

Abstract
The FDA Biopharmaceutical Classification System guidance allows waivers for in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms only for BCS class I. Extensions of the in vivo biowaiver for a number of drugs in BCS Class III and BCS class II have been proposed, particularly, BCS class II weak acids. However, a discrepancy between the in vivo- BE results and in vitro- dissolution results for a BCS class II acids was recently observed. The objectives of this study were to determine the oral absorption of BCS class II weak acids via simulation software and to determine if the in vitro dissolution test with various dissolution media could be sufficient for in vitro bioequivalence studies of ibuprofen and ketoprofen as models of carboxylic acid drugs.
The oral absorption of these BCS class II acids from the gastrointestinal tract was predicted by GastroPlus™. Ibuprofen did not satisfy the bioequivalence criteria at lower settings of intestinal pH=6.0. Further the experimental dissolution of ibuprofen tablets in the low concentration phosphate buffer at pH 6.0 (the average buffer capacity 2.2 mmol L-1/pH) was dramatically reduced compared to the dissolution in SIF (the average buffer capacity 12.6 mmol L -1/pH). Thus these predictions for oral absorption of BCS class II acids indicate that the absorption patterns largely depend on the intestinal pH and buffer strength and must be carefully considered for a bioequivalence test. Simulation software may be very useful tool to aid the selection of dissolution media that may be useful in setting an in vitro bioequivalence dissolution standard.
Keywords: weak acid, ibuprofen, ketoprofen, pH, simulation, GastroPlus, in vitro dissolution, dissolution media
Introduction
The U.S. Food and Drug Administration (FDA) released guidelines for the pharmaceutical industry based on the Biopharmaceutics Classification System (BCS) in 2000 [1]. The BCS classifies drugs according to the biopharmaceutical properties governing their absorption and the controlling factors; permeability, solubility and product dissolution. The European Medicines Agency (EMA) and FDA have released guidances for the investigation of bioequivalence (BE) and requirements of the BCS-based biowaiver for immediate release (IR) drug products [2]. The BCS classifies drugs as highly soluble when their highest IR dose strength is soluble at 37 °C in 250 mL or less of aqueous media over the pH range of 1.0-7.5 [3-4]. On the other hand, the EMA has defined highly soluble at 37 °C in 250 mL or less of buffer over a range of 1.0 to 6.8 at the highest drug dose [2, 5]. The current FDA criteria to be highly soluble drugs in order to be granted a biowaver are likely conservative, while both the WHO and the EMA extend biowaivers to some drugs when they meet the specific criterion for weak acids (rapid dissolution at pH 6.8) and are categorized as BCS class II [2, 6].
The feasibility of granting biowavers to some BCS class II drugs has previously been evaluated; many BCS class II drugs such as flurbiprofen, naproxen, ketoprofen, rifampicin, and carbamazepine exhibit good oral absorption, even though they are almost insoluble in acidic or basic pH conditions [7-10]. Some BCS class II weak acids, particularly the small molecule non-steroidal anti-inflammatory drugs (NSAIDs), are potential candidates for biowaivers [11]. These drugs can dissolve quickly and behave like BCS class I drugs at intestinal pH (6.5-7.0) in the GI tract, even though they exhibit low solubility at acidic pH [4]. Ibuprofen, an NSAID, has been considered as a BCS biowaiver candidate due to its high permeability and high solubility at gastrointestinal pH [12]. However, in a recent report, it was determined that it would be risky to biowaiver BCS class II acidic drugs using just in vitro dissolution tests [13]. Thus, this report raises questions of the system used for in vitro dissolution study and of the differences of in vitro- in vivo dissolution and, hence, in vivo absorption and systemic availability.
It is well known that buffer capacity, ionic strength, pH, and buffer composition of the dissolution media can have significant effects on the drug dissolution [14]. It has been suggested that the media of the in vitro dissolution media are not adequate to assess in vivo drug dissolution [15-16]. The USP simulated intestinal fluid (SIF) and fasted-state simulated small intestine fluid (FaSSIF) have been mainly used for in vitro dissolution studies even though the buffer species employed in those media are phosphate [17-18]. However, the main physiological buffer in the human intestine is predominantly bicarbonate and therefore, bicarbonate buffers may better reflect the in vivo environment and provide more suitable in vitro dissolution media to assess in vivo dissolution of test products [18]. Yet, SIF is predominantly used for in vitro dissolution study and was used for in vitro dissolution tests of ibuprofen for BE studies previously noted [13]. Sheng and the colleagues have reported that the drugs, whose pKa values are lower than 5.5, would exhibit a significant difference in their solubility and dissolution rates in SIF compared to bicarbonates and have suggested that lower concentration of phosphate for in vitro dissolution studies [18]. The adoption of inappropriate buffer media for in vitro BE studies could cause the failure in concluding the bioinequivalence for those noted ibuprofen tablets. Those dissolution media were constituted with either 70 – 80 mM phosphate buffer (pH 6.8 - 7.2) or 20 – 2270 mM acetate buffer (pH 4.5 - 6.0), not bicarbonate. The aim of this study is to determine the current in vitro dissolution methods are suitable to conclude the bioequivalence for test compounds and to predict the in vivo dissolution of test drug products and, hence, their absorption and systemic availability as a function of gastrointestinal variables especially pH and buffer capacity. When the buffer species for in vitro dissolution are not replicating in vivo buffer media, the results of in vitro dissolution tests may neither reflect in vivo dissolution nor, correctly determine bioequivalence/bioinequivalence of test drug products.
The two major objectives of this study were: 1) to investigate how the lowering pH of the intestinal media affects the oral absorption of BCS class II weak acids using the simulation software GastroPlus™ in order to understand the in vivo dissolution of those acids; and 2) to determine if the in vitro dissolution tests with SIF would be sufficient for BE studies. For these purposes, ibuprofen (pKa 4.5) and ketoprofen (pKa 3.7), which are lower than pKa 5.5, were selected as model drugs for in silico simulation. Their oral drug absorption, Cmax, AUC 0-inf and Fa %, were predicted while incorporating the effects of dissolution kinetics along with intestinal pH. For testing in vitro dissolution media, the in vitro dissolution rate of ibuprofen tablets was performed in the simulated intestinal fluid (SIF) and the low phosphate concentration of SIF (LSIF) at pH 6.0 and 6.8 in order to investigate the dissolution rate along with the pH changes of dissolution media.
Materials and Methods
Materials
Ibuprofen tablets were obtained from Amneal Pharmaceuticals (Hauppauge, NY). Potassium phosphate monobasic, sodium hydroxide, acetonitrile, high-performance liquid chromatography (HPLC) grade, and other reagents were obtained from Fisher Scientific (St. Louis, MO). All chemicals were either analytical or HPLC grade.
Computer Hardware and Software
GastroPlus™ version 7.0 (SimulationPlus, Inc., Lancaster, CA) was run using an IBM computer with Intel Core™ 2 Duo processors. This software allows the input of different dissolution velocities for pharmacokinetic predictions.
Simulation Design
The oral drug absorption was predicted based on the physicochemical, pharmacokinetic, and drug dissolution properties of two selected BCS class II drugs using a previously reported simulation method. Briefly, the default gastric emptying time (15 min) was used for this virtual trial. The virtual trials were performed with 24 h monitoring for ibuprofen and ketoprofen. In virtual trials, the variations in population physiology were defined as means with coefficients of variation in log space and were randomly selected within those ranges. Virtual trials were performed as references (n = 500) on each drug compound with an IR dosage form. Virtual trials (n=24) with randomly selected physiological conditions were performed as samples with an IR dosage form at the different intestinal pHs. The results of dissolution rates for those drug compounds with an IR dosage form were obtained by GastroPlus™ single simulation (Figure 1).
Figure 1. The delayed dissolution rates of an IR dosage from to the lowered pH of GI tract used in the simulations.
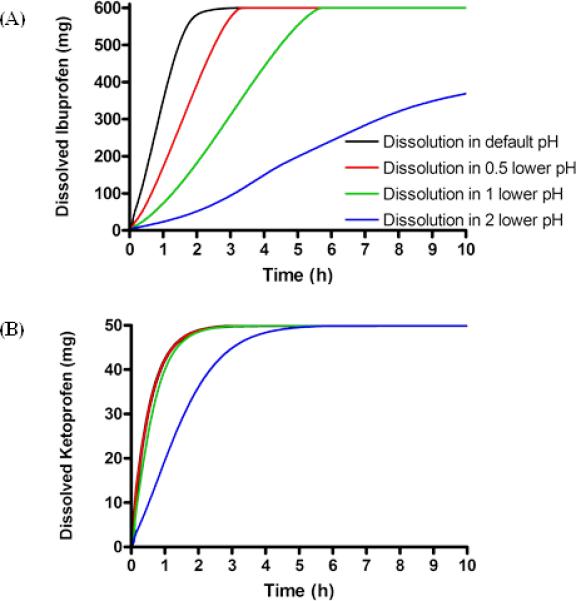
A) ibuprofen and B) ketoprofen. Black line represents the drug dissolution of an IR dosage form in default pH at the small intestine, red line represents the drug dissolution of an IR dosage form in half point lower pH at the small intestine, green line represents the drug dissolution of an IR dosage form in 1point lower pH at the small intestine, blue line the drug dissolution of an IR dosage form in 2 point lower pH at the small intestine.
Input Parameters for Pharmacokinetic Simulations
The physicochemical and biopharmaceutical properties of ibuprofen and ketoprofen used in the GastroPlus™ simulations, as well as the chemical, physiological, and pharmacological parameters for drugs cited in the literature are presented in Table 1 [13, 19-25]. The Johnson model was adopted as a dissolution model for this set of simulation [26]. Since the size of drug particles affects drug dissolution rate, a default radius of 25 μm was set as the mean with 10% log-normal distribution as its coefficient of variation [27]. Virtual trials were performed using the GastroPlus™ standard physiological conditions: the averaged human intestinal pH at each intestinal segment as a default setting. The pH at each intestinal compartment except caecum and colon was lowered from the default setting and was used as the lower pH setting (Table 2). In those virtual trials, dose, dose volume, molecular weight, log P, pKa, particle density, and diffusion coefficient of drug compounds were fixed. All other parameters for virtual trials such as effective human permeability, intestinal transit time and pharmacokinetic clearance were defined as variables, which were randomly created within a 5-20 % log-normal distribution based on their mean values.
Table 1.
Chemical/Physiological/Pharmacological Parameters of BCS class II drugs for GastroPlus™ Simulation
| Ibuprofen | Ketoprofen | ||
|---|---|---|---|
| MW | 206.3 | 254.3 | |
| Dose | mg | 600a | 50f |
| Dose number | 63# | 197# | |
| Dose volume | mL | 250 | 250 |
| Solubility (pH1.2) | mg/mL | 2.1 × 10-2b | 5.1 × 10-2b |
| logP | 4.0c | 2.8g | |
| pKa | 4.5d | 3.7b | |
| Mean precipitation time | sec | 900 | 900 |
| Human Peff | × 10-4 cm2/sec | 4.10* | 8.70h |
| Body weight | kg | 70 | 70 |
| Vc | L/kg | 0.2e | 0.4f |
| Total clearance | L/h/kg | 0.10a | 0.06f |
Table 2.
Absorption scale factor (ASF), fluid volume (mL) and pH settings at different intestinal sites for simulation
| pH condition for the simulation | ||||||
|---|---|---|---|---|---|---|
| Intestinal | Default pH | 0.5 lower pH | 1.0 lower pH | 2.0 lower pH* | Fluid volume | ASF |
| Duodenum | 6.0 | 5.5 | 5.0 | 4.0 | 41.6 | 7.0 |
| Jejunum1 | 6.2 | 5.7 | 5.2 | 4.2 | 154.2 | 10.0 |
| Jejunum2 | 6.4 | 5.9 | 5.4 | 4.4 | 122.3 | 7.0 |
| Ileum1 | 6.6 | 6.1 | 5.6 | 4.6 | 94.3 | 2.7 |
| Ileum2 | 6.9 | 6.4 | 5.9 | 4.9 | 70.5 | 2.6 |
| Ileum3 | 7.4 | 6.9 | 6.4 | 5.4 | 49.8 | 2.5 |
| Caecum | 6.4 | 6.4 | 6.4 | 6.4 | 47.5 | 0.1 |
| Asc Colon | 6.8 | 6.8 | 6.8 | 6.8 | 50.3 | 0.3 |
Simulations with lowered pH numbers were strictly hypothetical to see the effect of intestinal pH for drug absorption.
In vitro dissolution of ibuprofen immediate release (IR) tablets and pH of dissolution media
The dissolution profile of ibuprofen IR tablet was determined using a Hanson SR6 Dissolution Test Station (Chatsworth, CA) equipped with USP apparatus II (paddles). The paddle speed was set to 100 rpm.
Dissolution tests were conducted on single dosage unit (400 mg ibuprofen tablet) at 37±0.5 °C in 500 mL of either simulated intestinal fluid (SIF) (50 mM phosphate buffer) in pH 6.0 or pH 6.8 or low phosphate concentration of SIF (LSIF) (10 mM phosphate buffer; buffer capacity 1.6-2.8 mmol L-1 /pH) in pH 6.0 or pH 6.8. Tablets were introduced to the vessel by adding them directly to the medium without sinkers. Sampling was performed over 3 h and was filtered to remove any undissolved solid particles. Drug release profiles were determined on an Agilent HPLC system (Agilent Technologies, Santa Clara, CA). The HPLC system consisted of Agilent pumps (1100 series), an Agilent autosampler (1200 series), and an Agilent UV-Vis detector (1100 series) controlled by Chemstation® 32 software (version B.01.03). Samples were resolved in an Agilent Eclipse Plus C18 reverse-phase column (3.5 μm, 4.6 × 75 mm) equipped with a guard column. The mobile phase consisted of 0.1% TFA/water (Solvent A) and 0.1% TFA/acetonitrile (Solvent B) with the solvent B gradient changing from 0– 56% at a rate of 2%/min during a 20 min run for ibuprofen. A standard curve generated for ibuprofen was utilized for quantitation of integrated area under peaks. The detection wavelength was 254 nm and spectra were acquired in the 220–380 nm range.
The pH was monitored at each time point by SevenEasy Mettler Toledo (Columbus, OH) equipped with InLab 413 SG/2m probe. All experiments were performed in triplicate and average values were calculated.
Results
The oral absorption of BCS class II drugs was predicted using GastroPlus™ virtual trials. Those predicted numbers, mean Cmax, AUC 0-inf and Fa % ± standard deviation (SD), were obtained with 500 virtual trials using an IR dosage form as a reference and with 24 virtual trials as samples using the same dosage at different intestinal pHs.
ibuprofen
The impact of release rate and gastrointestinal pHs on oral drug absorption
Ibuprofen exhibited good oral absorption (81.6 – 99.9 %) throughout all tested release rates. The dissolved drug amounts of an IR dosage form at different intestinal pHs were calculated by a GastroPlus™ single simulation and were plotted as a function of time for the first 10 h (Figure 1A). Figure 1A exhibited the delayed dissolution rates of ibuprofen due to the lowering of intestinal pHs. Ibuprofen would dissolve 100 % in 5 h after oral administration except in the condition of lowering intestinal pH 2.0 points. The IR dosage form of ibuprofen exhibited similar AUC 0-inf but did not maintain a similar Cmax (Figure 2), as expected. The ibuprofen tablets did not demonstrate BE in any simulation in lower intestinal pH conditions even though those simulations were performed with the same formulation. As expected the gastrointestinal pH would have an enormous impact on the dissolution and, hence, the oral absorption of ibuprofen due to its pH-dependent solubility in the physiological range. With the lowest tested gastrointestinal pHs in the GI tract, the value of Cmax was reduced 58.1 % compared to one in the average physiological intestinal pHs (pH range of 6.0 – 7.4) in human, while the AUC 0-inf of ibuprofen did exhibited the BE with similar predicted Fa as expected (Figure 2 and Table 5).
Figure 2.
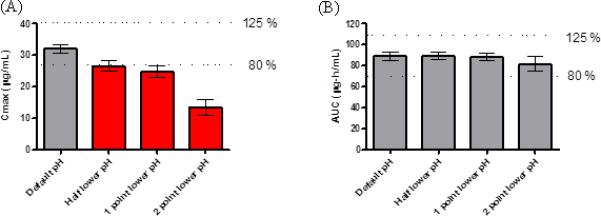
Cmax (A) and AUC 0-inf (B) of ibuprofen predicted by computer simulations. Data reported as mean ± SD, 90% confidence interval (CI) of Cmax (A) and AUC 0-inf (B), the simulation being taken with lowered intestinal pHs corresponding to an IR dosage form in default intestinal pHs as comparator. Black bars represent bioequivalence, and red bars represent outside of bioequivalence criteria (outside of 80% and 125% of the comparator).
Table 5.
Predicted Fa of ibuprofen and ketoprofen at different intestinal pHs
| Release Rate | An IR dose |
|||
|---|---|---|---|---|
| Default pH | 0.5 lower pH | 1.0 lower pH | 2.0 lower pH | |
| Ibuprofen (%) | 99.9 ± 0.0 | 99.7 ± 0.2 | 98.9± 0.4 | 81.6 ± 7.1 |
| ketoprofen (%) | 100.0 ± 0.0 | 100.0 ± 0.0 | 100.0 ± 0.0 | 99.9 ± 0.0 |
Data reported as mean ± SD.
ketoprofen
The impact of release rate and gastrointestinal pHs on oral drug absorption
Ketoprofen exhibited complete oral absorption (99.9 – 100.0 %) throughout all tested release rates (Table 5). Ketoprofen exhibited similar AUC 0-inf values throughout all the different conditions (the difference; AUC 0-inf: 0.0 – 4.3 %). The dissolved drug amounts of an IR dosage form at different intestinal pHs were calculated by a GastroPlus™ single simulation and were plotted as a function of time for the first 10 h (Figure 1B). Figure 1B exhibited the delayed dissolution rates of ketoprofen due to the setting of lower intestinal pHs. However, the difference in dissolution rates was minimal until pH was lowered 2.0 points. The dissolved amount of ketoprofen in each intestinal pH condition reached 100 % at 5 h after oral administration. Unlike ibuprofen, an IR dosage form of ketoprofen at each lowered pH condition in human intestine exhibited BE when the result was compared with the reference intestinal pH (Figure 3). Oral absorption of ketoprofen was not affected as much as one of ibuprofen by lowering intestinal pH due to its lower pKa. With the lowest intestinal pH setting in this simulation (pH range of 4.0 – 5.4), the value of Cmax was only reduced 8.4 % compared to the reference result. AUC 0-inf and predicted Fa of ketoprofen in all tested conditions satisfied BE and exhibited similar results (Figure 3 and Table 5).
Figure 3.
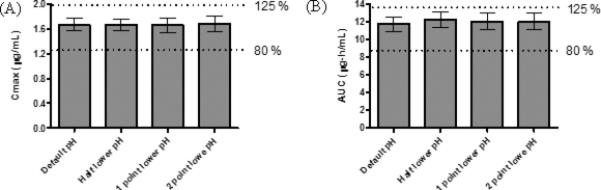
Cmax (A) and AUC 0-inf (B) of ketoprofen predicted by computer simulations. Data reported as mean ± SD, 90% confidence interval (CI) of Cmax (A) and AUC 0-inf (B), the simulation being taken with lowered intestinal pHs corresponding to an IR dosage form in default intestinal pHs as comparator. Black bars represent bioequivalence, and red bars represent outside of bioequivalence criteria (outside of 80% and 125% of the comparator).
In vitro dissolution test of ibuprofen
The dissolution of ibuprofen tablets in SIF and LSIF at pH 6.8 exhibited the similar profile with LSIF exhibiting the slower dissolution rate of ibuprofen (Figure 4A). Ibuprofen tablets in SIF at pH 6.8 were completely dissolved at 30 min while those in LSIF at pH 6.8 exhibited complete dissolution at 60 min. Although the pH of SIF media did not change, pH of LSIF was decreased from 6.8 to 6.1 during the experiment over 3 h (Figure 4A). Figure 4B demonstrated that the dissolution (average 22 %) of ibuprofen in LSIF was dramatically reduced in pH 6.0 compared to one (average 74 %) in SIF over 3 h. The pH in SIF was slightly lowered from 6.0 to 5.7 over 3 h, while pH in LSIF was decreased from 6.0 to 5.0 in 45 min in accordance with the dissolution of ibuprofen (Figure 4B). At pH 6.0, the dissolved ibuprofen in LSIF was enough to lower the pH of dissolution media and, as a result, ibuprofen only dissolved 22 % or 88 mg of 400 mg dose in LSIF, due to the lower buffer capacity compared to SIF (the average buffer capacity ; LSIF 2.2 mmol L -1/pH vs. 12.6 mmol L-1/pH).
Figure 4.
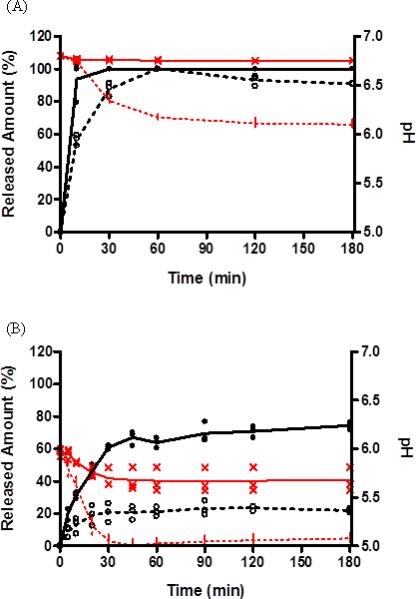
Release profile of ibuprofen tablets in different buffer concentration of SIF at pH 6.8 (A) and pH 6.0 (B) with a fixed rotation speed of 100 rpm as well as monitoring pH change. Data reported (n=3). ( • ) Ibuprofen in SIF (50mM phosphate buffer), ( ○ ) ibuprofen in LSIF (10mM phosphate buffer). ( ) Average of released ibuprofen in SIF, (
) Average of released ibuprofen in SIF, ( ) Average of released ibuprofen in LSIF (10mM phosphate buffer). (
) Average of released ibuprofen in LSIF (10mM phosphate buffer). ( ) pH of SIF (50mM phosphate buffer), (
) pH of SIF (50mM phosphate buffer), ( ) pH of LSIF (10mM phosphate buffer). (
) pH of LSIF (10mM phosphate buffer). ( ) Average pH of SIF (50mM phosphate buffer), (
) Average pH of SIF (50mM phosphate buffer), ( ) Average pH of diluted SIF (10mM phosphate buffer).
) Average pH of diluted SIF (10mM phosphate buffer).
Discussion
The FDA guidelines currently allow biowaivers only for BCS class I drugs, while the EMA has discussed allowing biowaivers for some BCS Class II and III drug products [2, 28-29]. The possibility of extending biowaivers to BCS class II has been discussed due to the conservative FDA requirement for drugs to be highly soluble [30]. It has been argued that biowaivers should be extended to some BCS class II compounds that have specific biopharmaceutical characteristics, such as weak acids, particularly the small molecule nonsteroidal anti-inflammatory drugs (NSAIDs) [8-9, 31]. The proposed rationale is that poorly soluble weak acids with pKa values ≤ 5.0, such as ibuprofen, ketoprofen, and naproxen would have solubilities of ≥ 1 mg/mL at around pH 6.5, which is the average pH value of the fasted state in the jejunum, resulting in rapid (less than 0.5 h) and reliable dissolution of those drugs [32]. Weak acids will exhibit high solubility and act like BCS class I drugs at the small intestine even though those drugs are classified as low solubility because of in vitro dissolution tests performed in a wide pH range (FDA; pH 1.0 – 7.5, EMA; 1.0 – 6.8). The pH-regulating excipients and surfactants are often used in the formulation of low soluble BCS class II drugs, in order to increase the rate of dissolution and, hence, absorption [6, 12, 30, 33]. Indeed, it is not evident that all ibuprofen IR dosage forms with a marketing authorization achieve bioequivalence [12, 34]. However, the bioinequivalence between those formulations could be detected by the right in vitro dissolution studies. The BCS class currently categorizes the high and low solubility on API itself and would not consider the solubility changes by its formulation. Extending biowaivers to BCS class II drug products must be carefully evaluated to ensure the safety of drug products and to understand the formulation effects on drug dissolution and absorption.
The demonstration of BE by in vitro dissolution studies seems feasible for a biowaiver based approval. However, it has been reported that in vitro dissolution studies failed to detect the bioinequivalence between the comparator and its test compound of BCS class II [13]. This discrepancy between in vitro BE dissolution studies and in vivo BE studies for a BCS class II drug raises the questions regarding the current in vitro BE dissolution tests. In order to understand the difference between in vivo and in vitro behaviors of BCS class II weak acids, the in vivo dissolution and absorption of ibuprofen and ketoprofen were predicted by the simulation software GastroPlus™. Additionally, the in vitro dissolution studies were performed in two different dissolution media to understand the importance of the selection in dissolution media for BE studies. The small-intestinal transit time, approximately 3 - 4 h in the fasted state, is longer than the gastric residence time of approximately 15 - 60 min [35-37]. It has been reported that the residence times in the caecum and colon are around 3 - 7 h and 12 - 24 h, respectively, which are much longer than the 3 h residence time reported for the entire small intestine [38-39]. In our simulations, the residence times used for the stomach, small intestine (duodenum, jejunum, and ileum), caecum, and ascending colon were 0.2 h, 3.2 h, 4.2 h, and 12.6 h within a 10 % log-normal distribution based on those mean values, respectively.
The measured pH of human intestine in the fasted state reportedly ranged from 5.5 to 7.5 in the duodenum, from 6.2 to 6.7 in the proximal small intestine, from 6.3 to 7.3 in the middle small intestine, from 6.7 to 7.7 in the distal small intestine, and from 5.5 to 7.6 in the ascending colon [38, 40-41]. The pH values of the duodenum, jejunum, and ileum were set to 6.0, 6.2-6.4, and 6.6-7.4, respectively. Those starting pH values were gradually lowered in this set of simulations within a 6 - 10 % log-normal distribution based on those mean values (Table 2). At those starting pHs, a weakly acidic drug product such as ibuprofen (pKa 4.5) and ketoprofen (pKa 3.7) would exhibit high solubility in the duodenum, jejunum, ileum, caecum, and colon and have longer residence time at the small intestine, caecum and colon for a total of 18 – 35 h. As a result, it is highly likely that ibuprofen and ketoprofen may behave similar to a BCS class I drug in the small and large intestines even though it is still classified as a low solubility drug due to their poor solubility at gastric pH. Therefore, weak acids of BCS class II drugs may be completely absorbed because of their high solubility in small and large intestines and have sufficient residence time throughout the whole intestine. BCS class II acidic drugs, ibuprofen and ketoprofen, exhibited complete absorption (99.9 – 100.0 %) in this prediction study with an IR dosage form. Indeed, it has been reported that weakly acidic BCS class II drug products have been shown to be completely orally absorbed due to their high solubility at the pH range (pH 6.0 – 6.4) of the small intestine [31-32]. Thus, drugs with pKa values ≤ 5.0 of IR oral dosage forms would have sufficient time for complete dissolution and absorption in the small intestine due to their high permeability. The average volumes of human intestinal fluid from the duodenum and jejunum in a fasted state are reportedly 184 mL and 63 mL, respectively [41]. With those volumes in a fasted state at the duodenum and jejunum, more than 50 mg of ibuprofen and ketoprofen would be dissolved at those small intestinal segments at pH 6.0 and 6.2, respectively, indicating that about 10 % and 100 % of orally dosed ibuprofen (600 mg dose) and ketoprofen (50 mg dose) were dissolved without any pH change. Therefore, the ranges of dissolved ibuprofen and ketoprofen at the duodenum and jejunum would be 2 - 4 mmol/L and 1 - 3 mmol/L, respectively. The buffer capacity values obtained for fasted-state human intestinal fluid in several reports ranged from 2.4 – 5.6 mmol L-1/pH [42-43]. This suggests that the buffer capacity at the duodenum and proximal jejunum is relatively lower than in vitro dissolution media [44-45]. As a result, the dissolved amount of weak acids in vivo could significantly lower the intestinal pH and, hence, slow the dissolution rate. Indeed, Lee and colleagues have reported the significant decrease of duodenal pH by the presence of acid in humans [40]. In our simulations and in vitro dissolution studies, drastically slowed dissolution rates of an IR dosage form of ibuprofen and ketoprofen were observed when the intestinal pH and buffer concentration were reduced (the range of 0.5 – 2 pH at each segment of the small intestine and 10 – 50 mM buffer concentration of SIF at pH 6.0 and 6.8) (Figures 1 and 4). The slowed dissolution rates of ibuprofen and ketoprofen reduced the rate of absorption and, therefore, the values of Cmax (Tables 3 and 4). However, the changes of AUC 0-inf for ibuprofen and ketoprofen in the lowered intestinal pH condition were minimal, 0.8 – 8.4 % for ibuprofen and 0.9 – 4.3 % for ketoprofen (Tables 3 and 4). The predicted Fa values of ibuprofen and ketoprofen were unchanged and exhibited the ranges of 81.6 – 99.9 % and 99.9 – 100.0 %, respectively (Table 5). Those results indicate that BCS class II acidic drugs have long enough transit time in the small intestine to be completely absorbed because of their high permeability and high solubility in the physiological pHs. The lowered pHs in the intestinal segments slowed the dissolution rate and, hence, the absorptive rate for acidic drugs like ibuprofen and ketoprofen.
Table 3.
Simulated Cmax and AUC0-inf of IR ibuprofen at different intestinal pHs
| An IR dose |
||||
|---|---|---|---|---|
| Release Rate | Default pH | 0.5 lower pH | 1.0 lower pH | 2.0 lower pH |
| Cmax (μg/mL) | 32.2 ± 1.3 | 26.6 ± 1.6 | 24.8 ± 1.9 | 13.5 ± 2.5 |
| AUC0-inf (μg-h/mL) | 89.1 ± 3.9 | 90.0 ± 3.5 | 88.4 ± 3.9 | 81.6 ± 5.8 |
Data reported as mean ± SD.
Table 4.
Simulated Cmax and AUC0-inf of IR ketoprofen at different intestinal pHs
| An IR dose |
||||
|---|---|---|---|---|
| Release Rate | Default pH | 0.5 lower pH | 1.0 lower pH | 2.0 lower pH |
| Cmax (× 10-1 μg/mL) | 16.7 ± 1.1 | 16.6 ± 1.0 | 16.5 ± 1.2 | 15.3 ± 1.5 |
| AUC0-inf (μg-h/mL) | 11.7 ± 0.8 | 12.2 ± 0.9 | 12.0 ± 1.0 | 11.7 ± 1.0 |
Data reported as mean ± SD.
The results of in vitro dissolution study for ibuprofen tablets in different pH and buffer strength supported the simulation outcomes. The lowered pH of dissolution media attributed the slower dissolution rate for ibuprofen in LSIF (Figure 4B), suggesting that the dissolution rate of acidic drugs would be affected more at the proximal small intestine such as duodenum and proximal jejunum (pH 5.5-6.2) than one at the distal small intestine such as ileum (pH 6.7-7.7). At pH 6.8, the starting pH was high enough not to affect the dissolution rate of acids even though the pH in LSIF was reduced to pH 6.1. The difference of the dissolution rates in LSIF at pH 6.0 and one in SIF at pH 6.0 was significant and as the pH of dissolution media decreases, the solubility of ibuprofen would be largely affected at pH 5.0, which is closer to the pKa of ibuprofen. Based on this result, ketoprofen, which possesses similar physiological characteristics to ibuprofen, would exhibit a similar result of in vitro dissolution at the similar condition. However, the pKa of ibuprofen is higher than ketoprofen's (ibuprofen; pKa 4.5 vs. ketoprofen; pKa 3.7). Therefore, the dissolution rate of ibuprofen would be more sensitive to the pH changes in the human intestine. Indeed, our simulation results indicated that ibuprofen would be more sensitive to the change of intestinal pH than ketoprofen (Figures 2 and 3). The in vivo dissolution rates of acidic drugs would be altered by the lowered pHs of buffer in the human intestine. Shown in Figures 4A and B, the pH of dissolution media would shift especially when the buffer capacity of media is low. The shift of pH in in vivo dissolution media would gradually occur in accordance with the dissolved amount of acidic drugs. The limitation of this phenomenon in in silico simulation is that the gradually decreasing pH of dissolution medium in each intestinal compartment due to low buffer capacity cannot be accomplished. GastroPlus™ has a function to create a gradually decreasing/increasing pH profile at any given time in GI tract. However, this function cannot be used for this series of simulations with an IR dosage form without knowing the in vivo dissolution rate, specific intestinal pH and the buffer capacity. Therefore, in these simulations, the physiological pHs in gastrointestinal segments adjusted as an initial setting and those pHs were altered by the dissolved amount of acidic or basic drugs. The assumption in the simulation software is that the buffer capacity of in vivo media is high enough to maintain the pH independent to how much of acidic or basic drugs dissolved. The initial pH settings in those simulations assumed to be the final pH of in vivo media when the drug is completely dissolved at a given transit time at specific intestinal segments. Therefore, those simulations give a progressive prediction by setting the lowest pHs in the GI segments as the initial settings.
The results of in silico and in vitro studies clearly demonstrate that the possibility of slow dissolution rates of acidic drugs in vivo can cause the bioinequivalence even though test products show similar in vitro USP type dissolution. Our in silico results demonstrated that ibuprofen dosage forms could fail the BE test when the oral absorption, Cmax and AUC 0-inf, of ibuprofen with the default intestinal pH setting was compared with one with the lowered intestinal pH setting. The simulation and in vitro dissolution results suggest that dissolved ibuprofen and ketoprofen could lower the regional intestinal pH up to 1-3 units and, hence, the in vivo dissolution rate of ibuprofen and ketoprofen might be slower due to this lowered solubility. Ketoprofen (pKa 3.7), which has the similar chemical and physiological characteristics to ibuprofen, exhibited slower in vivo dissolution rate than in vitro dissolution rate, which is attributed to a lower pH and poorer buffering capacity in vivo [44]. In vitro dissolution media, fasted USP SIF, FaSSIF, FaSSIFm, and FaSSIF-V2, retain strong buffer capacities, 18.4, 12, 12, and 10 mmol L-1/pH, which are 1.8 – 7.7-fold higher than the reported buffer capacity in in vivo fluid, 2.4 – 5.6 mmol L-1/pH [16, 44-47]. Buffer capacity and pH of dissolution medium clearly has a significant effect on the dissolution rate of these NISAID compound. Those differences between in vivo and in vitro would produce the different dissolution results. Thus, in order to perform in vitro BE studies, the rational selection of buffer media with appropriate buffering capacity and pH is extremely important. Real human intestinal fluid is the most relevant media for in vitro dissolution studies. However, the supply of human intestinal fluid is very limited. Therefore, simulated intestinal fluid (SIF) as an alternative to human intestinal fluid has been mainly utilized for in vitro dissolution. While the compendial simulated intestinal fluid (50 mM phosphate buffer) has been accepted as the in vitro dissolution media, the main physiological buffer in the human intestine is bicarbonate. Therefore, the in vitro dissolution studies in bicarbonate buffer would predict better in vivo dissolution rates. Further the concentration of phosphate buffer (50 mM) very likely is too high for BE studies reflecting in vivo dissolution media compared to the bicarbonates [18]. The bicarbonate concentration was reported in the range of 4 – 21 mM and the average of 15 mM [18, 48]. In our calculation, the 5 – 15 mM bicarbonate buffers at pH 6.0 and pH 6.8 are equivalent to 8 – 45 mM phosphate buffers. The in vitro dissolution studies with 50 mM phosphate buffer may not be suitable for in vitro BE dissolution medium, especially for BCS class II acidic drugs. The importance of the selection in dissolution media for BE studies was demonstrated by in silico studies and in vitro dissolution studies with the same dosage form. Indeed, the importance of selecting the right in vitro dissolution media and methods for the test compounds has been indicated for development of solid oral dosage forms [49-50]. We hypothesize that the slower drug dissolution of ibuprofen in vivo caused bioinequivalence due to the lower buffer capacity, different pKa of the in vivo buffer and the pH shift of intestinal fluid in vivo, which would not be represented by in vitro dissolution studies with SIF due to its high buffer capacity. We expect that if the dissolution studies were performed with media reflecting human intestinal fluids and bicarbonate buffers, the bioinequivalence of ibuprofen products could be detected by the in vitro dissolution studies. Therefore, the biorelevant dissolution media such as bicarbonate buffers and pH's should be considered for use as in vitro dissolution media for in vitro BE dissolution studies. The selection of dissolution media for in vitro BE studies is a key to predict in vivo dissolution of test compounds and would depend on the physiochemical property of test compounds. With our simulation results, the simulation software can be a valuable tool to predict the in vivo dissolution of test compounds and can aid in selection of the suitable in vitro dissolution media for BE studies.
Conclusions
The in vitro dissolution rate is clearly dependent on the pH, buffer species, and buffer capacity of the medium for NSAID drugs products. The USP test media clearly do not reflect the human intestinal in vivo environment and may not suitable for in vitro BE dissolution study. The results of this principally computational study can provide a guide to selecting a dissolution method and media that can be used for establishing an in vitro bioequivalence standard.
Acknowledgement
We are thankful to Mr. Brian Krieg for his excellent insight and advice. This work was supported by NIH Grant NIGMD-2R01GM037188.
References
- 1.U.S. FDA Department of Health and Human Services Food and Drug Administration Center for Evaluation and Research Guidances for industry: Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a Biopharmaceutics Classification System. 2000.
- 2.EMA Guideline on the investigation of bioequivalence. 2010.
- 3.Amidon GL, Lennernas H, Shah VP, et al. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 4.Yazdanian M, Briggs K, Jankovsky C, et al. The “high solubility” definition of the current FDA Guidance on Biopharmaceutical Classification System may be too strict for acidic drugs. Pharm Res. 2004;21:293–299. doi: 10.1023/b:pham.0000016242.48642.71. [DOI] [PubMed] [Google Scholar]
- 5.Gupta E, Barends DM, Yamashita E, et al. Review of global regulations concerning biowaivers for immediate release solid oral dosage forms. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences. 2006;29:315–324. doi: 10.1016/j.ejps.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Polli JE, Abrahamsson BS, Yu LX, et al. Summary workshop report: bioequivalence, biopharmaceutics classification system, and beyond. The AAPS journal. 2008;10:373–379. doi: 10.1208/s12248-008-9040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agrawal S, Panchagnula R. Implication of biopharmaceutics and pharmacokinetics of rifampicin in variable bioavailability from solid oral dosage forms. Biopharmaceutics & drug disposition. 2005;26:321–334. doi: 10.1002/bdd.464. [DOI] [PubMed] [Google Scholar]
- 8.Davies NM. Clinical pharmacokinetics of flurbiprofen and its enantiomers. Clinical pharmacokinetics. 1995;28:100–114. doi: 10.2165/00003088-199528020-00002. [DOI] [PubMed] [Google Scholar]
- 9.Faassen F, Vromans H. Biowaivers for oral immediate-release products: implications of linear pharmacokinetics. Clinical pharmacokinetics. 2004;43:1117–1126. doi: 10.2165/00003088-200443150-00004. [DOI] [PubMed] [Google Scholar]
- 10.Kovacevic I, Parojcic J, Homsek I, et al. Justification of Biowaiver for Carbamazepine, a Low Soluble High Permeable Compound, in Solid Dosage Forms Based on IVIVC and Gastrointestinal Simulation. Molecular pharmaceutics. 2008;6:40–47. doi: 10.1021/mp800128y. [DOI] [PubMed] [Google Scholar]
- 11.Tubic-Grozdanis M, Bolger MB, Langguth P. Application of gastrointestinal simulation for extensions for biowaivers of highly permeable compounds. The AAPS journal. 2008;10:213–226. doi: 10.1208/s12248-008-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Potthast H, Dressman JB, Junginger HE, et al. Biowaiver monographs for immediate release solid oral dosage forms: ibuprofen. J Pharm Sci. 2005;94:2121–2131. doi: 10.1002/jps.20444. [DOI] [PubMed] [Google Scholar]
- 13.Alvarez C, Nunez I, Torrado JJ, et al. Investigation on the possibility of biowaivers for ibuprofen. J Pharm Sci. 2011;100:2343–2349. doi: 10.1002/jps.22472. [DOI] [PubMed] [Google Scholar]
- 14.Mooney KG, Mintun MA, Himmelstein KJ, et al. Dissolution kinetics of carboxylic acids II: effect of buffers. J Pharm Sci. 1981;70:22–32. doi: 10.1002/jps.2600700104. [DOI] [PubMed] [Google Scholar]
- 15.Fallingborg J. Intraluminal pH of the human gastrointestinal tract. Danish medical bulletin. 1999;46:183–196. [PubMed] [Google Scholar]
- 16.Vertzoni M, Fotaki N, Kostewicz E, et al. Dissolution media simulating the intralumenal composition of the small intestine: physiological issues and practical aspects. J Pharm Pharmacol. 2004;56:453–462. doi: 10.1211/0022357022935. [DOI] [PubMed] [Google Scholar]
- 17.The United States Pharmacopeia USP 24 The National Formulary NF19. 2000.
- 18.Sheng JJ, McNamara DP, Amidon GL. Toward an in vivo dissolution methodology: a comparison of phosphate and bicarbonate buffers. Molecular pharmaceutics. 2009;6:29–39. doi: 10.1021/mp800148u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abernethy DR, Greenblatt DJ. Ibuprofen disposition in obese individuals. Arthritis and rheumatism. 1985;28:1117–1121. doi: 10.1002/art.1780281006. [DOI] [PubMed] [Google Scholar]
- 20.Avdeef A, Box KJ, Comer JE, et al. PH-metric log P 11. pKa determination of water-insoluble drugs in organic solvent-water mixtures. Journal of pharmaceutical and biomedical analysis. 1999;20:631–641. doi: 10.1016/s0731-7085(98)00235-0. [DOI] [PubMed] [Google Scholar]
- 21.Avdeef A, Box KJ, Comer JE, et al. pH-metric logP 10. Determination of liposomal membrane-water partition coefficients of ionizable drugs. Pharm Res. 1998;15:209–215. doi: 10.1023/a:1011954332221. [DOI] [PubMed] [Google Scholar]
- 22.Geisslinger G, Menzel S, Wissel K, et al. Pharmacokinetics of ketoprofen enantiomers after different doses of the racemate. British journal of clinical pharmacology. 1995;40:73–75. doi: 10.1111/j.1365-2125.1995.tb04537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herzfeldt CDK,R. Dissociation constants, solubilities and dissolution rates of some selected nonsteroidal antiinflammatories. Drug development and industrial pharmacy. 1983;9:767–793. [Google Scholar]
- 24.Kasim NA, Whitehouse M, Ramachandran C, et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Molecular pharmaceutics. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 25.Sangster J. Octanol-water partition coefficients: 1993.
- 26.Lu AT, Frisella ME, Johnson KC. Dissolution modeling: factors affecting the dissolution rates of polydisperse powders. Pharm Res. 1993;10:1308–1314. doi: 10.1023/a:1018917729477. [DOI] [PubMed] [Google Scholar]
- 27.Mutalik S, Anju P, Manoj K, et al. Enhancement of dissolution rate and bioavailability of aceclofenac: a chitosan-based solvent change approach. Int J Pharm. 2008;350:279–290. doi: 10.1016/j.ijpharm.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 28.US FDA Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a Biopharmaceutics Classification System. FDA Guidance for Insudtry. 2002 [Google Scholar]
- 29.EMA Concept Paper on BCS- Based Biowaiver. 2007.
- 30.Yu LX, Amidon GL, Polli JE, et al. Biopharmaceutics classification system: the scientific basis for biowaiver extensions. Pharm Res. 2002;19:921–925. doi: 10.1023/a:1016473601633. [DOI] [PubMed] [Google Scholar]
- 31.Davies NM. Clinical pharmacokinetics of ibuprofen. The first 30 years. Clinical pharmacokinetics. 1998;34:101–154. doi: 10.2165/00003088-199834020-00002. [DOI] [PubMed] [Google Scholar]
- 32.Dressman JB, Hempenstall J, Reppas, C. J. The BCS: Where do we go from here? Pharmaceutical Technology. 2001;25:68–76. [Google Scholar]
- 33.Aburub A, Risley DS, Mishra D. A critical evaluation of fasted state simulating gastric fluid (FaSSGF) that contains sodium lauryl sulfate and proposal of a modified recipe. Int J Pharm. 2008;347:16–22. doi: 10.1016/j.ijpharm.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh LK, Ghosh NC, Chatterjee M, et al. Product development studies on the tablet formulation of ibuprofen to improve bioavailability. Drug development and industrial pharmacy. 1998;24:473–477. doi: 10.3109/03639049809085645. [DOI] [PubMed] [Google Scholar]
- 35.Kaus LC, Fell JT. Effect of stress on the gastric emptying of capsules. Journal of clinical and hospital pharmacy. 1984;9:249–251. doi: 10.1111/j.1365-2710.1984.tb01083.x. [DOI] [PubMed] [Google Scholar]
- 36.Kortejarvi H, Shawahna R, Koski A, et al. Very rapid dissolution is not needed to guarantee bioequivalence for biopharmaceutics classification system (BCS) I drugs. J Pharm Sci. 2010;99:621–625. doi: 10.1002/jps.21879. [DOI] [PubMed] [Google Scholar]
- 37.Oberle RL, Chen TS, Lloyd C, et al. The influence of the interdigestive migrating myoelectric complex on the gastric emptying of liquids. Gastroenterology. 1990;99:1275–1282. doi: 10.1016/0016-5085(90)91150-5. [DOI] [PubMed] [Google Scholar]
- 38.Ibekwe VC, Fadda HM, McConnell EL, et al. Interplay between intestinal pH, transit time and feed status on the in vivo performance of pH responsive ileo-colonic release systems. Pharm Res. 2008;25:1828–1835. doi: 10.1007/s11095-008-9580-9. [DOI] [PubMed] [Google Scholar]
- 39.Madsen JL, Krogsgaard OW. Gastrointestinal scintiscanning: dosimetry. European journal of nuclear medicine. 1989;15:260–261. doi: 10.1007/BF00257544. [DOI] [PubMed] [Google Scholar]
- 40.Lee KJ, Vos R, Janssens J, et al. Influence of duodenal acidification on the sensorimotor function of the proximal stomach in humans. American journal of physiology. Gastrointestinal and liver physiology. 2004;286:G278–284. doi: 10.1152/ajpgi.00086.2003. [DOI] [PubMed] [Google Scholar]
- 41.Perez de la Cruz Moreno M, Oth M, Deferme S, et al. Characterization of fasted-state human intestinal fluids collected from duodenum and jejunum. J Pharm Pharmacol. 2006;58:1079–1089. doi: 10.1211/jpp.58.8.0009. [DOI] [PubMed] [Google Scholar]
- 42.Kalantzi L, Goumas K, Kalioras V, et al. Characterization of the human upper gastrointestinal contents under conditions simulating bioavailability/bioequivalence studies. Pharm Res. 2006;23:165–176. doi: 10.1007/s11095-005-8476-1. [DOI] [PubMed] [Google Scholar]
- 43.Persson EM, Gustafsson AS, Carlsson AS, et al. The effects of food on the dissolution of poorly soluble drugs in human and in model small intestinal fluids. Pharm Res. 2005;22:2141–2151. doi: 10.1007/s11095-005-8192-x. [DOI] [PubMed] [Google Scholar]
- 44.Corrigan OI, Devlin Y, Butler J. Influence of dissolution medium buffer composition on ketoprofen release from ER products and in vitro-in vivo correlation. Int J Pharm. 2003;254:147–154. doi: 10.1016/s0378-5173(03)00004-8. [DOI] [PubMed] [Google Scholar]
- 45.Levis KA, Lane ME, Corrigan OI. Effect of buffer media composition on the solubility and effective permeability coefficient of ibuprofen. Int J Pharm. 2003;253:49–59. doi: 10.1016/s0378-5173(02)00645-2. [DOI] [PubMed] [Google Scholar]
- 46.Galia E, Nicolaides E, Horter D, et al. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm Res. 1998;15:698–705. doi: 10.1023/a:1011910801212. [DOI] [PubMed] [Google Scholar]
- 47.Jantratid E, Janssen N, Reppas C, et al. Dissolution media simulating conditions in the proximal human gastrointestinal tract: an update. Pharm Res. 2008;25:1663–1676. doi: 10.1007/s11095-008-9569-4. [DOI] [PubMed] [Google Scholar]
- 48.Karr WG, Abbott WO, Sample AB. Intubation Studies of the Human Small Intestine. Iv. Chemical Characteristics of the Intestinal Contents in the Fasting State and as Influenced by the Administration of Acids, of Alkalies and of Water. The Journal of clinical investigation. 1935;14:893–900. doi: 10.1172/JCI100739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anand O, Yu LX, Conner DP, et al. Dissolution testing for generic drugs: an FDA perspective. The AAPS journal. 2011;13:328–335. doi: 10.1208/s12248-011-9272-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Siewert M, Dressman J, Brown CK, et al. FIP/AAPS guidelines to dissolution/in vitro release testing of novel/special dosage forms. AAPS PharmSciTech. 2003;4:E7. doi: 10.1208/pt040107. [DOI] [PMC free article] [PubMed] [Google Scholar]