

Abstract
Highly specialized, but exceedingly small populations of cells play important roles in many tissues. The identification of cell-type specific markers and gene expression programs for extremely rare cell subsets has been a challenge using standard whole-tissue approaches. Gene expression profiling of individual cells allows for unprecedented access to cell types that comprise only a small percentage of the total tissue1-7. In addition, this technique can be used to examine the gene expression programs that are transiently expressed in small numbers of cells during dynamic developmental transitions8.
This issue of cellular diversity arises repeatedly in the central nervous system (CNS) where neuronal connections can occur between quite diverse cells9. The exact number of distinct cell types is not precisely known, but it has been estimated that there may be as many as 1000 different types in the cortex itself10. The function(s) of complex neural circuits may rely on some of the rare neuronal types and the genes they express. By identifying new markers and helping to molecularly classify different neurons, the single-cell approach is particularly useful in the analysis of cell types in the nervous system. It may also help to elucidate mechanisms of neural development by identifying differentially expressed genes and gene pathways during early stages of neuronal progenitor development.
As a simple, easily accessed tissue with considerable neuronal diversity, the vertebrate retina is an excellent model system for studying the processes of cellular development, neuronal differentiation and neuronal diversification. However, as in other parts of the CNS, this cellular diversity can present a problem for determining the genetic pathways that drive retinal progenitors to adopt a specific cell fate, especially given that rod photoreceptors make up the majority of the total retinal cell population11. Here we report a method for the identification of the transcripts expressed in single retinal cells (Figure 1). The single-cell profiling technique allows for the assessment of the amount of heterogeneity present within different cellular populations of the retina2,4,5,12. In addition, this method has revealed a host of new candidate genes that may play role(s) in the cell fate decision-making processes that occur in subsets of retinal progenitor cells8. With some simple adjustments to the protocol, this technique can be utilized for many different tissues and cell types.
Keywords: Neuroscience, Issue 62, Single-cells, transcriptomics, gene expression, cell-type markers, retina, neurons, genetics
Protocol
1. Cell Dissociation
A flowchart outlining the protocol is shown is Figure 1. For the catalog numbers of the particular reagents used throughout this protocol, please refer to Table 1. Dissect the retina in a PBS bath. During the dissection, it is best to remove the vitreous and the lens since keeping them with the retina can impede the dissociation. It is not always critically important to remove all of the retinal pigment epithelium (RPE) and in some instances it may be impossible to completely remove it. However, for single cell profiling experiments of photoreceptors, the RPE should be removed. Failure to remove the RPE can lead to contamination of the rod cell profiles with RPE expressed genes. This is presumably due to the connection between the photoreceptors and the RPE. For the dissociation, whole adult murine retinas are incubated at 37 °C for 10 min in a 1.5 ml microcentrifuge tube containing 20 μl activated papain, 20 μl 25 mM Cysteine in 5 mM EDTA, and 360 μl Hank's Balanced Salt Solution (HBSS) supplemented with 10 mM HEPES. Filter-tipped pipette tips are used for all steps throughout the protocol to minimize potential contamination. The papain concentration and the time of incubation will vary according to the age and the nature of the tissue. For example, to dissociate developing retinas isolated at postnatal day 0 (P0), incubate the retina in 10 μl activated papain, 10 μl 25 mM Cysteine in 5 mM EDTA, and 380 μl Hank's Balanced Salt Solution (HBSS) supplemented with 10 mM HEPES for 10 min at 37 °C. Retinas from different species also require slightly different conditions. Chicken retinas are thinner than mouse retinas at most stages of development. Using 10 μl activated papain, 10 μl 25 mM Cysteine in 5 mM EDTA, and 380 μl Hank's Balanced Salt Solution (HBSS) supplemented with 10 mM HEPES for 5 min at 37 °C is sufficient to dissociate these retinas.
Gently tap the tube to dislodge any settled cells, then triturate gently 10-20 times with a p1000 pipettor. Incubate for an additional 10 min at 37 °C if large clumps of tissue persist and then re-triturate 10-20 times.
Add 5 μl of DNase I (10 U/μl) and incubate for 5 min at room temperature. Triturate gently with a p1000 pipettor. The exact number of times will need to be determined empirically, but generally falls between 10-20.
Centrifuge in a table-top centrifuge at 3000 rpm for 3 min Remove the supernatant, leaving 100-200 μl of liquid at the bottom, and tap the tube to dislodge the pellet. Add 1 ml of HBSS and resuspend the pellet with gentle trituration. Tapping the tube is critical here as simply resuspending the pellet with lyse many of the retinal cells. This is especially true with adult murine retinas.
Centrifuge at 3000 rpm for 3 min Carefully remove the supernatant and resuspend in 450 μl of PBS containing 0.1% BSA. It is important to remove as much of the supernatant as possible to minimize the presence of mRNAs from lysed cells and any products from the dissociation that could inhibit future reactions.
2. Harvesting Single Cells
Prepare two 6 cm dishes containing 5 ml of PBS supplemented with 0.1% BSA. Plate the dissociated cells onto one dish and allow the cells to settle for at least 5 min. The cell density used depends greatly on the nature of the single cells being profiled. For example, for very rare cells labeled with a fluorescent marker such a GFP, entire dissociated retinas are plated on a single 6 cm dish. For cells that are somewhat more abundant, fewer cells are initially plated to make it easier to harvest one (or very close to one) cell with the first micropipette.
Place the plate of dissociated cells on an inverted microscope. We use the IMT-2 model from Olympus. Using micropipettes that have been drawn to a fine tip (inner diameter 0.5 mm, outer diameter 1.04 mm) (Figure 2) and together with an aspirator tube, harvest a single cell. The cells readily enter the micropipette when it is placed close to them on the dish. It is critical that the aspirator tube is neither too long nor too short for the distance to the stage associated with the inverted microscope (The aspirator tubes we use with our inverted microscope are 38.1 cm long). If the tube is too long, cells may not be expelled efficiently from the micropipette into the collection tube. The primary reason we use the older IMT-2 model inverted microscope is that we have found it has the ideal distance to the stage for our aspirator assembly tubes. After entering the micropipette, the cell (or cells) is expelled onto a second plate (wash plate) to ensure that one and only one cell is harvested for gene expression profiling. On the second plate it is often necessary to either remove extraneous cells with a new micropipette or move the cell of interest to a different location to ensure that only a single cell enters the micropipette.
When an individual cell is completely isolated from neighboring cells, use a new micropipette to aspirate the cell of interest as in section 2.2 and then expel it directly into a 0.2 ml PCR tube containing 4.5 μl cell lysis buffer. The cell is expelled onto the side of the tube, being careful not to break off the tip of the micropipette into the tube. The samples can be spun briefly in a bench top microcentrifuge to immerse the cell in lysis buffer. This spinning is usually done after every 5th cell and then again at the end of collection.Tubes containing single cells in lysis buffer should be kept on ice for the duration of the single cell isolation. For optimal results, single cells are collected within a two-hour window post-dissociation. After this time has elapsed, it is best to proceed to the reverse transcription step since additional time has been observed to increase the chances of RNA degradation.
3. Reverse Transcription
Briefly spin the samples in a benchtop minicentrifuge to ensure all the single cells are immersed in the cell lysis buffer. To promote cell lysis, incubate the sample at 70 °C for 90 sec. in a thermocycler. Immediately place the tubes back on ice.
Leave the tubes on ice for 2 min. For all reagent additions, use filter-tip pipette tips to prevent contamination. To perform the reverse transcription, add 0.33 μl Superscript III (200 U/μl), 0.05 μl RNase Inhibitor (40 U/μl), and 0.07 μl T4 gene 32 protein. Incubate the reaction mixture for 50 min at 50 °C in a thermocycler. Inactivate the enzyme at 70 °C for 15 min and place on ice.
To remove the free primer, add 0.1 μl Exonuclease Buffer (10X), 0.8 μl dH20 (molecular biology grade), and 0.1 μl Exonuclease I (20 U/μl). Incubate at 37 °C for 30 min in a thermocycler. Inactivate the enzyme by incubating the reaction at 80 °C for 25 min and then placing the tubes on ice.
4. Tailing and Single-Cell PCR
Add 6 μl of the tailing reaction mixture and use a thermocycler to incubate the sample at 37 °C for 20 min, 70 °C for 10 min, and hold at 4 °C
- Add 10 μl of the tailed sample to the PCR reaction mixture and perform the second strand synthesis and PCR amplification using the following conditions:
- 95 °C for 2 min
- 37 °C for 5 min
- 72 °C for 16 min
- 93 °C for 40 sec
- 67 °C for 1 min
- 72 °C for 6 min, adding 6 sec per cycle
- Go to step 4 34 times
- 72 °C for 10 min
- Hold at 4 °C
5. Labeling for Affymetrix Chips
Label 10-20 μg of cDNA (the concentration of the amplified cDNA resulting from a single cell is usually ~1 μg/μl) to obtain a robust hybridization on Affymetrix microarrays. First, fragment the cDNA by adding 10-20 μl of the single-cell cDNA to 8 μl 1X One-Phor-All buffer and 1 μl of diluted (1:10 in 1X One-Phor-All buffer) DNaseI in an 80 μl reaction. Incubate in a thermocycler for 13 min at 37 °C. Inactivate the DNase I for 15 min at 100 °C and place on ice.
Add 20 μl 5X TdT buffer, 2.5 μl Biotin N6-ddATP (Enzo Biosciences), and 1 μl TdT (diluted 1:8 in TdT buffer).
Incubate for 90 min at 37 °C, then 5 min at 65 °C. Store at -20 °C or hybridize immediately to an Affymetrix microarray. The hybridization is performed using standard Affymetrix protocols.
6. Representative Results
To assess the quality of the cDNA, 10 μl of the cDNA sample was loaded on a 1% agarose gel. The cDNA is mainly judged by the size range (500-2000 bp) and intensity of the cDNA smear (Figure 1 and Figure 3a). In the gel depicted in Figure 1, every other lane shows a cDNA smear from retinal cells isolated E16.5. The lanes in between show the resulting smears when only media is aspirated into the needle and deposited into the PCR tube. These lanes are never completely devoid of a faint smear, but they do not show any results when gene-specific PCR primers are used to amplify genes from them. Additionally, the intensity of the cDNA smear can vary somewhat (compare Figure 1 and Figure 3a). In Figure 3a lanes a, f, g, and h contain robust cDNA smears while lanes b, c, d, and e are considerably less robust.
Gene-specific PCR is used as a secondary screen of the quality of the cDNA from a single cell. The cDNAs in Figure 3 were isolated from fluorescent retinal ganglion cells and they were tested using PCR primers for the retinal ganglion cell markers Brn3b and Pax6. For this assay, 1 μl of the cDNA was subjected to PCR for 30 cycles. Real-time quantitative PCR would be a preferable assay, but it can be prohibitively expensive and is not necessary for merely assessing the cDNA quality. All 8 of the single cells tested positive for Brn3b and 6 out 8 for Pax6 (Figure 3b). Even the cDNA smears that were less robust produced bands for Brn3b and Pax6. Despite these PCR results, it is our experience that cDNA smears such as those in lanes b, c, d, and e do not yield robust hybridizations on Affymetrix arrays and are generally avoided. Gene specific PCR for the photoreceptor marker Crx was used to determine the level of contamination in the single cell cDNAs (Figure 3b). A photoreceptor marker was chosen because these cells make up the majority of the retina (~70%) and, therefore, any cell lysis that occurred would most likely involve rod photoreceptors. There was only a faint amount of Crx present in these cDNA preparations even after 30 cycles of PCR. Finally, this PCR-based screen can be used to begin to determine which subsets of a particular cell type the cDNAs were derived from before subjecting them to a full microarray profiling. This can help to prioritize the cells that are profiled, as this is the most expensive step in the process. For example, we have identified subsets of retinal ganglion cells by screening them for the presence of the gene Tachykinin1 (Figure 3b).
From the small PCR-based screen, particular single cells are chosen and their cDNAs hybridized to an Affymetrix microarray. The microarray data is compared and patterns can be discovered using standard clustering algorithms. Heatmaps, such as in Figure 4, are generated to graphically represent the data. There are two main conclusions regarding the single cell profiling data in Figure 4. First, genes expressed in specific cell types are found almost exclusively in those cell types after the single cell protocol is performed. In most instances where marker genes of one cell type are found also in a second cell type, such as the observation that bipolar cells express some "rod" genes at lower levels, this expression in the second cell type is readily confirmed by either in situ hybridization or antibody staining. Second, within the more diverse amacrine and ganglion cell profiles, heterogeneity of gene expression is immediately apparent. Pou4f1 is only expressed in about 1/4 of the developing ganglion cells, while Nr4a2 and Scube2 are two examples of heterogeneously expressed genes in different amacrine cells. In fact, the genes shown in the heatmap are just a small sample as several hundred genes have been identified and confirmed as markers of developing ganglion cells or as markers of different populations of amacrine cells2,4.
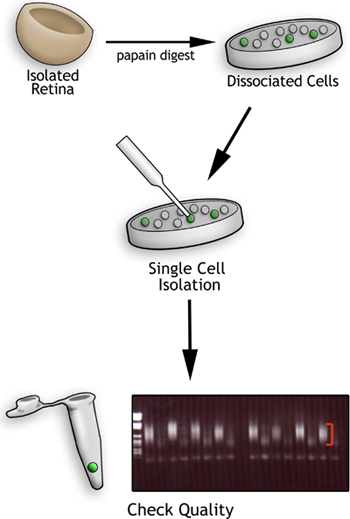 Figure 1. Flowchart of the single cell profiling method. First retinas are isolated and dissociated into individual cells using papain. Second, single cells are harvested with a pulled glass needle and deposited into PCR tubes. The cells are lysed and the cDNA amplified using reverse transcription followed by PCR. The resulting cDNA quality is assessed on an agarose gel such as the one shown. After the DNA ladder in the first lane, the lanes show cDNA smears from single cells (indicated by the red brackets) alternating with amplification products from media controls. Smears of this quality (red brackets) are hybridized to Affymetrix microarrays.
Figure 1. Flowchart of the single cell profiling method. First retinas are isolated and dissociated into individual cells using papain. Second, single cells are harvested with a pulled glass needle and deposited into PCR tubes. The cells are lysed and the cDNA amplified using reverse transcription followed by PCR. The resulting cDNA quality is assessed on an agarose gel such as the one shown. After the DNA ladder in the first lane, the lanes show cDNA smears from single cells (indicated by the red brackets) alternating with amplification products from media controls. Smears of this quality (red brackets) are hybridized to Affymetrix microarrays.
 Figure 2. Photos of the needles and aspirator tube assembly. Single cells are isolated using the capillary action of a pulled glass micropipette (inner diameter 0.5 mm, outer diameter 1.04 mm) and are transferred by the pressure of blowing into the aspirator. It is important that the aspirator tube is both long enough to easily manipulate and short enough to reliably discharge individual cells. A close-up view of the capillary tube pulled into a needle is shown in B.
Figure 2. Photos of the needles and aspirator tube assembly. Single cells are isolated using the capillary action of a pulled glass micropipette (inner diameter 0.5 mm, outer diameter 1.04 mm) and are transferred by the pressure of blowing into the aspirator. It is important that the aspirator tube is both long enough to easily manipulate and short enough to reliably discharge individual cells. A close-up view of the capillary tube pulled into a needle is shown in B.
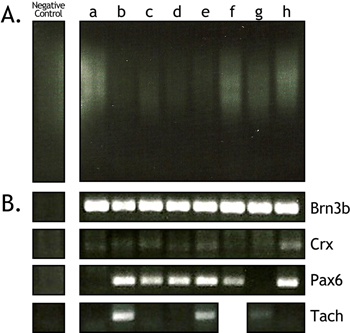 Figure 3. Representative example of single cell cDNA smears and gene specific PCRs. To determine the quality of the single cell cDNA after amplification, 10 μl of each single cell sample were run on a 1% agarose gel along with a negative control (A). A smear should appear between 500-2000 bp. Additional PCR-based screening for specific genes can help to identify/confirm the type of cell that was isolated and the amount of contamination present in the sample (B). Primers specific for the retinal ganglion cell markers Brn3b and Pax6 were tested to confirm the identity of these cells (Rows 1 and 2). To assess the amount of photoreceptor contamination in the preparation, primers specific for the photoreceptor marker Crx were used (Row 3). Subsets of ganglion cells may also be identified through screening for markers such as Tachykinin1 (Row 4).
Figure 3. Representative example of single cell cDNA smears and gene specific PCRs. To determine the quality of the single cell cDNA after amplification, 10 μl of each single cell sample were run on a 1% agarose gel along with a negative control (A). A smear should appear between 500-2000 bp. Additional PCR-based screening for specific genes can help to identify/confirm the type of cell that was isolated and the amount of contamination present in the sample (B). Primers specific for the retinal ganglion cell markers Brn3b and Pax6 were tested to confirm the identity of these cells (Rows 1 and 2). To assess the amount of photoreceptor contamination in the preparation, primers specific for the photoreceptor marker Crx were used (Row 3). Subsets of ganglion cells may also be identified through screening for markers such as Tachykinin1 (Row 4).
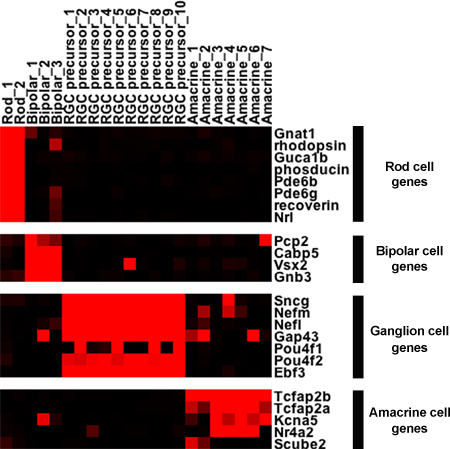 Figure 4. Single cell expression of marker genes. The microarray results for selected genes expressed in 22 distinct single cells are shown in a heatmap format. The intensities from the microarray signals have been scaled to correspond with the intensity of the red color. Black indicates the absence of signal on the microarray. The retinal ganglion cell (RGC) precursors shown were isolated from embryonic time points, while the other cells were isolated from adult retinas.
Figure 4. Single cell expression of marker genes. The microarray results for selected genes expressed in 22 distinct single cells are shown in a heatmap format. The intensities from the microarray signals have been scaled to correspond with the intensity of the red color. Black indicates the absence of signal on the microarray. The retinal ganglion cell (RGC) precursors shown were isolated from embryonic time points, while the other cells were isolated from adult retinas.
Discussion
An ever-expanding number of studies are revealing robust cell-to-cell variability in populations that were believed to be more homogeneous with regard to their gene expression6,8. In at least one instance, this gene expression "noise" has been shown to play an important biological function13. Gene expression differences between individual cells are obscured using traditional whole-tissue methods. These experiments generate the expression profile of an "average" cell, which may not be representative14. Here we present a method for the isolation and identification of the gene expression patterns from single retinal cells. The study of individual cells allows researchers to probe the cellular heterogeneity underlying complex tissues, which is critical for determining distinctive gene expression patterns of various cell types. Additionally, single-cell isolation can provide insight into the gene programs driving the activity of individual cells in time points throughout development. Though especially useful in studying the nervous system, where the functioning of complex cellular networks can depend on the unique gene expression of rare cell types, this protocol can be adapted to isolate individual cells from various tissues.
In the early-developing murine retina, ganglion cells, horizontal cells, cone photoreceptor cells and amacrine cells are all generated in an overlapping window of time15. Additionally, each cell that has exited the cell cycle is in a different stage of maturation and this fact only adds to the complexity of the developing retina. Finally, for some types of retinal neurons, there exist numerous different morphologies (up to ~30 in the case of amacrine cells) in the mature retina16. Since the retina is such a mixture of cell types, microarray and serial analysis of gene expression (SAGE) based studies17-19 that relied on the homogenization of the entire retina are unable to uncover markers genes expressed in rare subtypes and unable to resolve dynamic gene expression differences between cell types, especially during development. To begin to understand the complexity of different cell types both in the adult retina and during retinal development, the single cell gene expression profiles of ~200 individual cells from many different time points have been analyzed2-5,8. The resulting data has provided a host of new markers for individual cell types and provided a window into important transitions in developmental time that were previously unappreciated.
The single cell expression profiles have revealed considerable heterogeneity in gene expression in amacrine cells4, where it was expected, and in Müller glia cells, where it was unexpected5. While an examination of the single amacrine cell data revealed more than 450 genes that were expressed in amacrine cells and excluded from other retinal cell types, none of these genes were expressed in all the amacrine cells4. These results most likely arise from the fact that the amacrine cell class is extremely diverse and the underlying heterogeneity is a reflection of the distinct functions of these cells. These findings would not have been possible using whole tissue-based approaches. Finally, cycling retinal progenitor cells (RPCs) displayed the highest degree of heterogeneity in gene expression of any of the retinal cell types profiled8. Transcription factors were the class of genes with the highest degree of heterogeneity among the progenitor cells, suggesting that dynamic gene expression patterns could play a major role in the development of the different retinal cell types8. Again, these results would not have been possible without the use of the single cell gene expression profiling technique.
Single cell transcriptome studies can also be utilized to gain insight into disease mechanisms. In many neurodegenerative diseases, including retinal degenerative diseases, some neurons die while the neighboring neurons survive20. Understanding why some cells undergo apoptosis requires the identification of genes or gene programs that are altered in individual cells in these disease models. Whole-tissue models will again potentially obscure the changes since cells often do not die at the same time and, therefore, at any given time the tissue is a mixture of dying and surviving cells. Additionally, for many cancers the cell of origin is an open question. This is particularly the case with the childhood eye tumor retinoblastoma. Recently using the single cell profiling protocol detailed here, it was shown that individual cells from retinoblastomas possess gene expression programs of multiple cell types21. It appears that these cells are hybrids of undifferentiated progenitor cells and neurons21. These experiments required an analysis at the single cell level and would not have been possible with whole tissue approaches.
Alternative approaches
Linear amplification is an alternative to the PCR-based amplification protocol detailed here. While PCR-based amplification is believed to result in a skewing of abundance relationships, linear amplification is thought to maintain these relationships22. The technique of linear amplification has been used to profile several neuronal types23. However, direct comparisons between the two methods have indicated that the linear amplification technique can be associated with a high false negative rate24,25. We favor the PCR-based method detailed in this report for two reasons. First, in our experiments we focus on genes with robust expression differences between cells. Second, we have found a very good correlation between our single cell microarray profiling experiments and in situ hybridization studies for the same genes. In fact, to date we have performed in situ hybridizations for hundreds of genes and have observed at least 75% match the predicted pattern from the microarrays. This is most likely a conservative estimate since the most common reason for a discrepancy is a lack of signal from the in situ probe.
Disclosures
No conflicts of interest declared.
References
- Tietjen I. Single-cell transcriptional analysis of neuronal progenitors. Neuron. 2003;38:161–175. doi: 10.1016/s0896-6273(03)00229-0. [DOI] [PubMed] [Google Scholar]
- Trimarchi JM. Molecular heterogeneity of developing retinal ganglion and amacrine cells revealed through single cell gene expression profiling. J. Comp. Neurol. 2007;502:1047–1065. doi: 10.1002/cne.21368. [DOI] [PubMed] [Google Scholar]
- Trimarchi JM, Cho SH, Cepko CL. Identification of genes expressed preferentially in the developing peripheral margin of the optic cup. Dev. Dyn. 2009;238:2327–2327. doi: 10.1002/dvdy.21973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry TJ, Trimarchi JM, Stadler MB, Cepko CL. Development and diversification of retinal amacrine interneurons at single cell resolution. Proc. Natl. Acad. Sci. U.S.A. 2009;106:9495–9500. doi: 10.1073/pnas.0903264106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch K. The transcriptome of retinal Muller glial cells. J. Comp. Neurol. 2008;509:225–238. doi: 10.1002/cne.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos CA. Evidence for diversity in transcriptional profiles of single hematopoietic stem cells. PLoS Genet. 2006;2:e159. doi: 10.1371/journal.pgen.0020159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang MK, Melton DA. Single-cell transcript analysis of pancreas development. Dev. Cell. 2003;4:383–393. doi: 10.1016/s1534-5807(03)00035-2. [DOI] [PubMed] [Google Scholar]
- Trimarchi JM, Stadler MB, Cepko CL. Individual retinal progenitor cells display extensive heterogeneity of gene expression. PLoS ONE. 2008;3:e1588. doi: 10.1371/journal.pone.0001588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SB, Sugino K, Hempel CM. The problem of neuronal cell types: a physiological genomics approach. Trends Neurosci. 2006;29:339–345. doi: 10.1016/j.tins.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Stevens CF. Neuronal diversity: too many cell types for comfort. Curr. Biol. 1998;8:R708–R710. doi: 10.1016/s0960-9822(98)70454-3. [DOI] [PubMed] [Google Scholar]
- Cepko CL, Austin CP, Yang X, Alexiades M, Ezzeddine D. Cell fate determination in the vertebrate retina. Proc. Natl. Acad. Sci. U.S.A. 1996;93:589–595. doi: 10.1073/pnas.93.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS. Identification of molecular markers of bipolar cells in the murine retina. J. Comp. Neurol. 2008;507:1795–1810. doi: 10.1002/cne.21639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–547. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levsky JM, Singer RH. Gene expression and the myth of the average cell. Trends Cell Biol. 2003;13:4–6. doi: 10.1016/s0962-8924(02)00002-8. [DOI] [PubMed] [Google Scholar]
- Livesey FJ, Cepko CL. Vertebrate neural cell-fate determination: lessons from the retina. Nat. Rev. Neurosci. 2001;2:109–118. doi: 10.1038/35053522. [DOI] [PubMed] [Google Scholar]
- Masland RH, Raviola E. Confronting complexity: strategies for understanding the microcircuitry of the retina. Annu. Rev. Neurosci. 2000;23:249–284. doi: 10.1146/annurev.neuro.23.1.249. [DOI] [PubMed] [Google Scholar]
- Blackshaw S. Genomic analysis of mouse retinal development. PLoS Biol. 2004;2:e247. doi: 10.1371/journal.pbio.0020247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey FJ, Young TL, Cepko CL. An analysis of the gene expression program of mammalian neural progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1374–1379. doi: 10.1073/pnas.0307014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowers I. Identification of novel genes preferentially expressed in the retina using a custom human retina cDNA microarray. Invest. Ophthalmol. Vis. Sci. 2003;44:3732–3741. doi: 10.1167/iovs.02-1080. [DOI] [PubMed] [Google Scholar]
- Clarke GC, Leavitt RA, Andrews BR, Hayden DF, Lumsden MR, J C, McInnes RR. A one-hit model of cell death in inherited neuronal degenerations. Nature. 2000;406:195–199. doi: 10.1038/35018098. [DOI] [PubMed] [Google Scholar]
- McEvoy JF-O, Zhang J, Nemeth J, Brennan K, Bradley R, Krafcik C, Rodriguez-Galindo F, Wilson C, Xiong M, Lozano S. Coexpression of normally incompatible developmental pathways in retinoblastoma genesis. Cancer Cell. 2011;20:260–275. doi: 10.1016/j.ccr.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelder RNVan. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Che S. Expression profile analysis within the human hippocampus: comparison of CA1 and CA3 pyramidal neurons. J. Comp. Neurol. 2005;487:107–118. doi: 10.1002/cne.20535. [DOI] [PubMed] [Google Scholar]
- Iscove NN. Representation is faithfully preserved in global cDNA amplified exponentially from sub-picogram quantities of mRNA. Nat. Biotechnol. 2002;20:940–943. doi: 10.1038/nbt729. [DOI] [PubMed] [Google Scholar]
- Subkhankulova T, Livesey FJ. Comparative evaluation of linear and exponential amplification techniques for expression profiling at the single-cell level. Genome Biol. 2006;7:R18. doi: 10.1186/gb-2006-7-3-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]