem. 2009;146:833–8. doi: 10.1093/jb/mvp126. [DOI] [PubMed] [Google Scholar]
- 36.Geusz ME, Blakely KT, Hiler DJ, Jamasbi RJ. Elevated mPer1 gene expression in tumor stroma imaged through bioluminescence. Int J Cancer. 2010;126:620–30. doi: 10.1002/ijc.24788. [DOI] [PubMed] [Google Scholar]
- 37.Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–37. doi: 10.1016/S0092-8674(00)81199-X. [DOI] [PubMed] [Google Scholar]
- 38.Vitaterna MH, Selby CP, Todo T, Niwa H, Thompson C, Fruechte EM, et al. Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc Natl Acad Sci USA. 1999;96:12114–9. doi: 10.1073/pnas.96.21.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng B, Albrecht U, Kaasik K, Sage M, Lu W, Vaishnav S, et al. Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell. 2001;105:683–94. doi: 10.1016/S0092-8674(01)00380-4. [DOI] [PubMed] [Google Scholar]
- 40.Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, et al. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999;98:193–205. doi: 10.1016/S0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- 41.Wani AA, D’Ambrosio SM, Alvi NK. Quantitation of pyrimidine dimers by immunoslot blot following sublethal UV-irradiation of human cells. Photochem Photobiol. 1987;46:477–82. doi: 10.1111/j.1751-1097.1987.tb04798.x. [DOI] [PubMed] [Google Scholar]
- 42.Gaddameedhi S, Kemp MG, Reardon JT, Shields JM, Smith-Roe SL, Kaufmann WK, et al. Similar nucleotide excision repair capacity in melanocytes and melanoma cells. Cancer Res. 2010;70:4922–30. doi: 10.1158/0008-5472.CAN-10-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.